3.5. Chemical Experimental Data
2-(1H-Indol-1-yl)-ethanol (3a).
To the boiling suspension of LiAlH4 (15.2 g, 0.4 mol) in THF (500 mL), the solution of 2-(1H-indol-1-yl)acetic acid 1a (17.56 g, 0.1 mol) in THF (100 mL) was gradually added, then the reaction mixture was refluxed for 5 h. After cooling to RT, the reaction mixture was quenched with KOH (20% aqueous solution), then was filtered and diluted with EtOAc (300 mL) and aqueous solution of citric acid (10.0 g in 100 mL) was added. The organic layer was separated, washed with water and brine, and evaporated in vacuo. The residue was purified by flash chromatography (50 g of silica gel) using EtOAc-hexane (1:10 to 1:1) as an eluent, to give 3a (13.2 g, 82%) as a colorless oil.
1H NMR: δ 7.52 (dt, 1H, J = 7.8, 1.1 Hz), 7.48–7.41 (m, 1H), 7.33 (d, 1H, J = 3.1 Hz), 7.10 (ddd, 1H, J = 8.2, 7.0, 1.3 Hz), 6.99 (ddd, 1H, J = 8.0, 7.0, 1.0 Hz), 6.40 (dd, 1H, J = 3.1, 0.9 Hz), 4.93 (t, 1H, J = 5.3 Hz), 4.19 (t, 2H, J = 5.7 Hz), 3.70 (q, 2H, J = 5.5 Hz). 13C NMR: δ 136.33, 129.59, 128.53, 121.26, 120.73, 119.24, 110.32, 100.67, 60.76, 48.66, 48.64. HRMS (EI) m/z [M + H]+ Calcd for C10H12NO+ 162.0913; Found 162.0923.
3-(1H-Indol-1-yl)propan-1-ol (3b).
The same procedure as above was carried out using 3-(1H-indol-1-yl)propanoic acid (18.9 g, 0.1 mol), to give 3b (13.8 g, 79%) as a colorless oil.
1H NMR: δ 7.53 (dt, 1H, J = 7.8, 1.0 Hz), 7.44 (dd, 1H, J = 8.3, 1.0 Hz), 7.32 (d, 1H, J = 3.1 Hz), 7.14–7.07 (m, 1H), 7.04–6.97 (m, 1H), 6.41 (dd, 1H, J = 3.2, 0.9 Hz), 4.69 (s, 1H), 4.21 (t, 2H, J = 6.9 Hz), 3.37 (d, 2H, J = 5.2 Hz), 1.88 (t, 2H, J = 6.6 Hz). 13C NMR: δ 136.08, 129.12, 128.53, 121.38, 120.85, 119.27, 110.17, 100.82, 58.27, 42.84, 33.43. HRMS (EI) m/z [M + H]+ Calcd for C11H14NO+ 176.1070; Found 176.1073.
4-(1H-Indol-1-yl)butan-1-ol (3c).
To the suspension of KOH (20 g, 0.35 mol) in DMSO (100 mL), indole (11.7 g, 0.1 mol) and 4-bromobutan-1-ol (16.8 g, 0.11 mol) were added. After intensive stirring at RT for 5 h, the reaction mixture was filtered, diluted with EtOAc (300 mL), and washed with an aqueous solution of citric acid (10.0 g, 100 mL). The organic layer was separated, washed with water and brine, and evaporated in vacuo. The residue was purified by flash chromatography (50 g of silica gel) using EtOAc-Hexane (1:10 to 1:1) as an eluent, to give 3c (17.5 g, 93%) as a colorless oil.
1H NMR: δ 7.53 (dt, 1H, J = 7.8, 1.0 Hz), 7.44 (dd, 1H, J = 8.3, 1.1 Hz), 7.33 (d, 1H, J = 3.1 Hz), 7.11 (ddd, 1H, J = 8.2, 6.9, 1.2 Hz), 7.00 (ddd, 1H, J = 8.0, 7.0, 1.0 Hz), 6.41 (dd, 1H, J = 3.1, 0.9 Hz), 4.47 (t, 1H, J = 5.1 Hz), 4.15 (t, 2H, J = 7.1 Hz), 3.39 (td, 2H, J = 6.4, 4.9 Hz), 1.77 (dq, 2H, J = 9.6, 7.2 Hz), 1.43–1.32 (m, 2H). 13C NMR: δ 136.11, 129.00, 128.57, 121.34, 120.85, 119.23, 110.20, 100.79, 60.80, 45.84, 30.21, 27.09. HRMS (EI) m/z [M + H]+ Calcd for C12H18NO+ 190.1226; Found 190.1228.
5-(1H-Indol-1-yl)pentan-1-ol (3d).
The same procedure as above was carried out using indole (11.7 g, 0.1 mol) and 5-bromopentan-1-ol (18.3 g, 0.11 mol), to give 3d (18.1 g, 89%) as a colorless oil.
1H NMR: δ 7.54 (dt, 1H, J = 7.9, 1.0 Hz), 7.42 (dd, 1H, J = 8.2, 1.1 Hz), 7.31 (d, 1H, J = 3.2 Hz), 7.12 (ddd, 1H, J = 8.2, 7.0, 1.3 Hz), 7.01 (ddd, 1H, J = 8.0, 6.9, 1.0 Hz), 6.41 (dd, 1H, J = 3.1, 0.9 Hz), 4.43 (s, 1H), 4.11 (t, 2H, J = 7.0 Hz), 3.37 (t, 2H, J = 6.5 Hz), 1.73 (p, 2H, J = 7.2 Hz), 1.49–1.37 (m, 2H), 1.33–1.19 (m, 2H). 13C NMR: δ 136.10, 128.97, 128.57, 121.35, 120.86, 119.23, 110.14, 100.80, 61.06, 45.95, 32.54, 30.23, 23.37. HRMS (EI) m/z [M + H]+ Calcd for C13H18NO+ 204.1383; Found 204.1381.
6-(1H-Indol-1-yl)hexan-1-ol (3e).
The same procedure as above was carried out using indole (11.7 g, 0.1 mol) and 6-bromohexan-1-ol (20.0 g, 0.11 mol), to give 3e (19.5 g, 90%) as a colorless oil.
1H NMR: δ 7.22–7.15 (m, 1H), 7.09–6.98 (m, 3H), 3.52 (t, 1H, J = 6.8 Hz), 2.85 (t, 1H, J = 7.2 Hz), 2.20 (d, 3H, J = 11.3 Hz), 1.77 (p, 1H, J = 6.9 Hz). 13C NMR: δ 170.04, 165.67, 151.95, 136.37, 135.88, 134.80, 132.56, 130.15, 129.63, 125.82, 42.19, 37.21, 30.97, 28.14, 20.96, 20.84. HRMS (EI) m/z [M + H]+ Calcd for C14H20NO+ 218.1539; Found 218.1540.
2-(1H-Indol-1-yl)ethyl acetate (4a).
To the solution of 2-indol-1-yl-ethanol 3a (12 g, 75 mmol) in pyridine (100 mL), Ac2O (8 mL, 10 mmol) and DMAP (100 mg, 0.08 mmol) were added. After stirring at rt for 5 h, the reaction mixture was evaporated in vacuo, diluted with EtOAc (300 mL) and washed with an aqueous solution of citric acid (1.0 g in 100 mL). The organic layer was separated, washed with water and brine and evaporated in vacuo. The residue was purified by flash chromatography (100 g of silica gel) using EtOAc-Hexane (1:10 to 1:3) as an eluent, to give 4a (14.7 g, 97%) as a colorless oil. 1H NMR: δ 7.63 (d, 1H, J = 7.8 Hz), 7.50 (dd, 1H, J = 8.3, 0.6 Hz), 7.35 (d, 1H, J = 3.2 Hz), 7.26–7.16 (m, 1H), 7.11 (td, 1H, J = 7.5, 0.9 Hz), 6.52 (dd, 1H, J = 3.1, 0.7 Hz), 4.37 (qd, 4H, J = 6.2, 1.5 Hz), 1.93 (s, 3H). 13C NMR: δ 170.53, 136.43, 129.16, 128.75, 121.63, 120.96, 119.58, 110.09, 101.50, 63.40, 44.93, 20.86.
3-(1H-Indol-1-yl)propyl acetate (4b).
The same procedure as above was carried out using 3-indol-1-yl-propan-1-ol 3b (7.0 g, 40 mmol), to give 4b (8.1 g, 94%) as a colorless oil. 1H NMR: δ 7.62 (d, 1H, J = 7.8 Hz), 7.46 (d, 1H, J = 8.2 Hz), 7.31 (d, 1H, J = 3.1 Hz), 7.24–7.15 (m, 1H), 7.15–7.06 (m, 1H), 6.55 – 6.47 (m, 1H), 4.22 (t, 2H, J = 6.8 Hz), 3.96 (t, 2H, J = 6.4 Hz), 2.14–1.96 (m, 5H). 13C NMR: δ 170.75, 136.20, 128.81, 128.74, 121.54, 120.97, 119.43, 110.00, 101.23, 61.71, 42.73, 29.29, 20.92.
4-(1H-Indol-1-yl)butyl acetate (4c).
The same procedure as above was carried out using 4-(1H-indol-1-yl)butan-1-ol 3c (10.0 g, 53 mmol), to give 4c (11.7 g, 96%) as a colorless oil. 1H NMR: δ 7.53 (d, 1H, J = 7.8 Hz), 7.47–7.39 (m, 1H), 7.32 (d, 1H, J = 3.1 Hz), 7.15–7.05 (m, 1H), 7.04–6.96 (m, 1H), 6.41 (dd, 1H, J = 3.1, 0.7 Hz), 4.15 (t, 2H, J = 7.0 Hz), 3.95 (t, 2H, J = 6.6 Hz), 1.94 (s, 3H), 1.92–1.69 (m, 2H), 1.69–1.42 (m, 2H). 13C NMR: δ 170.85, 136.06, 128.93, 128.55, 121.40, 120.86, 119.28, 110.12, 100.92, 100.89, 85.49, 63.84, 45.44, 26.84, 25.99, 21.06, 21.05.
5-(1H-Indol-1-yl)pentyl acetate (4d).
The same procedure as above was carried out using 5-(1H-indol-1-yl)pentan-1-ol 3d (10.0 g, 49 mmol), to give 4d (11.4 g, 95%) as a colorless oil. 1H NMR: δ 7.52 (d, J = 7.8 Hz, 2H), 7.47–7.38 (m, 2H), 7.32 (d, J = 3.1 Hz, 2H), 7.15–7.05 (m, 2H), 7.04–6.94 (m, 2H), 6.40 (dd, J = 3.0, 0.6 Hz, 2H), 4.12 (t, J = 7.0 Hz, 4H), 3.92 (t, J = 6.6 Hz, 4H), 2.48 (s, 1H), 1.94 (s, 6H), 1.82–1.64 (m, 4H), 1.62–1.47 (m, 4H), 1.23 (dd, J = 9.2, 6.2 Hz, 4H). 13C NMR: δ 170.84, 136.03, 128.97, 128.51, 121.33, 120.83, 119.22, 110.14, 100.77, 64.07, 45.69, 29.86, 28.10, 23.15, 21.10.
6-(1H-Indol-1-yl)hexyl acetate (4e).
The same procedure as above was carried out using 6-(1H-indol-1-yl)hexan-1-ol 3e (10.0 g, 46 mmol), to give 4e (10.97 g, 93%) as a colorless amorphous solid. 1H NMR: δ 7.51 (d, 2H, J = 7.8 Hz), 7.42 (d, 2H, J = 8.2 Hz), 7.31 (d, 2H, J = 3.0 Hz), 7.09 (d, 2H, J = 7.3 Hz), 6.99 (d, 2H, J = 7.3 Hz), 6.39 (d, 2H, J = 2.7 Hz), 4.12 (t, 4H, J = 7.0 Hz), 3.92 (t, 4H, J = 6.6 Hz), 1.95 (s, 6H), 1.72 (d, 3H, J = 7.2 Hz), 1.69–1.25 (m, 10H), 1.25–0.98 (m, 5H). 13C NMR: δ 170.86, 136.05, 128.95, 128.51, 121.31, 120.82, 119.20, 110.11, 100.75, 64.16, 45.76, 30.15, 28.44, 26.34, 25.45, 21.11.
2-(3-Formyl-1H-indol-1-yl)ethyl acetate (5a).
2-(1H-Indol-1-yl)ethyl acetate 4a (14.7 g, 72 mmol) was dissolved in the solution of POCl3 (0.9 mL, 10 mmol) in DMF (50 mL) and intensively stirred at 5 °C for 5 h. The reaction mixture was quenched with Na2CO3 (10% aqueous solution), diluted with EtOAc (100 mL) and water (200 mL). The organic layer was separated and the water layer was re-extracted with EtOAc (100 mL). The combined extracts were washed with water and brine and evaporated in vacuo. The residue was purified by flash chromatography (100 g of silica gel) using EtOAc-Hexane (1:5 to 1:1) as an eluent to give 5a (11.9 g, 71%) as a colorless oil. 1H NMR: δ 9.94 (s, 1H), 8.27 (s, 1H), 8.16 (d, 1H, J = 7.3 Hz), 7.61 (d, 1H, J = 7.9 Hz), 7.36–7.21 (m, 2H), 4.51 (t, 2H, J = 5.0 Hz), 4.38 (t, 2H, J = 5.1 Hz), 1.88 (s, 3H). 13C NMR: δ 185.18, 170.48, 141.46, 137.60, 125.09, 124.04, 122.98, 121.56, 117.98, 111.35, 62.73, 45.81, 20.84.
3-(3-Formyl-1H-indol-1-yl)propyl acetate (5b).
The same procedure as above was carried out using 3-(1H-indol-1-yl)propylacetate 4b (8.0 g, 36.8 mmol), to give 5b (6.1 g, 68%) as a colorless amorphous solid. 1H NMR δ 9.91 (s, 1H), 8.27 (s, 1H), 8.19–8.11 (m, 1H), 7.58 (d, J = 8.0 Hz, 1H), 7.33–7.21 (m, 2H), 4.32 (t, J = 6.9 Hz, 2H), 3.96 (t, J = 6.2 Hz, 2H), 3.53 (s, 2H), 2.18–2.02 (m, 2H), 1.91 (s, 3H). 13C NMR: δ 184.98, 170.77, 170.76, 141.11, 137.43, 125.13, 124.00, 122.92, 121.56, 117.73, 111.29, 61.61, 43.80, 28.75, 20.91.
4-(3-Formyl-1H-indol-1-yl)butyl acetate (5c).
The same procedure as above was carried out using 4-(1H-indol-1-yl)butyl acetate 4c (11.7 g, 50 mmol), to give 5c (9.7 g, 74%) as a colorless amorphous solid. 1H NMR: δ 9.89 (s, 1H), 8.30 (s, 1H), 8.10 (d, 1H, J = 7.5 Hz), 7.61 (d, 1H, J = 8.1 Hz), 7.33–7.20 (m, 2H), 4.28 (t, 2H, J = 7.1 Hz), 3.98 (t, 2H, J = 6.6 Hz), 1.94 (s, 3H), 1.91–1.75 (m, 2H), 1.75–1.47 (m, 2H). 13C NMR: δ 184.99, 170.84, 141.12, 137.41, 125.11, 123.97, 122.91, 121.51, 117.55, 111.46, 85.48, 63.71, 46.32, 26.39, 25.83, 21.08.
5-(3-Formyl-1H-indol-1-yl)pentyl acetate (5d).
The same procedure as above was carried out using 5-(1H-indol-1-yl)pentyl acetate 4d (11.4 g, 46 mmol), to give 5d (9.78 g, 77%) as a colorless amorphous solid. 1H NMR: δ 9.90 (s, 1H), 8.30 (s, 1H), 8.12 (d, 1H, J = 7.4 Hz), 7.61 (d, 1H, J = 8.1 Hz), 7.34–7.22 (m, 2H), 4.26 (t, 2H, J = 7.1 Hz), 3.95 (t, 2H, J = 6.6 Hz), 1.94 (s, 3H), 1.88–1.73 (m, 2H), 1.64–1.51 (m, 2H), 1.34–1.06 (m, 2H). 13C NMR: δ 184.57, 170.46, 140.76, 137.06, 124.74, 123.57, 122.50, 121.13, 117.13, 111.09, 63.61, 46.18, 28.94, 27.63, 22.60, 20.70.
6-(3-Formyl-1H-indol-1-yl)hexyl acetate (5e).
The same procedure as above was carried out using 6-(1H-indol-1-yl)hexyl acetate 4e (10.9 g, 42 mmol), to give 5e (8.8 g, 73%) as a colorless amorphous solid. 1H NMR: δ 9.87 (s, 1H), 8.30 (s, 1H), 8.09 (d, 1H, J = 7.6 Hz), 7.61 (d, 1H, J = 8.2 Hz), 7.29 (dt, 2H, J = 8.2, 1.3 Hz), 7.23 (dt, 2H, J = 7.0, 1.0 Hz), 4.29 (t, 2H, J = 7.1 Hz), 3.92 (t, 2H, J = 7.0 Hz), 1.94 (s, 3H), 1.82–1.75 (m, 2H), 1.53–1.46 (m, 2H) 1.34–1.22 (m, 4H). 13C NMR: δ 184.89, 170.55, 140.86, 137.15, 124.88, 123.53, 122.59, 121.19, 117.20, 111.01, 64.14, 46.64, 29.59, 28.37, 26.14, 25.38, 21.13.
1-(2-Hydroxyethyl)-1H-indole-3-carbaldehyde (6a).
2-(3-Formyl-1H-indol-1-yl)ethyl acetate 5a (11.9 g, 51 mmol) was dissolved in the solution Na (200 mg, 0.8 mmol) in MeOH (50 mL) and stirred at 10 min. Then, the reaction mixture was evaporated in vacuo, quenched with an aqueous solution of citric acid (0.5 g, 50 mL), and EtOAc (200 mL). The organic layer was separated and the water layer was re-extracted with EtOAc (100 mL). The extracts were combined, washed with water and brine and evaporated in vacuo. The residue was purified by flash chromatography (50 g of silica gel) using EtOAc-Hexane (1:5 to 1:0) as an eluent, to give 6a (8.8 g, 91%) as a colorless amorphous solid. 1H NMR: δ 9.90 (s, 1H), 8.24 (s, 1H), 8.14–8.08 (m, 1H), 7.60 (d, 1H, J = 8.0 Hz), 7.33–7.20 (m, 2H), 5.03 (t, 1H, J = 5.2 Hz), 4.30 (t, 2H, J = 5.3 Hz), 3.76 (q, 2H, J = 5.2 Hz). 13C NMR: δ 185.08, 141.97, 137.70, 125.14, 123.83, 122.84, 121.42, 117.41, 111.63, 60.04, 49.51. HRMS (EI) m/z [M + H]+ Calcd for C11H12NO2+ 190.0863; Found 190.0872.
1-(3-Hydroxypropyl)-1H-indole-3-carbaldehyde (6b).
The same procedure as above was carried out using 3-(3-Formyl-1H-indol-1-yl)propyl acetate (5b) (6.0 g, 24 mmol), to give 6b (4.5 g, 92%) as a colorless amorphous solid. 1H NMR: δ 9.89 (s, 1H), 8.26 (s, 1H), 8.10 (d, 1H, J = 7.5 Hz), 7.59 (d, 1H, J = 8.1 Hz), 7.35–7.21 (m, 2H), 4.73 (t, 1H, J = 5.0), 4.32 (t, 2H, J = 7.0 Hz), 3.39 (dd, 2H, J = 11.2, 5.9 Hz), 1.94 (p, 2H, J = 6.5 Hz). 13C NMR: δ 184.99, 141.29, 137.45, 125.11, 123.96, 122.90, 121.50, 117.48, 111.44, 57.99, 43.82, 32.79. HRMS (EI) m/z [M + H]+ Calcd for C12H14NO2+ 204.1019; Found 204.1030.
1-(4-Hydroxybutyl)-1H-indole-3-carbaldehyde (6c).
The same procedure as above was carried out using 4-(3-formyl-1H-indol-1-yl)butyl acetate 5c (5.0 g, 19 mmol), to give 6c (3.7 g, 90%) as a colorless amorphous solid. 1H NMR: δ 9.89 (s, 1H), 8.29 (s, 1H), 8.10 (d, 1H, J = 7.6 Hz), 7.60 (d, 1H, J = 8.1 Hz), 7.33–7.21 (m, 2H), 4.47 (t, 1H, J = 5.1 Hz), 4.27 (t, 2H, J = 7.1 Hz), 1.90–1.76 (m, 2H), 1.47–1.33 (m, 2H). 13C NMR: δ 184.93, 141.11, 137.46, 125.14, 123.93, 122.86, 121.49, 117.49, 111.49, 60.64, 46.69, 29.94, 26.60. HRMS (EI) m/z [M + H]+ Calcd for C13H16NO2+ 218.1176; Found 218.1186.
1-(5-Hydroxypentyl)-1H-indole-3-carbaldehyde (6d).
The same procedure as above was carried out using 5-(3-formyl-1H-indol-1-yl)pentyl acetate 5d (5.0 g, 18 mmol), to give 6d (3.9 g, 93%) as a colorless amorphous solid. 1H NMR: δ 9.88 (s, 1H), 8.29 (s, 1H), 8.13–8.06 (m, 1H), 7.59 (d, 1H, J = 8.2 Hz), 7.32–7.20 (m, 2H), 4.41 (br, 1H), 4.24 (t, 2H, J = 7.0 Hz), 3.34 (t, 2H, J = 6.4 Hz), 1.78 (p, 2H, J = 7.2 Hz), 1.41 (p, 2H, J = 6.7 Hz), 1.31–1.20 (m, 2H). 13C NMR (100 MHz, dmso) δ 185.00, 141.21, 137.46, 125.12, 123.96, 122.89, 121.51, 117.47, 111.49, 106.48, 60.90, 46.79, 32.36, 29.66, 23.18. HRMS (EI) m/z [M + H]+ Calcd for C14H18NO2+ 232.1332; Found 232.1338.
1-(6-Hydroxyhexyl)-1H-indole-3-carbaldehyde (6e).
The same procedure as above was carried out using 6-(3-formyl-1H-indol-1-yl)hexyl acetate 5e (6.0 g, 20 mmol), to give 6e (4.8 g, 94%) as a colorless amorphous solid. 1H NMR: δ 9.88 (s, 1H), 8.29 (s, 1H), 8.09 (d, 1H, J = 7.7 Hz), 7.59 (d, 1H, J = 8.1 Hz), 7.33–7.19 (m, 2H), 4.38 (s, 1H), 4.24 (t, 2H J = 7.1 Hz), 3.33 (t, 2H, J = 6.3 Hz), 1.77 (p, 2H, J = 7.1 Hz), 1.41–1.19 (m, 6H). 13C NMR: δ 185.11, 140.95, 137.09, 125.33, 123.99, 122.77, 121.82, 117.40, 111.85, 61.02, 45.91, 32.65, 30.34, 26.75, 26.77. HRMS (EI) m/z [M + H]+ Calcd for C15H20NO2+ 246.1489; Found 246.1483.
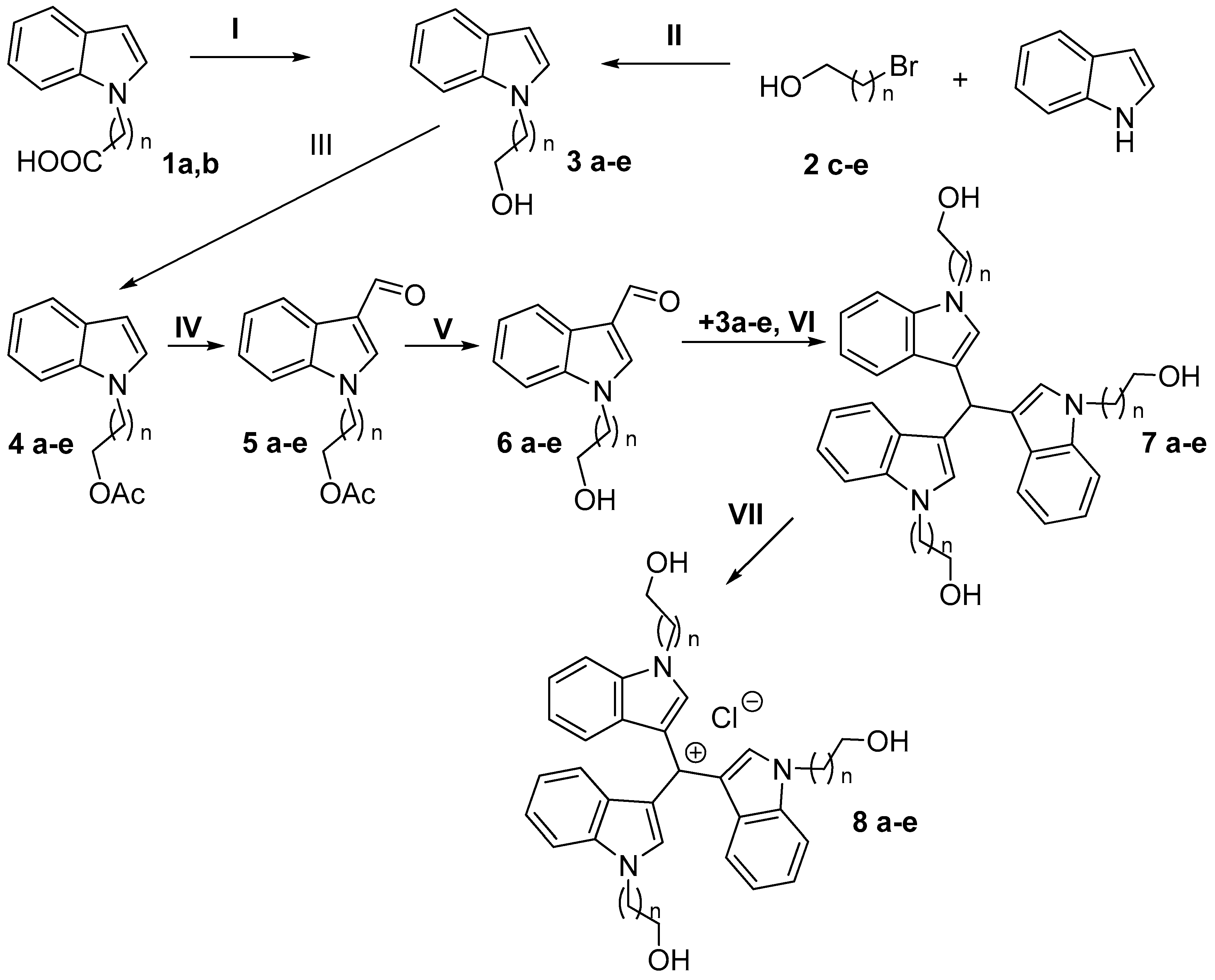
tris(1-[2-Hydroxyethyl]-1H-indol-3-yl)methane (7a).
To the solution of 2-indol-1-yl-ethanol 3a (3.0 g, 18.7 mmol) in MeOH (100 mL), 1-(2-hydroxyethyl)-1H-indole-3-carbaldehyde 6a (1.7 g, 9.1 mmol), AcOH (1 mL), and Dy(OTf)3 (10 mg, 16.4 μmol) were added. After refluxing for 12 h, the reaction mixture was cooled to RT. The resulting suspension was filtered, the precipitate washed with MeOH (2 × 50 mL) and Et2O (50 mL). The residue was dried in vacuo, to give 7a (3.9 g, 89%) as a colorless amorphous solid.
1H NMR: δ 7.52–7.32 (m, 6H), 7.05 (t, 3H, J = 7.5 Hz), 7.01 (s, 3H), 6.88 (t, 3H, J = 7.3 Hz), 6.03 (s, 1H), 4.80 (t, 3H, J = 5.0 Hz), 4.11 (t, 6H, J = 5.1 Hz), 3.64 (d, 6H, J = 5.3 Hz). 13C NMR: δ 136.48, 127.25, 127.11, 120.59, 119.39, 118.0, 117.33, 109.77, 60.36, 48.12. HRMS (EI) m/z [M − H]+ Calcd for C34H37N3O3+ 493.2365; Found 492.2282.
tris(1-[3-Hydroxypropyl]-1H-indol-3-yl)methane (7b).
The same procedure as above was carried out using 3-indol-1-yl-propan-1-ol 3b (3.0 g, 17.1 mmol) and 1-(3-hydroxypropyl)-1H-indole-3-carbaldehyde 6b (1.7 g, 8.5 mmol), to give 7b (3.8 g, 85%) as a colorless amorphous solid. 1H NMR: δ 7.39 (t, 6H, J = 7.5 Hz), 7.05 (d, 3H, J = 7.9 Hz), 6.98 (s, 3H), 6.88 (d, 3H, J = 7.7 Hz), 6.03 (s, 1H), 4.52 (t, 3H, J = 5.0 Hz), 4.13 (t, 6H, J = 6.8 Hz), 3.42–3.23 (m, 6H), 1.89–1.67 (m, 6H). 13C NMR: δ 136.2, 127.05, 126.69, 120.71, 119.55, 117.99, 117.24, 109.63, 57.79, 42.2, 33.03. HRMS (EI) m/z [M − H] + Calcd for C34H37N3O3+ 535.2835; Found 534.2747.
tris(1-[4-Hydroxybutyl]-1H-indol-3-yl)methane (7c).
The same procedure as above was carried out using 4-indol-1-yl-butan-1-ol 3c (3.0 g, 15.8 mmol) and 1-(4-Hydroxybutyl)-1H-indole-3-carbaldehyde 6c (1.6 g, 7.8 mmol), to give 7c (3.7 g, 82%) as a colorless amorphous solid. 1H NMR: δ 7.40 (d, J = 2.8 Hz, 3H), 7.38 (d, J = 3.3 Hz, 3H), 7.05 (t, J = 7.5 Hz, 3H), 6.96 (s, 3H), 6.86 (t, J = 7.4 Hz, 3H), 6.03 (s, 1H), 4.44 (t, J = 5.0 Hz, 3H), 4.07 (t, J = 6.8 Hz, 6H), 3.35 (q, J = 6.1 Hz, 6H), 1.90–1.48 (m, 6H), 1.48–1.11 (m, 6H). 13C NMR: δ 157.88, 144.03, 138.55, 124.44, 123.13, 120.55, 112.03, 59.90, 46.90, 29.14, 25.85. HRMS (EI) m/z [M − H] + Calcd for C37H43N3O3+ 577.3304; Found 576.3213.
tris(1-[5-Hydroxypentyl]-1H-indol-3-yl)methane (7d).
The same procedure as above was carried out using 5-indol-1-yl-pentan-1-ol 3d (3.0 g, 15.8 mmol) and 1-(5-hydroxypentyl)-1H-indole-3-carbaldehyde 5d (1.6 g, 7.8 mmol), to give 7d (4.2 g, 88%) as a colorless amorphous solid. 1H NMR: δ 7.38 (d, 6H, J = 5.3 Hz), 7.05 (t, 3H, J = 7.3 Hz), 6.95 (s, 3H), 6.86 (t, 3H, J = 7.3 Hz), 6.02 (s, 1H), 4.33 (t, 3H, J = 4.5 Hz), 4.06 (br, 6H), 1.65 (t, 6H, J = 6.8 Hz), 1.39 (t, 6H, J = 6.7 Hz), 1.37–0.95 (m, 6H). 13C NMR: δ 136.2, 127.05, 126.67, 120.68, 119.59, 117.91, 117.14, 109.64, 60.56, 45.24, 32.07, 29.69, 22.76. HRMS (EI) m/z [M] + Calcd for C40H49N3O3+ 619.3774; Found 618.3677.
tris(1-[6-Hydroxyhexyl]-1H-indol-3-yl)methane (7e).
The same procedure as above was carried out using 6-indol-1-yl-hexan-1-ol 3e (3.0 g, 13.8 mmol) and 1-(6-hydroxyhexyl)-1H-indole-3-carbaldehyde 6e (1.7 g, 6.9 mmol), to give 7e (3.7 g, 83%) as a colorless amorphous solid. 1H NMR: δ 7.38 (d, 6H, J = 8.6 Hz), 7.05 (t, 3H, J = 7.6 Hz), 6.93 (s, 3H), 6.85 (t, 3H, J = 7.7 Hz), 6.02 (s, 1H), 4.30 (t, 3H, J = 5.1 Hz), 4.06 (t, 6H, J = 6.7 Hz), 3.40–3.20 (m, 6H), 1.73–1.52 (m, 6H), 1.40–1.05 (m, 18H). 13C NMR (100 MHz, DMSO) δ = 136.18, 127.03, 126.67, 120.67, 119.56, 117.89, 117.09, 109.63, 60.54, 45.15, 32.43, 29.80, 26.07, 25.08. HRMS (EI) m/z [M]+ Calcd for C43H55N3O3+ 661.4243; Found 660.4160.
tris(1-(2-Hydroxyethyl)-1H-indol-3-yl)methylium chloride (8a).
To the solution of tris(1-[2-hydroxyethyl]-1H-indol-3-yl)methane 7a (2.0 g, 4.0 mmol) in THF (50 mL), DDQ (0.9 g, 4.0 mmol) was added. After stirring at rt for 1 h, the reaction mixture was quenched with conc. HCl (0.4 mL, 5 mmol) and evaporated in vacuo. The residue was purified by flash chromatography (100 g of silica gel) using CH2Cl2-MeOH (100:1 to 10:1) as an eluent to give 8a (1.6 g, 78%) as a red amorphous solid. Rt = 3.39 min. 1H NMR: δ 8.35 (s, 3H), 7.84 (d, 3H, J = 8.3 Hz), 7.40 (t, 3H, J = 7.9 Hz), 7.15–7.05 (m, 6H), 4.92 (s, 3H), 4.51 (t, 6H, J = 5.2 Hz), 3.93 (t, 6H). 13C NMR: δ 144.8, 138.69, 126.74, 124.22, 122.98, 120.60, 117.42, 112.00, 59.12, 49.57. HRMS (EI) m/z [M]+ Calcd for C31H30N3O3+ 492.2282; Found 492.2270.
tris(1-(3-Hydroxypropyl)-1H-indol-3-yl)methylium chloride (8b).
The same procedure as above was carried out using tris(1-[3-hydroxypropyl]-1H-indol-3-yl)methane 7b (2.0 g, 3.7 mmol), to give 8b (1.6 g, 73%) as a red amorphous solid. Rt = 3.55 min. 1H NMR: δ 8.38 (s, 3H), 7.83 (d, 3H, J = 8.3 Hz), 7.41 (t, 3H, J = 7.6 Hz), 7.13 (t, 3H, J = 7.6 Hz), 7.02 (s, 3H), 4.52 (t, 6H, J = 6.9 Hz), 4.46 (br, 3H), 3.56 (t, 6H, J = 5.9 Hz), 2.32–1.92 (m, 6H). 13C NMR: δ 157.77, 144.25, 138.49, 126.7, 124.35, 123.06, 120.49, 111.89, 57.47, 44.22, 31.89. HRMS (EI) m/z [M]+ Calcd for C34H36N3O3+ 534.2751; Found 534.2745.
tris(1-(4-Hydroxybutyl)-1H-indol-3-yl)methylium chloride (8c).
The same procedure as above was carried out using tris(1-[4-hydroxybutyl]-1H-indol-3- yl)methane 7c (2.0 g, 3.4 mmol), to give 8c (1.7 g, 81%) as a red amorphous solid. Rt = 8.76 min. 1H NMR: δ 8.42 (s, 3H), 7.83 (d, J = 8.3 Hz, 3H), 7.40 (t, J = 7.6 Hz, 3H), 7.12 (t, J = 7.5 Hz, 3H), 6.99 (d, J = 7.4 Hz, 3H), 4.49 (t, J = 7.1 Hz, 6H), 4.37 (s, 3H), 3.49 (t, J = 6.2 Hz, 6H), 2.15–1.90 (m, 6H), 1.68–1.47 (m, 6H). 13C NMR: δ 157.88, 144.03, 138.55, 126.78, 124.44, 123.13, 120.55, 117.4, 112.03, 59.90, 46.90, 29.14, 25.85. HRMS (EI) m/z [M]+ Calcd for C37H42N3O3+ 576.3221; Found 576.3227.
tris(1-(5-Hydroxypentyl)-1H-indol-3-yl)methylium chloride (8d).
The same procedure as above was carried out using tris(1-[5-hydroxypentyl]-1H-indol-3-yl)methane 7d (2.0 g, 3.2 mmol), to give 8d (1.8 g, 86%) as a red amorphous solid. Rt = 11.94 min. 1H NMR: δ 8.42 (s, 3H), 7.84 (d, 3H, J = 8.3 Hz), 7.41 (t, 3H, J = 7.6 Hz), 7.13 (t, 3H, J = 7.5 Hz), 7.01 (s, 3H), 4.46 (t, 6H, J = 7.0 Hz), 4.17 (s, 3H), 3.44 (br, 6H), 2.20–1.76 (m, 6H), 1.57–1.40 (m, 12H). 13C NMR: δ 138.47, 124.35, 123.06, 120.48, 111.94, 60.12, 46.93, 31.52, 28.73, 22.38. HRMS (EI) m/z [M]+ Calcd for C40H48N3O3+ 618.3690; Found 618.3682.
tris(1-(6-hydroxyhexyl)-1H-indol-3-yl)methylium chloride (8e).
The same procedure as above was carried out using tris(1-[6-hydroxyhexyl]-1H-indol-3- yl)methane 7e (2.0 g, 3.0 mmol), to give 8e (1.7 g, 81%) as a red amorphous solid. Rt = 16.08 min. 1H NMR: δ 8.42 (s, 3H), 7.84 (d, J = 8.3 Hz, 3H), 7.41 (t, J = 7.7 Hz, 3H), 7.12 (t, J = 7.5 Hz, 3H), 7.00 (d, J = 7.1 Hz, 3H), 4.45 (t, J = 7.1 Hz, 6H), 4.09 (br, 3H), 3.41 (t, J = 6.1 Hz, 6H), 2.17–1.75 (m, 6H), 1.50–1.35 (m, 18H). 13C NMR: δ 157.82, 143.94, 138.49, 126.76, 124.36, 123.03, 120.44, 111.93, 60.24, 46.89, 31.87, 28.87, 25.61, 24.68. HRMS (EI) m/z [M]+ Calcd for C43H54N3O3+ 660.4160; Found 660.4150.




{kind=link}
{kind=link}