The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors


1
Faculty of Kinesiology, University of Calgary, Calgary, AB T2N 4N1, Canada
2
McCaig Institute for Bone and Join Healthy, 3280 Hospital Drive NW, Calgary, AB T2N 4Z6, Canada
3
Department of Physiology and Pharmacology, Cumming School of Medicine, Foothills Hospital, 3330 Hospital Dr, Calgary, AB T2N 4N1, Canada
4
Centre for Critical Illness Research, Victoria Research Labs, Lawson Health Research Institute, A6-134, London, ON N6A 5W9, Canada
5
Division of Respirology, Department of Medicine, Western University, London, ON N6A 5W9, Canada
*
Author to whom correspondence should be addressed.
†
These authors contributed equally.
Pharmaceuticals 2021, 14(1), 31; https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010031
Submission received: 5 December 2020
/
Revised: 27 December 2020
/
Accepted: 28 December 2020
/
Published: 31 December 2020
(This article belongs to the Special Issue Novel Approaches for Targeting Metalloproteinases)
Abstract
:Matrix metalloproteinases (MMPs) have been demonstrated to have both detrimental and protective functions in inflammatory diseases. Several MMP inhibitors, with the exception of Periostat®, have failed in Phase III clinical trials. As an alternative strategy, recent efforts have been focussed on the development of more selective inhibitors or targeting other domains than their active sites through specific small molecule inhibitors or monoclonal antibodies. Here, we present some examples that aim to better understand the mechanisms of conformational changes/allosteric control of MMPs functions. In addition to MMP inhibitors, we discuss unbiased global approaches, such as proteomics and N-terminomics, to identify new MMP substrates. We present some examples of new MMP substrates and their implications in regulating biological functions. By characterizing the roles and substrates of individual MMP, MMP inhibitors could be utilized more effectively in the optimal disease context or in diseases never tested before where MMP activity is elevated and contributing to disease progression.
1. Introduction
Matrix metalloproteinases (MMPs) are zinc-dependent proteases that have been extensively studied in the context of extracellular matrix (ECM) breakdown and remodelling [1]. Increasingly, non-ECM substrates are being investigated for MMPs as ECM substrates only account for approximately 30% of all known MMP substrates [2,3]. The dysregulation of MMPs, their substrates, and the tissue inhibitor of metalloproteinases (TIMPs) often results in the progression of numerous diseases [1,3,4]. Various MMPs have been implicated in multiple cancers including pancreas, brain, lung, prostate, breast, skin and gastrointestinal tract [3,5]. MMP12 has been studied in chronic obstructive pulmonary disease (COPD) and the minor allele of a single nucleotide polymorphism in MMP12 (rs2276109) was associated with a beneficial effect on lung function in smokers and children with asthma [6,7]. Multiple MMPs have been investigated in rheumatoid arthritis and osteoarthritis yet the precise functions of individual MMP remains to be better characterized (reviewed in [8]). MMPs have also been studied in context of periodontal diseases [9,10]. It is not surprising that MMP inhibitors were tested in clinical trials. However, to date, the only MMP inhibitor that is currently approved is Periostat® (doxycycline hyclate), which is used for treating periodontitis (Figure 1a). Despite their biological roles in multiple cancers, in addition to inflammatory and autoimmune diseases, most MMP inhibitors failed due to a combination of factors including poor study design, a lack of understanding of biological roles of MMPs and the substrates they cleave, and the lack of specific inhibitors [1,3,5,11]. The structures and amino acid sequence of the catalytic domain of the 23 MMPs are highly conserved, which initially resulted in the design of broad spectrum MMP inhibitors. MMPs, however, have both detrimental and protective functions, limiting the use of these broad-spectrum inhibitors and increasing the complexity of developing MMP inhibitors to treat human diseases.
2. Regulation of MMP Activity
The catalytic activity of MMPs is tightly regulated by endogenous TIMPs [12]. TIMPs are secreted proteins that inhibit metalloproteinases [13] through the formation of 1:1 stoichiometric complexes [12]. The C-terminus of TIMPs interacts with the hemopexin like domain, found in all MMPs except MMP7 and MMP26, whereas the N-terminus interacts with the zinc ion within the catalytic domains of MMPs [14]. When an imbalance between MMPs and TIMPs occurs, it often results in inflammation and immune responses, as seen in many inflammatory diseases and cancers [15]. Therefore, the reestablishment of MMP-TIMP homeostasis is of pharmacological value and supports the need for the development of effective MMP inhibitors. Moreover, a better understanding of the biological functions of TIMPs is also needed to clarify their roles in human pathologies.
3. Non-Proteolytic Functions of MMPs
As demonstrated in previous clinical trials, broad spectrum targeting of the catalytic domain of MMPs is challenging. Thus, alternative methods for the inhibition of MMP functions have been investigated such as targeting exosites and ectosites. Not only would this potentially enable greater specificity between MMPs, but some exosites may have unique functions distinct from proteolysis. One example is the hemopexin (PEX) domain that contributes to protein-protein interactions and can initiate cell signalling and increased cell migration [16,17,18]. Since the amino acid sequences of the PEX domain across MMPs is more divergent and less conserved than the catalytic domain, the PEX domain is a potential site to target with inhibitors to increase selectivity. Interestingly, MMP7 and MMP26 do not contain a PEX domain, therefore not all MMPs require this domain. The MMP1 PEX domain is essential for binding to collagen and in the modulation of the triple helical structure of the substrate to allow access to the catalytic cleft [19]. Both the catalytic and PEX domain of MMP1 are necessary for the cooperative binding of triple helix collagen, demonstrating the importance of the PEX domain in substrate binding. Additionally, the conserved collagen residue P10 interacts with MMP1 via a hydrophobic pocket or exosite composed of Phe301, Ile271, and Arg27 within the PEX domain [19]. Further, when double mutants of Ile271Ala/Arg272Ala were generated, the collagenolytic function was significantly reduced. Thus, the inhibition of this hydrophobic pocket could potentially be a therapeutic approach to regulate MMP1 activity as it is important in not only the binding of triple helix collagen but in the processing of collagen [19]. The PEX domain of MMP12 plays a critical role in clearance of various bacteria such as Staphylococcus aureus, Klebsiella pneumoniae, Escherichia coli, and Salmonella enteriditis in the phagolysosome [18]. In Mmp12−/− mice, there was an increase in mortality at lower titer concentration when infected with S. aureus as compared with wild type mice [18]. Anti-bacterial properties of MMP12 were determined to be the result of disruption of the bacterial outer membrane by amino acids 344-363 in blade II of the PEX domain [18]. Conversely, the catalytic domain of MMP12 may contribute to the cleavage of bacterial toxins but did not demonstrate antibacterial properties against S. aureus α-toxins [18]. Therefore, a better characterization of the PEX domains of MMPs may reveal new exciting functions in other MMPs.
The PEX domain of MMPs is also implicated in homo-/hetero-dimerization and can form multimers [20]. The propeller structure of the PEX domain includes 4 blades composed of two alpha-helices and four beta strands [21]. In MMP9, a mutation in blade IV of the PEX domain resulted in a loss of homodimer formation [16]. Mutations in blade I of the MMP9 PEX domain resulted in a loss of interactions with the cell surface CD44 [16]. This interaction between the outer blade I of the MMP9 PEX domain and CD44 was shown to increase cell migration via the activation of epidermal growth factor receptor (EGFR) and downstream kinase signaling [16]. Peptides generated to mimic the outer beta strand of blade I or IV resulted in decreased levels of MMP9 dimers and also a reduction cell migration [16]. MMP9 can also increase angiogenesis [22]. Using an allosteric inhibitor to the PEX domain, Hariono et al. [22] demonstrated that inhibition of ECM proteolysis, which decreases the release of vascular endothelial growth factor (VEGF) from within the ECM, significantly reduces the binding of VEGF to its membrane receptor, and subsequently decreases angiogenesis.
The catalytic domain of membrane type 1-matrix metalloproteinase/MT1-MMP (MMP14) has been implicated in pro-tumorigenic functions by processing type I collagen, in addition to increasing cell migration, angiogenesis, and cell invasion [23,24,25]. The PEX domain of MT1-MMP also forms hetero- (with CD44) and homo-dimers via blades I and IV of the PEX domain, respectively [25]. Synthetic peptides mimicking the outermost strand motifs within the PEX domain (blades I and IV) of MT1-MMP were shown to specifically inhibit MT1-MMP-enhanced cell migration, although the ability to directly prevent MT1-MMP proteolytic activity was not shown [25]. The PEX domain contributes to the tumor promoting nature of MT1-MMP as tumour volume was significantly larger in cancer cells containing the PEX domain compared to those without [24]. MT1-MMP also contains transmembrane and cytoplasmic tail domains that have been shown to have distinct functions from the catalytic domain and could be targeted with inhibitors to interfere with the biological functions of MT1-MMP. Targeting the PEX domain of MMPs could provide non-competitive inhibition as compared with active site inhibition with broad-spectrum compounds [26]. Each MMP is likely to have unique exosites or “hotspots” that may be targeted individually due to divergence of their amino acid sequences, chemical potential and geometry [27]. However, the binding affinity of most exosites for substrate is typically low (10−6–10−7 M) making it potentially challenging to design an effective drug against that site [28,29].
4. Strategies for the Development of Protease Inhibitors
Multiple MMP inhibitors were originally designed with a substrate-based peptide, resembling the structure of type I collagen where MMPs cleave, aimed to interact with the necessary zinc ion in the MMPs’ active site [30]. This active site zinc ion is a required component of their catalytic site activity [31], coordinated by three histidine residues, and calcium ions, which stabilize conformation of the active protease [32]. Examples of chemical groups with zinc chelating agents used in the development of MMP inhibitors include hydroxamates, carboxylates, aminocarboxylates, phosphonate, and sulfhydryl groups [28,33]. Table 1, Table 2 and Table 3 provides a summary of MMP inhibitors that were tested in clinical trials and pre-clinical studies. One example is Batimastat, a peptidomimetic composed of a hydroxamate group, that was investigated for the treatment of breast cancer and was terminated in clinical trials due to its poor solubility and low oral bioavailability [34,35]. Later, a chemical analogue, marimastat, with improved oral bioavailability, was taken further along in clinical trials and was terminated due to musculoskeletal pain and lack of efficacy [34]. Another hydroxamate derivative, Prinomastat, was also unsuccessful in phase III clinical trials due to lack of efficacy in patients with late-stage disease [36]. While the use of MMP inhibitors in combination with traditional chemotherapy drugs was reported to improve adverse side effects, the chemotherapeutics, in turn, surprisingly lowered the therapeutic effects of the MMP inhibitors [37,38,39,40]. Despite termination of clinical trials for MMP inhibitors for the treatment of cancer and arthritis, another MMP inhibitor, Periostat® (CollaGenex Pharmaceuticals Inc.), was successfully approved for the treatment of periodontitis [41,42,43]. Periostat® is a synthetic tetracycline, (doxycycline hyclate) but its precise mechanism of action on MMPs activity remains unclarified [42]. Periostat®’s ability to bind to the calcified surfaces of tooth roots may potentiate its efficacy in periodontal disease [44]. The gradual release of doxycycline from teeth in active form also may contribute to increased exposure, the maintained effectiveness during the post-treatment period [42]. Periostat® also reduces the level of localized and systemic inflammatory mediators in osteopenic patients in addition to improving on the clinical measurements of periodontitis [45]. Additionally, it has showed therapeutic effects in multiple sclerosis and type II diabetes. In multiple sclerosis (MS), in a combination therapy with intramuscular interferon-β (IFNβ), oral doxycycline was found to be effective, safe and well-tolerated [46]. In this study, outcome measures included number of lesion changes, relapse rates, safety and tolerability of the combination therapy in patients with MS. Multiple parameters were recorded including the Expanded Disability Status Scale scores, MMP9 levels in the serum, and the transendothelial migration of monocytes exposed to serum from patients with relapsing-remitting multiple sclerosis (RRMS). The inhibitory effect of doxycycline was associated with decreased serum level of MMP9 and was found to be corelated with reduction in brain lesion activity as measured by gadolinium-enhancing lesion number change [46]. When serum from RRMS patients was incubated with monocytes, their transendothelial migration was significantly diminished. Importantly, in this study, the adverse effects were mild, and one out of fifteen patients relapsed. In another clinical trial with obese people with type II diabetes, doxycycline was tested over a 12-week timepoint resulting in decreased inflammation and improved insulin sensitivity [47]. This effect was associated with a decrease in C-reactive protein and myeloperoxidase comparing to the placebo; it also increased 3′- phosphoinositide kinease-1, protein kinase B, and glycogen synthase kinase 3 ß [47]. However, these clinical trials were performed only on a small number of patients and further studies on a larger number of patients are needed to further test their efficacy. The failure of all MMP inhibitors in clinical trials, with the exception of Periostat®, led to the investigation of the roles of MMPs beyond their recognized roles in ECM remodeling [5]. Recent studies using animal model of disease coupled with high-throughput methods for substrate discovery (will be further discussed in Section 8) have revealed important roles of MMPs in inflammation and viral/bacterial infections [3,11,18,48,49]. In fact, multiple MMPs, such as MMP2, -3, -8, -9, and -12, play important roles in maintaining tissue homeostasis and have been demonstrated to have protective effects (full list is reviewed in [3]). Therefore, the ideal MMP inhibitor should be able to interfere with detrimental MMPs while sparing the beneficial MMPs. The catalytic domains of MMPs are highly conserved, therefore, targeting other MMP domains, which are unique to a single MMP, represents an alternative method of MMP inhibitor design.
5. Small Molecule MMP Inhibitors
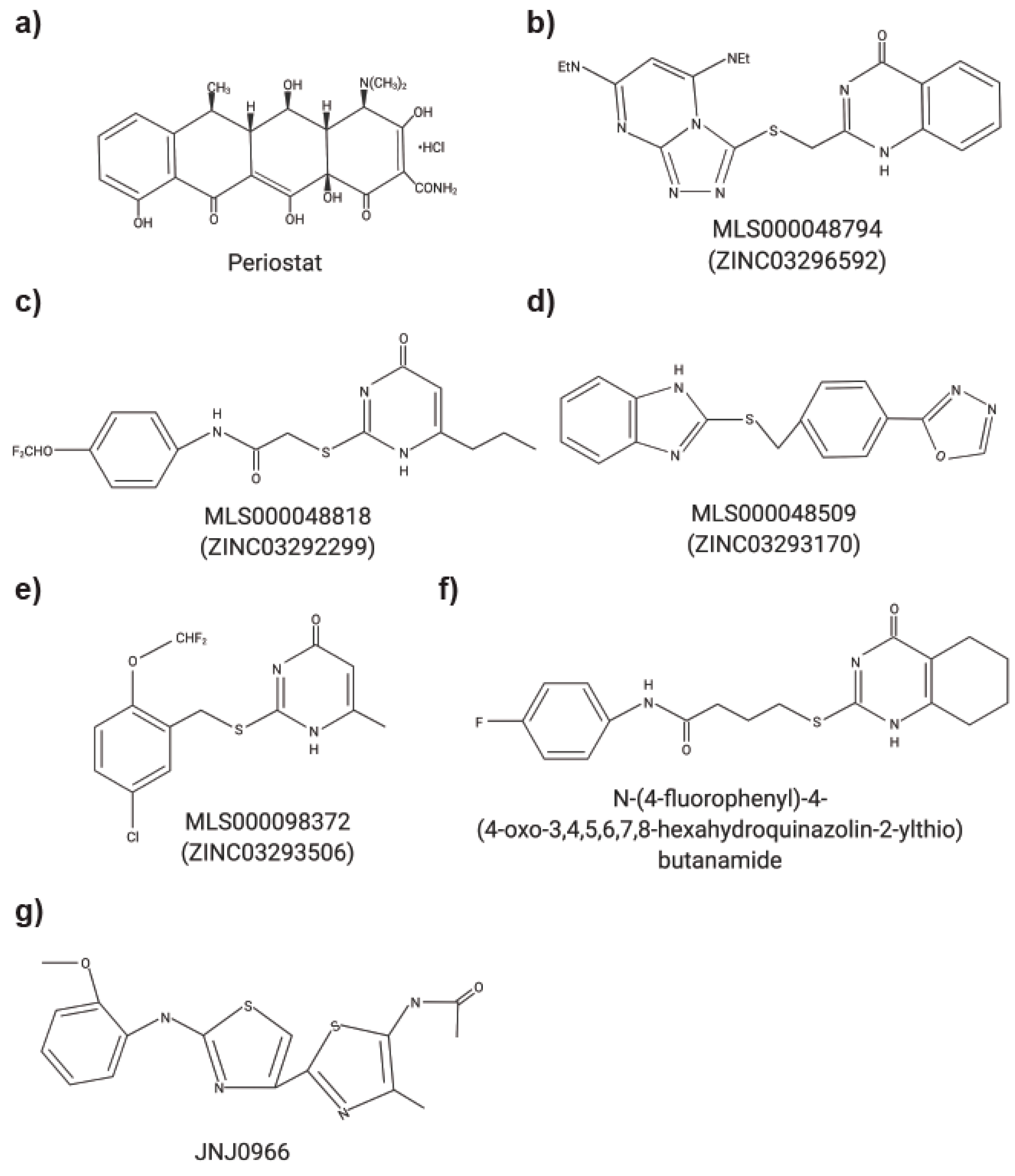
Strategies to inhibit MMP functions with small molecules has been explored (Table 2) [16,22,24,25,50]. Italicize in silico analysis of MMP9 in which molecular docking programs were utilized to map potential ligand binding sites in the PEX domain at the dimerization interface were successful at identifying compound that interfered with MMP9 homodimerization and blocked a downstream signaling pathway critical for MMP9 mediated cell migration and invasion [50]. These compounds (Figure 1b–e) spared MMP9′s proteolytic activity, and ‘compound 2′ (Figure 1c) from their study significantly diminished the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2). In a xenograft model of metastatic breast cancer cells (MDA-MB-435) stably transfected with a green fluorescent protein (GFP), a small molecule exosite MMP9 inhibitor, ‘compound 2′, significantly decreased the tumor size and reduced the number of lung metastasis [50]. A follow-up study [17] demonstrated that treatment with their newly identified compound (Figure 1f) disrupted MMP9 homodimerization and prevented association with α4β1 integrin, CD44 and decreased phosphorylation of Src and its downstream target proteins focal adhesion kinase and paxillin. In the in vivo model of chick chorioallantoic membrane, treatment with a PEX domain small molecule inhibitor resulted in a reduction of cancer cell invasion and angiogenesis [17]. Using previously published active compounds (Figure 1c,f), Hariono et al. [22] evaluated ten additional arylamide compounds. Using molecular dynamic simulations, the mechanism of MMP inhibition via the hemopexin domain of MMP9 was investigated. Two compounds, (3-bromo-N-(4-nitrophenyl)propanamide) and (3-bromo-N- {4-[(pyrimidine-2-yl)sulfamoyl]phenyl}propanamide), demonstrated significant inhibition and cytotoxicity against 4T1 murine breast cancer cells. Using a biochemical and structural screening workflow, the compound JNJ0966 (Figure 1g) was found to selectively impede activation of proMMP9 into its active form via an interaction with a structural pocket in proximity to the MMP9 zymogen cleavage site near Arg106 [29]. JNJ0966 was unable to interact with the active forms of MMP1, -2, -3, -9, or -14. In a mouse experimental autoimmune encephalomyelitis model, JNJ0966 reduced disease severity at a dose of 10 and 30 mg/kg and to the same levels of dexamethasone [29]. Italicize in silico analysis of MT1-MMP’s PEX domain identified a potentially targetable site that is distinct from the dimerization interface and located at the center of the PEX domain [24]. Subsequent docking studies of a small molecule inhibitor led to identification of a novel PEX inhibitor which is selective for MT1-MMP as compared to MMP2. It did not show any toxicity or interference to catalytic activities including MT1-MMP mediated activation of MMP2. This compound was effective in attenuating cancer cell migration and reduced tumor volume in vivo [24]. Despite promising results, there are still only a few studies that have tested small molecule MMP inhibitors in animal models and additional work in characterizing these inhibitors is needed to better understand whether this could be a viable approach to inhibit MMP function.
6. MMP Inhibition Using Selective Antibody
Monoclonal antibodies have emerged as potential enzyme inhibitors with numerous examples demonstrating them to be effective [51,52,53,54]. Initially, antibody generation was deemed challenging for the active site of MMPs due to the instability during presentation and lack of surface accessibility of the catalytic metal-protein cleft [55]. Sela-Passwell et al. [55] used a synthetic metal-baring mimicry complex and were able to generate a response not only to the metal-protein cleft, but also to the enzyme surface of MMP2 and MMP9 with high specificity which resulted in the generation of the monoclonal antibody SDS3. In a dextran sodium sulfate (DSS)-colitis mouse model, SDS3 was demonstrated to prevent colonic inflammation, release of proinflammatory cytokines, and tissue damage [55]. The applicability of SDS3 towards colitis and inflammatory bowel disorders was reinforced through clustering of the labelled SDS3 antibody in the intestine of the mice 24 h after injection. Another MMP9 monoclonal antibody inhibitor, REGA-3G12, was demonstrated to be selective towards MMP9 through recognition of the N-amino terminal catalytic domain subsequently inhibiting its catalysis [56,57]. REGA-3G12 recognizes the Trp116-Lys214 motif domain part of the Phe107-Gly223 outside of the Zn2+ binding site of MMP9, a site exploited by multiple MMP inhibitors. Although REGA-3G12 targets the catalytic domain, it was shown to be selective to MMP9 but not MMP2, therefore demonstrating the potential of the catalytic domain to be used to generate selective MMP inhibitors. The selectivity of REGA-3G12 is likely due to the vast number of interactions with the carboxy-terminal of the catalytic domain. REGA-3G12 did not show significant binding to synthetic linear fragments of the epitopes recognized by REGA-3G12, demonstrating the antibody may recognize a conformation instead of a linear residue. MT1-MMP inhibition is another attractive anti-cancer target as it is highly expressed in breast cancer and contributes to migration, invasion, and neovascularization [58]. Using a phage library display approach, an MT1-MMP active site inhibitor antibody, DX-2400, was developed to selectively inhibit active MT1-MMP. DX-2400 was also shown to inhibit pro-MMP2 activation due to the inhibition of MT1-MMP and reduced breast cancer cell invasion [58]. DX-2400 exhibited promising therapeutic results in an MDA-MB-231 breast cancer xenograft tumor mouse model associated with a decrease in cell growth and vascularization. Conversely, non-metastatic MCF-7 breast cancer cell that do not express MT1-MMP, did not show decreased growth in a xenograft tumor mouse model [58]. DX-2400 also delayed metastasis, and when in combination with bevacizumab delayed breast cancer cells’ tumor growth [58]. Similarly, when applied to a BT-474 xenograft tumor mouse model, DX-2400 in combination with paclitaxel resulted in tumour growth delay. Surprisingly, while this study demonstrated promising results related to using selective MT1-MMP inhibitors to treat breast cancer, DX-2400 was unsuccessful in clinical trials.
Selective inhibition of MT1-MMP was also demonstrated in a model of influenza infections that resulted in ECM dysregulation and increased susceptibility for a bacterial co-infection [48]. The selective, potent and allosteric MT1-MMP inhibitor, LEM-2/15, was generated via mouse immunization with a cyclic peptide from the sequence of the V-B loop (residues 218–233), which displays a unique sequence divergence within the MMP family members [59]. This loop of MT1-MMP is flexible and likely undergoes conformational changes when binding to LEM-2/15 causing a narrower substrate binding cleft and constraining the flexibility of the loop. Therefore, the Fab fragment of LEM-2/15 interacted with the MT1-MMP expressed on the cell surface and inhibited its collagenase activity while not interfering significantly with the activation of proMMP2 and MT1-MMP homodimerization on the cell surface [59]. ECM remodeling is usually independent of the viral burden but rather linked to the proteolysis driven by the immune response. LEM-2/15 reduced inflammation and ECM remodeling during influenza infection [48]. When used prophylactically or therapeutically during a coinfection of influenza and S. pneumoniae, LEM-2/15 significantly improved survival in mice. Interestingly, when used in combination with Tamiflu® (Oseltamivir) both prophylactically or therapeutically, an approved anti-influenza inhibitor, 100% survival was achieved in a mouse model of viral infection [59]. Importantly, Tamiflu® alone resulted in increased survival only when used prophylactically. Thus, promoting ECM stability and homeostasis via MT1-MMP inhibition during influenza infection is an attractive target.
7. MMP Substrates Extend Beyond Matrix Proteins
Contrary to what their name suggests, MMPs have been shown to cleave substrates other than matrix (ECM) proteins [1,2,11]. MMPs cleave chemokines and cytokines to regulate their functions [49,60,61,62,63]. For example, the processing of monocyte chemoattractant proteins, CCL-7 and CCL-13, reduced the inflammatory response, as demonstrated in mouse model of inflammatory edema [64]. In a mouse model of asthma, MMP2 and MMP9 were found to be protective via disruption of transepithelial chemokine gradients regulated by CCL7, CCL11, and CCL17 [65]. In macrophages, MMP12 cleaved the C-terminus of IFNγ, removing the receptor binding site and thereby decreasing JAK–STAT1 signaling and IFNγ activation within the proinflammatory macrophage [63]. Genetic ablation of MMP12 or therapeutic inhibition of MMP12 using Rxp470.1 in murine models of autoimmune inflammatory diseases resulted in elevated IFNγ mediated inflammatory signatures compared to the control groups [63]. In virus-infected cells, MMP12 was shown to be localized in the nucleus and promoted NFKBIA transcriptional activity resulting in INFα secretion, a key mechanisms for antiviral immunity [49]. In parallel, extracellular MMP12 attenuated systemic IFNα, and use of an MMP12 inhibitor, Rxp470.1, which is unable to enter the cells, significantly reduced the viral load [49]. MMP processing of CCL15 and CCL23, implicated in inflammatory arthritis, resulted in an increase in monocyte recruitment during inflammation [62]. Collectively, these four lines of evidence support the critical role for MMPs in regulating the inflammatory response through direct cleavage of chemokines/cytokines. Further, only 30% of MMP substrates are linked with the ECM [2]. Therefore, additional roles of MMPs are likely to be identified when looking beyond the matrix.
8. Identification of Novel MMP Substrates Using N-Terminomics/TAILS
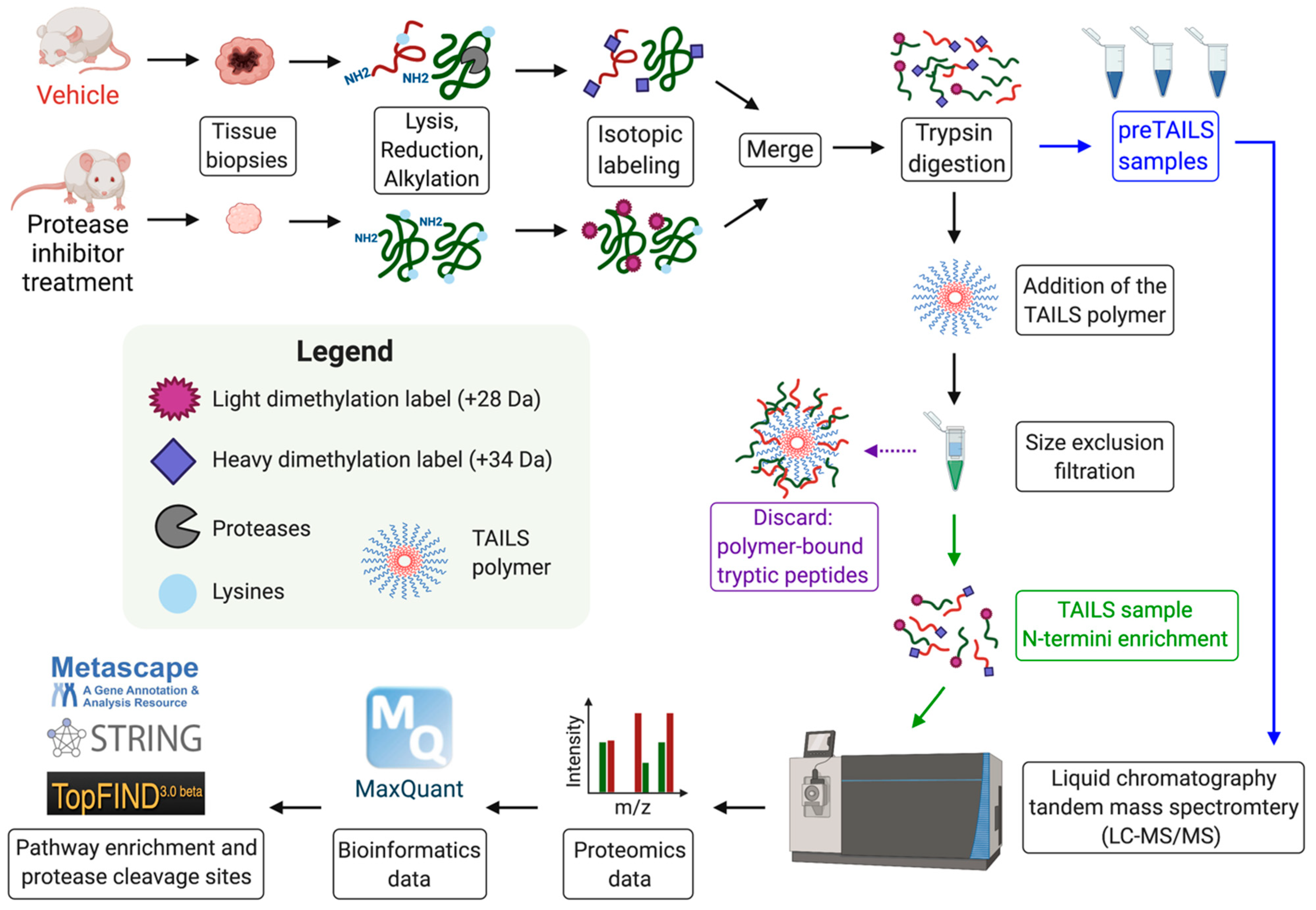
N-terminomics technologies have been used to profile and identify new MMP substrates in various cell systems and tissues [2,3,49,66,67]. One example is terminal amine isotopic labeling of substrates (TAILS), a high throughput quantitative proteomic platform that allows simultaneous quantitative analysis of the N-terminome and proteolysis on a proteome-wide scale, and hence allows for protease substrate discovery [68]. To study the substrate repertoire of a specific protease using TAILS, the protease of interest can be compared to an inactivated form of the protease or to a protease inhibitor-treated sample (Figure 2). Alternatively, tissue proteomes of protease knock-out mice can be compared to wild-type animals with or without induction of a specific infection, stress or disease. Once collected, proteomes are denatured, and it is important to avoid primary amine containing buffers. After the reduction and alkylation of cysteine residues, primary amines of both the N-termini and lysine residues are chemically labeled with formaldehyde. During this step, we incorporate stable isotope labeling in order to later compare the different conditions being tested. An example of isotopic labeling is light (+28 Da) and heavy (+34 Da) dimethylation used with the catalyst sodium cyanoborohydride (NaBH3CN) [69]. These isotopic modification can later be monitored using liquid chromatography and tandem mass spectrometry (LC-MS/MS) [68]. However, dimethylation reactions are limited to three distinct labels and other labelling such tandem mass tag (TMT) can label up to 11 different samples with a distinct isotope [67,70]. After isotopic labeling, the labeled proteomes are then mixed and digested with trypsin. During digestion, trypsin cleaves the peptide only after arginine (semi-ArgC specificity) as the blocked lysine residues are unreactive to trypsin. After trypsin cleavage, ~10% of the sample is collected and prepared for LC-MS/MS analysis; this is the pre-enrichment TAILS samples. Next, the TAILS aldehyde reactive polymer is used to remove the internal tryptic peptides that were generated during trypsin digestion. The unbound blocked and labeled peptides are recovered from the samples by size exclusion (10-kDa cut-off filters) filtration. The recovered peptides are then analyzed via LC-MS/MS analysis. The abundance ratio of blocked peptides from the TAILS samples can be compared to naturally blocked N-termini from the pre-enrichment TAILS samples. The neo-N-termini peptides specific to the protease of interest appear in higher ratios or only in the protease-treated sample and therefore show high protease/control abundance ratio. Therefore, the TAILS protocol is designed to identify new protease substrates and also identifies the precise cleavage site within the substrate sequence. TAILS has been used to profile the substrate repertoires of various MMPs and hundreds of new substrates have been identified in specific cell lines and tissues [66,71,72,73,74,75]. Using TAILS, the substrates of MMP2 and MMP9 were investigated in fibroblasts secretomes where 201 substrates were identified for MMP2 and only 19 for MMP9 [75]. Although, more MMP2 substrates were identified, most substrates can be cleaved by both MMP2 and MMP9 including thrombospondin-2, galectin-1, insulin-like growth factor-binding protein 4 (IGFBP4), dickkopf-related protein-3, and pyruvate kinase M1/M2 [75]. This study suggests that the regulation of MMP2 and MMP9’s activity could be linked with differences in genetic expression, different rates of TIMP inhibition, alternate activation mechanisms and/or distinct kinetic activities that could explain the differences in the phenotypes of Mmp2−/− and Mmp9−/− mice. Additional italicize in vivo studies comparing various cells, tissues, and organs of WT counterparts, Mmp2−/− and Mmp9−/− mice using disease models will help better characterize the unique roles and substrates of these two MMPs.
The substrates of macrophage MMP12 were investigated using TAILS by incubating murine MMP12 with secretomes from Mmp12−/− murine embryonic fibroblasts (MEFs), murine macrophage cell line RAW264.7 secretomes and also by comparing WT and Mmp12−/− peritoneal macrophages from a peritonitis model using thioglycollate stimulation for 4 days [71]. Hundreds of MMP12 substrates were identified including pyruvate kinase, biglycan, vimentin, renin-receptor and alpha-2-HS-glycoprotein (see [71] for the full list of substrates). Using TAILS, new functions for MMP12 in coagulation, complement activation/deactivation and resolution of inflammation were identified. To further confirm these MMP12 substrates in human diseases, TAILS was used to investigate nine COPD patients at exacerbation and recovery [7]. When comparing MMP12 substrates from murine peritonitis and joint inflammation model to the sputum of COPD patients, multiple identical substrates were identified including alpha-2-HS-glycoprotein, complement C3 (C3), complement C4-B (C4b), hemopexin, antithrombin III (SERPINC1), but also new substrates were identified such as transmembrane protease serine 7 (TMPRSS7) and DEP domain-containing mTOR-interacting protein (DEPTOR) [7,71]. MMP12 can cleave hundreds of substrates, therefore, the regulation of its activity is likely driven and impacted by other proteases, TIMPs, tissue specific microenvironment and other immune cells present such as neutrophils, eosinophils, natural killer cells, mast cells, T and B cells. Interestingly, MMP12 has been predominantly identified as a beneficial/protective MMP in inflammatory diseases although it has been implicated as a potential drug target in certain cancers. However, its precise role in inflammation needs to be further characterized [3,49,63,71].
Using TAILS, numerous non-ECM substrates have been identified further demonstrating new roles for MMPs (reviewed in [1,2,76]). For example, 58 new substrates were identified when MT6-MMP (MMP25) was added to fibroblasts secretomes, including vimentin, cystatin C, galectin-1, secreted protein acidic and rich in cysteine (SPARC), and insulin-like growth factor-binding protein 7 (IGFBP7) [72]. These identified substrates indicated a novel role for MT6-MMP for the clearance of apoptotic neutrophils. Cleavage of vimentin by MT6-MMP resulted in a decrease in chemoattraction of THP-1 monocytic cells but an increase in phagocytosis activity in an assay where fluorescent microbeads were coated with vimentin or cleaved vimentin and added to THP-1 cells [72]. The identification of new MT6-MMP substrates using TAILS supported a key biological role for this MMP in innate immunity and resolution of inflammation. Most MMPs have yet to be investigated using unbiased N-terminomics approaches to identify the extent of their substrates. It is anticipated that the cell type producing MMPs and specific microenvironments where MMPs are present will greatly impact what substrates can be cleaved by MMPs.
9. Conclusions and Perspective: Next Generation of MMP Inhibitors
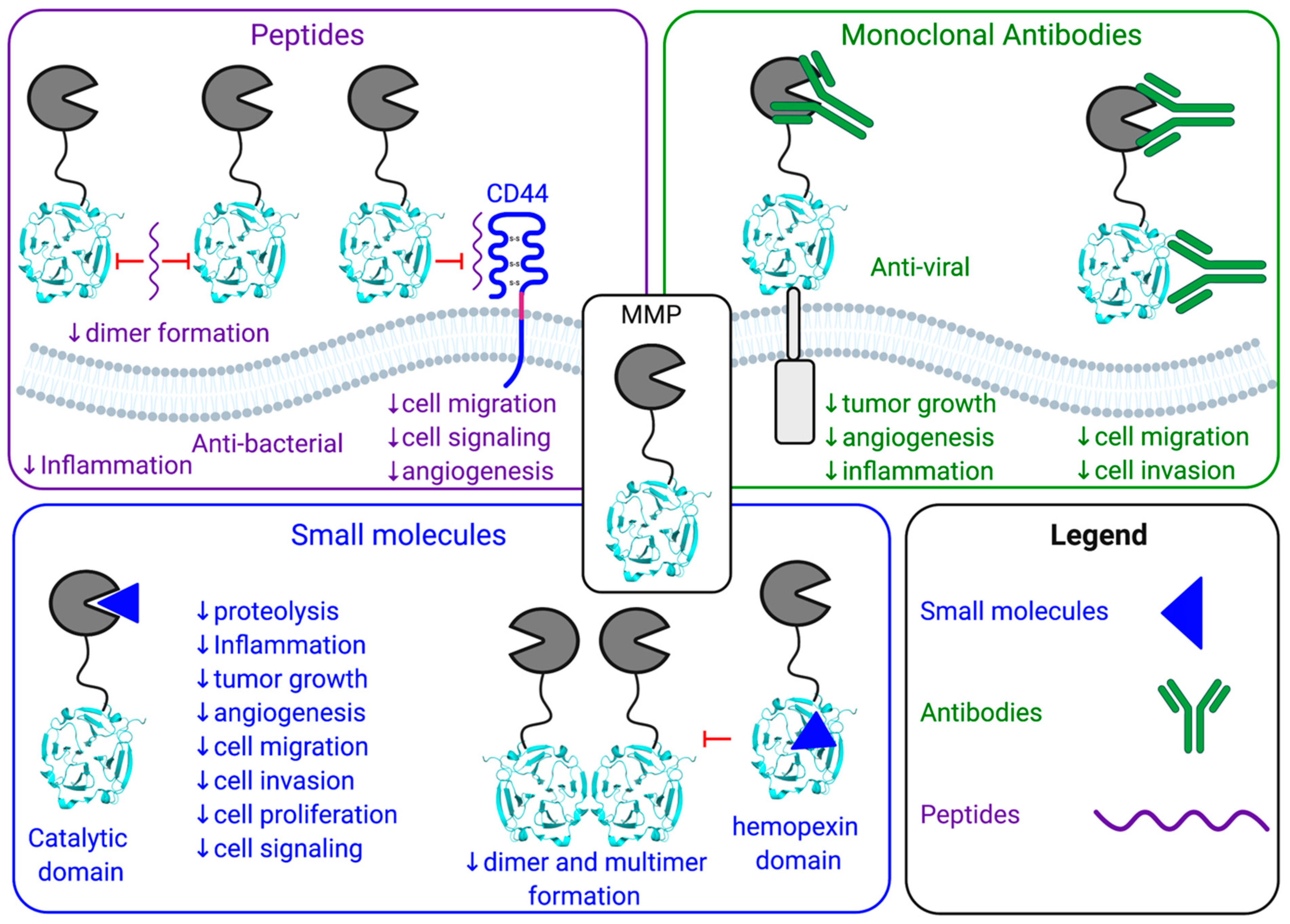
MMPs have been shown to play a key biological role in numerous pathologies, however, the broad-spectrum inhibitors targeting multiple MMPs has potentially impeded their therapeutic applicability and use in the clinic to treat joint diseases or cancers. There are various pharmacological approaches that have been utilized to inhibit both the proteolytic and non-proteolytic functions of MMPs: peptides, monoclonal antibodies, and small molecule inhibitors (Figure 1 and Figure 3, Table 1, Table 2 and Table 3). With the discovery and development of novel specific methods of targeting individual MMPs, there is renewed hope for other MMP inhibitors and a revival in better characterizing MMP functions. Moreover, additional opportunities for the use of MMP inhibitors may become apparent. For example, given the success of MMP inhibition in animal models, MMP inhibitors could play an emerging role in veterinary medicine. For example, MMP2 and MMP9 inhibitors may be beneficial for the treatment of canine chronic enteropathy [77]. Elevated levels of active MMP2 and MMP9 are found throughout the intestines of dogs with chronic enteropathy and are associated with increased inflammation and neutrophil infiltration. MMP2/9 inhibitors could be effective in lowering intestinal inflammation and decreased disease severity [77]. Another example is the use of MMP inhibitors to treat the elevated expression of MMP2 and MT1-MMP in the narrowing of myocardial vessels in canine myxomatous mitral valve disease [78]. When considering application to human health, one option could be to only inhibit MMPs in diseases requiring short-term treatments, therefore limiting the period over which the treatment is received effectively minimizing any detrimental consequences that could arise. For example, sepsis could be further investigated for the short-term implementation of MMP inhibitors. In sepsis, MMPs cleave and regulate cytokine storm and chemokine activity, thus, playing a role in many of the pathways resulting in complications [79]. Another example is MMP12 with its anti-bacterial functions and its involvement in the inflammatory pathway leading to reduced lethality of lipopolysaccharide (LPS) induced inflammation [18,80]. Selective MT1-MMP inhibitors alone or in combination with Tamiflu® also demonstrated efficacy in influenza infection models and could be further investigated for other viral infections. The utilization of shorter dosing and altering administration may reduce patient exposure to previously identified side effects of MMP inhibitors [28]. A more complete investigation into the protective and detrimental properties of MMPs and the subsequent development of therapeutics with high affinity for distinct MMPs may provide greater applications of MMP inhibition in the future.
Funding
Antoine Dufour and Sean Gill received funding from NSERC Discovery grants for this work.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Young, D.; Das, N.; Anowai, A.; Dufor, A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. Int. J. Mol. Sci. 2019, 20, 3847. [Google Scholar] [CrossRef] [Green Version]
- Dufour, A.; Overall, C.M. Subtracting Matrix Out of the Equation: New Key Roles of Matrix Metalloproteinases in Innate Immunity and Disease. In Matrix Metalloproteinase Biology, 1st ed.; Sagi, I., Gaffney, J., Eds.; John Wiley & Sons, Inc.: Franklin Township, NJ, USA, 2015; pp. 131–152. [Google Scholar] [CrossRef]
- Dufour, A.; Overall, C.M. Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol. Sci. 2013, 34, 233–242. [Google Scholar] [CrossRef]
- Mainoli, B.; Hirota, S.; Edgington-Mitchell, L.E. Proteomics and Imaging in Crohn’s Disease: TAILS of Unlikely Allies. Trends Pharmacol. Sci. 2020, 41, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef]
- Hunninghake, G.M.; Cho, M.H.; Tesfaigzi, Y.; Soto-Quiros, M.E.; Avila, L.; Lasky-Su, J.; Stidley, C.; Melén, E.; Söderhäll, C.; Hallberg, J.; et al. MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. 2009, 361, 2599–2608. [Google Scholar] [CrossRef] [Green Version]
- Mallia-Milanes, B.; Dufour, A.; Philp, C.; Solis, N.; Klein, T.; Fischer, M.; Bolton, C.E.; Shapiro, S.; Overall, C.M.; Johnson, S.R. TAILS proteomics reveals dynamic changes in airway proteolysis controlling protease activity and innate immunity during COPD exacerbations. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L1003–L1014. [Google Scholar] [CrossRef]
- Murphy, G.; Nagase, H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: Destruction or repair? Nat. Clin. Pract. Rheumatol. 2008, 4, 128–135. [Google Scholar] [CrossRef]
- Checchi, V.; Maravic, T.; Bellini, P.; Generali, L.; Consolo, U.; Breschi, L.; Mazzoni, A. The role of matrix metalloproteinases in periodontal disease. Int. J. Environ. Res. Public Health 2020, 17, 4923. [Google Scholar] [CrossRef]
- Franco, C.; Patricia, H.-R.; Timo, S.; Claudia, B.; Marcela, H. Matrix metalloproteinases as regulators of periodontal inflammation. Int. J. Mol. Sci. 2017, 18, 440. [Google Scholar] [CrossRef] [Green Version]
- Chopra, S.; Overall, C.M.; Dufour, A. Matrix metalloproteinases in the CNS: Interferons get nervous. Cell. Mol. Life Sci. 2019, 76, 3083–3095. [Google Scholar] [CrossRef]
- Moore, C.S.; Crocker, S.J. An alternate perspective on the roles of TIMPs and MMPs in pathology. Am. J. Pathol. 2012, 180, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Crocker, S.J.; Pagenstecher, A.; Campbell, I.L. The TIMPs tango with MMPs and more in the central nervous system. J. Neurosci. Res. 2004, 75, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Riedl, R. Challenges with matrix metalloproteinase inhibition and future drug discovery avenues. Expert Opin. Drug Discov. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- de Bruyn, M.; Vandooren, J.; Ugarte-Berzal, E.; Arijs, I.; Vermeire, S.; Opdenakker, G. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 295–358. [Google Scholar] [CrossRef]
- Dufour, A.; Zucker, S.; Sampson, N.S.; Kuscu, C.; Cao, J. Role of matrix metalloproteinase-9 dimers in cell migration design of inhibitory peptides. J. Biol. Chem. 2010, 285, 35944–35956. [Google Scholar] [CrossRef] [Green Version]
- Alford, V.M.; Kamat, A.; Ren, X.; Kumar, K.; Gan, Q.; Awwa, M.; Tong, M.; Seeliger, M.A.; Cao, J.; Ojima, I.; et al. Targeting the hemopexin-like domain of latent matrix metalloproteinase-9 (proMMP-9) with a small molecule inhibitor prevents the formation of focal adhesion junctions. ACS Chem. Biol. 2017, 12, 2788–2803. [Google Scholar] [CrossRef]
- Houghton, A.M.; Hartzell, W.O.; Robbins, C.S.; Gomis-Rüth, F.X.; Shapiro, S.D. Macrophage elastase kills bacteria within murine macrophages. Nature 2009, 460, 637–641. [Google Scholar] [CrossRef]
- Manka, S.W.; Carafoli, R.; Visse, R.; Bihan, D.; Raynal, N.; Farndale, R.W.; Murphy, G.; Enghild, J.J.; Hohenester, E.; Nagase, H. Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc. Natl. Acad. Sci. USA 2012, 109, 12461–12466. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Cha, H.; Kopetzki, E.; Huber, R.; Lanzendörfer, M.; Brandstetter, H. Structural basis of the adaptive molecular recognition by MMP9. J. Mol. Biol. 2002, 320, 1065–1079. [Google Scholar] [CrossRef]
- Hariono, M.; Nuwarda, R.F.; Yusuf, M.; Rollando, R.; Jenie, R.I.; Al-Najjar, B.; Julianus, J.; Putra, K.C.; Nugroho, E.S.; Wisnumurti, Y.K.; et al. Arylamide as Potential Selective Inhibitor for Matrix Metalloproteinase 9 (MMP9): Design, Synthesis, Biological Evaluation, and Molecular Modeling. J. Chem. Inf. Model. 2019, 60, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, T.Y.; Zheng, H.; Lui, R.; Wicha, M.S.; Yu, S.M.; Weiss, S.J. Divergent matrix-remodeling strategies distinguish developmental from neoplastic mammary epithelial cell invasion programs. Dev. Cell 2018, 47, 145–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remacle, A.G.; Golubkov, V.S.; Shiryaev, S.A.; Dahl, R.; Stebbins, J.L.; Chernov, A.V.; Cheltsov, A.V.; Pellecchia, M.; Strongin, A.Y. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res. 2012, 72, 2339–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarrabi, K.; Dufour, A.; Li, J.; Kuscu, C.; Pulkoski-Gross, A.; Zhi, J.; Hu, Y.; Sampson, N.S.; Zucker, S.; Cao, J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem. 2011, 286, 33167–33177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Li, S.; Zhang, J. Harnessing allostery: A novel approach to drug discovery. Med. Res. Rev. 2014, 34, 1242–1285. [Google Scholar] [CrossRef]
- Levin, M.; Udi, Y.; Solomonov, I.; Siga, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Overall, C.M.; Kleifeld, O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 2006, 94, 941–946. [Google Scholar] [CrossRef] [Green Version]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.; Zhang, Y.M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef] [Green Version]
- Fields, G.B. The rebirth of matrix metalloproteinase inhibitors: Moving beyond the dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef] [Green Version]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef] [Green Version]
- Wetmore, D.R.; Hardman, K.D. Roles of the propeptide and metal ions in the folding and stability of the catalytic domain of stromelysin (matrix metalloproteinase 3). Biochemistry 1996, 35, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Pirard, B. Insight into the structural determinants for selective inhibition of matrix metalloproteinases. Drug Discov. Today 2007, 12, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer—Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [Green Version]
- Hande, K.R.; Collier, M.; Paradiso, L.; Stuart-Smith, J.; Dixon, M.; Clendeninn, N.; Yeun, G.; Alberti, D.; Binger, K.; Wilding, G. Phase I and pharmacokinetic study of prinomastat, a matrix metalloprotease inhibitor. Clin. Cancer Res. 2004, 10, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Leighl, N.B.; Paz-Ares, L.; Douillard, J.-Y.; Peschel, C.; Arnold, A.; Depierre, A.; Santoro, A.; Betticher, D.C.; Gatzemeier, U.; Jassem, J.; et al. Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR. 18. J. Clin. Oncol. 2005, 23, 2831–2839. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Peschel, C.; Shepherd, F.; Paz-Ares, L.; Arnold, A.; Davis, M.; Tonato, M.; Smylie, M.; Tu, D.; Voi, M.; et al. Randomized phase II feasibility study of combining the matrix metalloproteinase inhibitor BMS-275291 with paclitaxel plus carboplatin in advanced non-small cell lung cancer. Lung Cancer 2004, 46, 361–368. [Google Scholar] [CrossRef]
- Lu, C.; Lee, J.; Komaki, R.; Herbst, R.S.; Feng, L.; Evans, W.K.; Choy, H.; Desjardins, P.; Esparaz, B.T.; Truong, M.T.; et al. Chemoradiotherapy with or without Ae-941 in Stage III Non–Small cell lung cancer: A randomized Phase III trial. JNCI J. Natl. Cancer Inst. C 2020, 102, 859–865. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Schulz, J.; Nemunaitis, J.; Brown, P.D.; Baillet, M.; Buckels, J.A. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br. J. Cancer 2002, 87, 161–167. [Google Scholar] [CrossRef]
- Preshaw, P.M.; Hefti, A.F.; Novak, M.J.; Michalowicz, B.S.; Pihlstrom, B.L.; Schoor, R.; Trummel, C.L.; Dean, J.; Van Dyke, T.E.; Walker, C.B.; et al. Subantimicrobial dose doxycycline enhances the efficacy of scaling and root planing in chronic periodontitis: A multicenter trial. J. Periodontol. 2004, 75, 1068–1076. [Google Scholar] [CrossRef]
- Peterson, J.T. Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail. Rev. 2004, 9, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Golub, L.M.; McNamara, T.F.; Ryan, M.E.; Kohut, B.; Blieden, T.; Payonk, G.; Sipos, T.; Baron, H.J. Adjunctive treatment with subantimicrobial doses of doxycycline: Effects on gingival fluid collagenase activity and attachment loss in adult periodontitis. J. Clin. Periodontol. 2001, 28, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.J.; Evans, R.T.; Coburn, R.A.; Genco, R.J. Tetracycline and its derivatives strongly bind to and are released from the tooth surface in active form. J. Periodontol. 1983, 54, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.B.; Stoner, J.A.; Nummikoski, P.V.; Reinhardt, R.A.; Goren, A.D.; Wolff, M.S.; Lee, H.M.; Lynch, J.C.; Valente, R.; Golub, L.M. Subantimicrobial dose doxycycline effects on alveolar bone loss in post-menopausal women. J. Clin. Periodontol. 2007, 34, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Alexander, J.S.; Schwendimann, R.N.; Kelley, R.E.; Gonzalez-Toledo, E.; Jimenez, J.J.; Mauro, L.; Jy, W.; Smith, S.J. Combination therapy with interferon beta-1a and doxycycline in multiple sclerosis: An open-label trial. Arch. Neurol. 2008, 65, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Frankwich, K.; Tibble, C.; Torres-Gonzalez, M.; Bonner, M.; Lefkowitz, R.; Tyndall, M.; Schmid-Schönbein, G.W.; Villarreal, F.; Heller, M.; Herbst, K. Proof of Concept: Matrix metalloproteinase inhibitor decreases inflammation and improves muscle insulin sensitivity in people with type 2 diabetes. J. Inflamm. 2012, 9, 35. [Google Scholar] [CrossRef] [Green Version]
- Talmi-Frank, D.; Altboum, Z.; Solomonov, I.; Udi, Y.; Jaitin, D.A.; Klepfish, M.; David, E.; Zhuravlev, A.; Keren-Shaul, H.; Winter, D.R.; et al. Extracellular matrix proteolysis by MT1-MMP contributes to influenza-related tissue damage and mortality. Cell Host Microbe 2016, 20, 458–470. [Google Scholar] [CrossRef] [Green Version]
- Marchant, D.J.; Bellac, C.L.; Moraes, T.J.; Wadsworth, S.J.; Dufour, A.; Butler, G.S.; Bilawchuk, L.M.; Hendry, R.G.; Robertson, A.G.; Cheung, C.T.; et al. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat. Med. 2014, 20, 493–502. [Google Scholar] [CrossRef]
- Dufour, A.; Sampson, N.S.; Li, J.; Kuscu, C.; Rizzo, R.C.; Deleon, J.L.; Zhi, J.; Jaber, N.; Liu, E.; Zucker, S.; et al. Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 2011, 71, 4877–4888. [Google Scholar] [CrossRef] [Green Version]
- Lipsky, P.E.; van der Heijde, D.M.; Clair, E.W.S.; Furst, D.E.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Weisman, M.; Emery, P.; Feldmann, M.; et al. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. Infliximab and methotrexate in the treatment of rheumatoid arthritis. N. Engl. J. Med. 2000, 343, 1594–1602. [Google Scholar] [CrossRef] [Green Version]
- Starnes, H.; Pearce, M.K.; Tewari, A.; Yim, J.H.; Zou, J.C.; Abrams, J.S. Anti-IL-6 monoclonal antibodies protect against lethal Escherichia coli infection and lethal tumor necrosis factor-alpha challenge in mice. J. Immunol. 1990, 145, 4185–4191. [Google Scholar] [PubMed]
- Hudziak, R.M.; Lewis, G.D.; Winget, M.; Fendly, B.M.; Shepard, H.M.; Ullrich, A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol. Cell. Biol. 1989, 9, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Milgrom, H.; Fick, R.B.; Su, J.Q.; Reimann, J.D.; Bush, R.K.; Watrous, M.L.; Metzger, W.J. Treatment of allergic asthma with monoclonal anti-IgE antibody. N. Engl. J. Med. 1999, 341, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T.; et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2012, 18, 143–147. [Google Scholar] [CrossRef]
- Martens, E.; Leyssen, A.; Aelst, I.V.; Fiten, P.; Piccard, H.; Hu, J.; Descamps, F.J.; van den Steen, P.E.; Proost, P.; van Damme, J.; et al. A monoclonal antibody inhibits gelatinase B/MMP-9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim. Biophys. Acta (BBA) Gen. Subj. 2007, 1770, 178–186. [Google Scholar] [CrossRef]
- Paemen, L.; Martens, E.; Masure, S.; Opdenakker, G. Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two-site ELISA and inhibition of enzymatic activity. Eur. J. Biochem. 1995, 234, 759–765. [Google Scholar] [CrossRef]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009, 69, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Udi, Y.; Grossman, M.; Solomonov, I.; Dym, O.; Rozenberg, H.; Moreno, V.; Cuniasse, P.; Dive, V.; Arroyo, A.G.; Sagi, I. Inhibition mechanism of membrane metalloprotease by an exosite-swiveling conformational antibody. Structure 2015, 23, 104–115. [Google Scholar] [CrossRef] [Green Version]
- Dean, R.A.; Cox, J.H.; Bellac, C.L.; Doucet, A.; Starr, A.E.; Overal, C.M. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR + CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: Potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 2008, 112, 3455–3464. [Google Scholar] [CrossRef] [Green Version]
- McQuibban, G.A.; Gong, J.-H.; Tam, E.M.; McCulloch, C.A.; Clark-Lewis, I.; Overall, C.M. Inflammation dampened by gelatinase a cleavage of monocyte chemoattractant protein-3. Science 2000, 289, 1202–1206. [Google Scholar] [CrossRef]
- Starr, A.E.; Dufour, A.; Maier, J.; Overall, C.M. Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J. Biol. Chem. 2012, 287, 5848–5860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufour, A.; Bellac, C.L.; Eckhard, U.; Solis, N.; Klein, T.; Kappelhoff, R.; Fortelny, N.; Jobin, P.; Rozmus, J.; Mark, J.; et al. C-terminal truncation of IFN-γ inhibits proinflammatory macrophage responses and is deficient in autoimmune disease. Nat. Commun. 2018, 9, 2416. [Google Scholar] [CrossRef] [PubMed]
- McQuibban, G.A.; Gong, J.-H.; Wong, J.P.; Wallace, J.L.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood J. Am. Soc. Hematol. 2002, 100, 1160–1167. [Google Scholar] [CrossRef]
- Corry, D.B.; Kiss, A.; Song, L.-Z.; Song, L.; Xu, J.; Lee, S.; Werb, Z.; Kheradmand, F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004, 18, 995–997. [Google Scholar] [CrossRef]
- Kleifeld, O.; Doucet, A.; auf dem Keller, U.; Prudova, A.; Schilling, O.; Kainthan, R.K.; Starr, A.E.; Foster, L.J.; Kizhakkedathu, J.N.; Overall, C.M. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 2010, 28, 281–288. [Google Scholar] [CrossRef]
- Prudova, A.; Gocheva, V.; auf dem Keller, U.; Eckhard, U.; Olson, O.C.; Akkari, L.; Butler, G.S.; Fortelny, N.; Lange, P.F.; Mark, J.C.; et al. TAILS N-terminomics and proteomics show protein degradation dominates over proteolytic processing by cathepsins in pancreatic tumors. Cell Rep. 2016, 16, 1762–1773. [Google Scholar] [CrossRef] [Green Version]
- Kleifeld, O.; Doucet, A.; Prudova, A.; Keller, U.a.d.; Gioia, M.; Kizhakkedathu, J.N.; Overall, C.M. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 2011, 6, 1578–1611. [Google Scholar] [CrossRef]
- Wold, F. In vivo chemical modification of proteins (post-translational modification). Annu. Rev. Biochem. 1981, 50, 783–814. [Google Scholar] [CrossRef]
- Biancur, D.E.; Paulo, J.A.; Matachowska, B.; del Rey, M.Q.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Bellac, C.L.; Dufour, A.; Krisinger, J.M.; Loonchanta, A.; Starr, A.E.; Keller, U.A.d.; Lange, P.F.; Goebeler, V.; Kappelhoff, R.; Butler, G.S.; et al. Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. 2014, 9, 618–663. [Google Scholar] [CrossRef] [Green Version]
- Starr, A.E.; Bellac, C.L.; Dufour, A.; Goebeler, V.; Overall, C.M. Biochemical characterization and N-terminomics analysis of leukolysin, the membrane-type 6 matrix metalloprotease (MMP25) chemokine and vimentin cleavages enhance cell migration and macrophage phagocytic activities. J. Biol. Chem. 2012, 287, 13382–13395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlage, P.; Egli, F.E.; Nanni, P.; Wang, L.W.; Kizhakkedathu, J.N.; Apte, S.S.; Keller, U.a.d. Time-resolved analysis of the matrix metalloproteinase 10 substrate degradome. Mol. Cell. Proteom. 2014, 13, 580–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- auf dem Keller, U.; Bellac, C.L.; Li, Y.; Lou, Y.; Lange, P.F.; Ting, R.; Harwig, C.; Kappelhoff, R.; Dedhar, S.; Adam, M.J.; et al. Novel matrix metalloproteinase inhibitor [18F] marimastat-aryltrifluoroborate as a probe for in vivo positron emission tomography imaging in cancer. Cancer Res. 2010, 70, 7562–7569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudova, A.; auf dem Keller, U.; Butler, G.S.; Overall, C.M. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteom. 2010, 9, 894–911. [Google Scholar] [CrossRef] [Green Version]
- Butler, G.S.; Overall, C.M. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 2009, 8, 935–948. [Google Scholar] [CrossRef]
- Hanifeh, M.; Rajamaki, M.M.; Makitalo, L.; Syrjä, P.; Sankari, S.; Kilpinen, S.; Spillmann, T. Identification of matrix metalloproteinase-2 and-9 activities within the intestinal mucosa of dogs with chronic enteropathies. Acta Vet. Scand. 2018, 60, 16. [Google Scholar] [CrossRef] [Green Version]
- Moesgaard, S.G.; Aupperle, H.; Rajamaki, M.M.; Falk, T.; Rasmussen, C.E.; Zois, N.E.; Olsen, L.H. Matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs) and transforming growth factor-β (TGF-β) in advanced canine myxomatous mitral valve disease. Res. Vet. Sci. 2014, 97, 560–567. [Google Scholar] [CrossRef]
- Qiu, Z.; Hu, J.; Van den Steen, P.E.; Opdenakker, G. Targeting matrix metalloproteinases in acute inflammatory shock syndromes. Comb. Chem. High Throughput Screen. 2012, 15, 555–570. [Google Scholar] [CrossRef]
- Xia, L.; Zhu, Z.; Zhang, L.; Xu, Y.; Chen, G.; Luo, J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol. Appl. Biochem. 2019. [Google Scholar] [CrossRef] [Green Version]
- Wojtowicz-Praga, S.; Low, J.; Marshall, J.; Ness, E.; Dickson, R.; Barter, J.; Sale, M.; McCann, P.; Moore, J.; Cole, A.; et al. Phase I trial of a novel matrix metalloproteinase inhibitor batimastat (BB-94) in patients with advanced cancer. Invest. New Drugs 1996, 14, 193–202. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Poole, C.; Primrose, J.; Rosemurgy, A.; Malfetano, J.; Brown, P.; Berrington, A.; Cornish, A.; Lynch, K.; Rasmussen, H.; et al. Combined analysis of studies of the effects of the matrix metalloproteinase inhibitor marimastat on serum tumor markers in advanced cancer: Selection of a biologically active and tolerable dose for longer-term studies. Clin. Cancer Res. 1998, 4, 1101–1109. [Google Scholar] [PubMed]
- Bramhall, S.R.; Rosemurgy, A.; Brwon, P.D.; Bowry, C.; Buckels, J.A.; Marimastat Pancreatic Cancer Study Group. Marimastat as first-line therapy for patients with unresectable pancreatic cancer: A randomized trial. J. Clin. Oncol. 2001, 19, 3447–3455. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Hallissey, M.T.; Whiting, J.; Scholefield, J.; Tierney, G.; Stuart, R.C.; Hawkins, R.E.; McCulloch, P.; Maughan, T.; Brown, P.D.; et al. Marimastat as maintenance therapy for patients with advanced gastric cancer: A randomised trial. Br. J. Cancer 2002, 86, 1864–1870. [Google Scholar] [CrossRef] [PubMed]
- Sparano, J.A.; Bernardo, P.; Stephenson, P.; Gradishar, W.J.; Ingle, J.N.; Zucker, S.; Davidson, N.E. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J. Clin. Oncol. 2004, 22, 4683–4690. [Google Scholar] [CrossRef]
- Levitt, N.C.; Eskens, F.A.L.M.; O’Byrne, K.J.; Propper, D.J.; Denis, L.J.; Owen, S.J.; Choi, L.; Foekens, J.A.; Wilner, S.; Wood, J.M.; et al. Phase I and pharmacological study of the oral matrix metalloproteinase inhibitor, MMI270 (CGS27023A), in patients with advanced solid cancer. Clin. Cancer Res. 2001, 7, 1912–1922. [Google Scholar] [PubMed]
- Bissett, D.; O’Byrne, K.J.; von Pawel, J.; Gatzemeier, U.; Price, A.; Nicolson, M.; Mercier, R.; Mazabel, E.; Penning, C.; Zhang, M.H.; et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J. Clin. Oncol. 2004, 23, 842–849. [Google Scholar] [CrossRef]
- Heath, E.I.; Burtness, B.A.; Kleinberg, L.; Salem, R.R.; Yang, S.C.; Heitmiller, R.F.; Canto, M.I.; Knisely, J.P.S.; Topazian, M.; Montgomery, E.; et al. Phase II, parallel-design study of preoperative combined modality therapy and the matrix metalloprotease (mmp) inhibitor prinomastat in patients with esophageal adenocarcinoma. Invest. New Drugs 2006, 24, 135–140. [Google Scholar] [CrossRef]
- Erlichman, C.; Adjei, A.A.; Alberts, S.R.; Sloan, J.A.; Goldberg, R.M.; Pitot, H.C.; Rubin, J.; Atherton, P.J.; Klee, G.G.; Humphrey, R. Phase I study of the matrix metal loproteinase inhibitor, BAY 12–9566. Ann. Oncol. 2001, 12, 389–395. [Google Scholar] [CrossRef]
- Moore, M.J.; Hamm, J.; Dancey, J.; Eisenberg, P.D.; Dagenais, M.; Fields, A.; Hagan, K.; Greenberg, B.; Colwell, B.; Zee, B.; et al. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2003, 21, 3296–3302. [Google Scholar] [CrossRef]
- Hirte, H.; Vergote, I.B.; Jeffrey, J.R.; Grimshaw, R.N.; Coppieters, S.; Schwartz, B.; Tu, D.; Sadura, A.; Brundage, M.; Seymour, L. A phase III randomized trial of BAY 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: A National Cancer Institute of Canada Clinical Trials Group Study. Gynecol. Oncol. 2006, 102, 300–308. [Google Scholar] [CrossRef]
- Rudek, M.A.; Figg, W.D.; Dyer, V.; Dahut, W.; Turner, M.L.; Steinberg, S.M.; Liewehr, D.J.; Kohler, D.R.; Pluda, J.M.; Reed, E. Phase I clinical trial of oral COL-3, a matrix metalloproteinase inhibitor, in patients with refractory metastatic cancer. J. Clin. Oncol. 2001, 19, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, M.; Cooley, T.P.; Lee, J.Y.; Rudek, M.A.; Scadden, D.T.; Ratner, L.; Pluda, J.M.; Figg, W.D.; Krown, S.E.; Dezube, B.J. Matrix metalloproteinase inhibitor COL-3 in the treatment of AIDS-related Kaposi’s sarcoma: A phase I AIDS malignancy consortium study. J. Clin. Oncol. 2001, 20, 153–159. [Google Scholar]
- Chu, Q.S.C.; Forouzesh, B.; Syed, S.; Mita, M.; Schwartz, G.; Cooper, J.; Curtright, J.; Rowinsky, E.K. A phase II and pharmacological study of the matrix metalloproteinase inhibitor (MMPI) COL-3 in patients with advanced soft tissue sarcomas. Invest. New Drugs 2007, 25, 359. [Google Scholar] [CrossRef] [PubMed]
- Baxter, B.T.; Pearce, W.H.; Waltke, E.A.; Littooy, F.N.; Hallett, J.W., Jr.; Kent, K.C.; Upchurch, G.R., Jr.; Chaikof, E.L.; Mills, J.L.; Fleckten, B.; et al. Prolonged administration of doxycycline in patients with small asymptomatic abdominal aortic aneurysms: Report of a prospective (Phase II) multicenter study. J. Vasc. Surg. 2002, 36, 1–12. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Humphrey, J.S.; Ness, E.A.; Johnson, M.D.; Gupta, E.; Williams, K.; Daly, D.J.; Sonnichsen, D.; Conway, D.; Marshall, J.; et al. A phase I study of oral BMS-275291, a novel nonhydroxamate sheddase-sparing matrix metalloproteinase inhibitor, in patients with advanced or metastatic cancer. Clin. Cancer Res. 2004, 10, 1963–1970. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.D.; Saphner, R.J.; Waterhouse, D.M.; Chen, T.-T.; Rush-Taylor, A.; Sparano, J.A.; Wolff, A.C.; Cobleigh, M.A.; Galbraith, S.; Sledge, G.W. A randomized phase II feasibility trial of BMS-275291 in patients with early stage breast cancer. Clin. Cancer Res. 2004, 10, 1971–1975. [Google Scholar] [CrossRef] [Green Version]
- Chiappori, A.A.; Eckhardt, S.G.; Bukowski, R.; Sullivan, D.M.; Ikeda, M.; Yano, Y.; Yamada-Sawada, T.; Kambayashi, Y.; Tanaka, K.; Javle, M.M.; et al. A phase I pharmacokinetic and pharmacodynamic study of s-3304, a novel matrix metalloproteinase inhibitor, in patients with advanced and refractory solid tumors. Clin. Cancer Res. 2007, 13, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Magnussen, H.; Watz, H.; Kirsten, A.; Wang, M.; Wray, H.; Samuelsson, V.; Mo, J.; Kay, R. Safety and tolerability of an oral MMP-9 and-12 inhibitor, AZD1236, in patients with moderate-to-severe COPD: A randomised controlled 6-week trial. Pulm. Pharmacol. Ther. 2011, 24, 563–570. [Google Scholar] [CrossRef]
- Dahl, R.; Titlestad, I.; Lindqvist, A.; Wielders, P.; Wray, H.; Wang, M.; Samuelsson, V.; Mo, J.; Holt, A. Effects of an oral MMP-9 and-12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: A randomised controlled trial. Pulm. Pharmacol. Ther. 2012, 25, 169–177. [Google Scholar] [CrossRef]
- Botkjaer, K.A.; Kwok, H.F.; Terp, M.G.; Karatt-Vellatt, A.; Santamaria, S.; McCafferty, J.; Andreasen, P.A.; Itoh, Y.; Ditzel, H.J.; Murphy, G. Development of a specific affinity-matured exosite inhibitor to MT1-MMP that efficiently inhibits tumor cell invasion in vitro and metastasis in vivo. Oncotarget 2016, 7, 16773. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Chemical structures of small molecule MMP inhibitors. (a) Periostat®: doxycycline hyclate. (b) MLS000048794: 2-[[3,5bis(ethylamino)-2,4,6,8,9-pentazabicyclo[4 .3.0]nona-2,4,7,9-tetraen-7yl] sulfanyl-methyl]-1H-quinazolin4-one). (c) MLS000048818: N-[4-(difluoromethoxy)phenyl]-2-[(4-oxo-6-propyl-1H-pyrimidin-2-yl)sulfanyl]-acetamide. (d) MLS000048509: 2-[[4-(1,3,4-oxadiazol-2-yl)phenyl] methylsulfanyl]-1Hbenzoimidazole). (e) MLS000098372: 2-[[5-chloro-2 (difluoromethoxy)phenyl]methylsulfanyl]-6-methyl-1H-pyrimidin-4-one). (f) N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide. (g) JNJ0966: N-{2-[(2-methoxyphenyl)amino]-4′-methyl-4,5′-bi-1,3-thiazol-2′-yl}acetamide.
Figure 1.
Chemical structures of small molecule MMP inhibitors. (a) Periostat®: doxycycline hyclate. (b) MLS000048794: 2-[[3,5bis(ethylamino)-2,4,6,8,9-pentazabicyclo[4 .3.0]nona-2,4,7,9-tetraen-7yl] sulfanyl-methyl]-1H-quinazolin4-one). (c) MLS000048818: N-[4-(difluoromethoxy)phenyl]-2-[(4-oxo-6-propyl-1H-pyrimidin-2-yl)sulfanyl]-acetamide. (d) MLS000048509: 2-[[4-(1,3,4-oxadiazol-2-yl)phenyl] methylsulfanyl]-1Hbenzoimidazole). (e) MLS000098372: 2-[[5-chloro-2 (difluoromethoxy)phenyl]methylsulfanyl]-6-methyl-1H-pyrimidin-4-one). (f) N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio)butanamide. (g) JNJ0966: N-{2-[(2-methoxyphenyl)amino]-4′-methyl-4,5′-bi-1,3-thiazol-2′-yl}acetamide.

Figure 2.
Experimental workflow of the N-terminomics/TAILS protocol using dimethylation isotopic labeling in a cancer mouse model treated with a protease inhibitor or a vehicle control. Tumors are lysed, reduced and alkylated before being isotopically labeled with light (+28 Da) or deuterated (+34 Da) formaldehyde. Proteins are then digested with trypsin and a pre-enrichment TAILS sample is collected. The remaining peptides are subjected to the TAILS polymer and N-termini are added to a size exclusion filter. Samples are subjected to liquid chromatography and tandem mass spectrometry (LC-MS/MS). The proteomics data is analyzed by bioinformatics software (e.g., MaxQuant). Data interpretation is analyzed with pathway enrichment tools (Metascape, STRING) or protease analysis software (TopFIND).
Figure 2.
Experimental workflow of the N-terminomics/TAILS protocol using dimethylation isotopic labeling in a cancer mouse model treated with a protease inhibitor or a vehicle control. Tumors are lysed, reduced and alkylated before being isotopically labeled with light (+28 Da) or deuterated (+34 Da) formaldehyde. Proteins are then digested with trypsin and a pre-enrichment TAILS sample is collected. The remaining peptides are subjected to the TAILS polymer and N-termini are added to a size exclusion filter. Samples are subjected to liquid chromatography and tandem mass spectrometry (LC-MS/MS). The proteomics data is analyzed by bioinformatics software (e.g., MaxQuant). Data interpretation is analyzed with pathway enrichment tools (Metascape, STRING) or protease analysis software (TopFIND).

Figure 3.
Schematic of three pharmacological approaches to inhibit the biological functions of MMPs: peptides (purple), monoclonal antibodies (green) and small molecules (blue).
Figure 3.
Schematic of three pharmacological approaches to inhibit the biological functions of MMPs: peptides (purple), monoclonal antibodies (green) and small molecules (blue).


{kind=link}
{kind=link}
{kind=link}
Table 1.
MMPs inhibitors in clinical trials.
| Inhibitor Names | Class/Structure | Selectivity | Diseases | Clinical Trial | Outcomes/ Side Effects | References |
|---|---|---|---|---|---|---|
| Batimastat | Peptidomimetic/ Hydroxamate | Broad spectrum | Malignant tumor | Phase I | Local toxicities i.e., abdominal discomfort | [81] |
| Marimastat | Peptidomimetic/ Hydroxamate | Broad spectrum | Progressive ovarian, prostatic, pancreatic and colorectal cancer | Phase III | Adverse musculoskeletal (MS) syndrome | [82] |
| Pancreatic cancer | Phase III | Musculoskeletal pain and inflammation | [83] | |||
| Pancreatic cancer | Phase III, in combination with gemcitabine | Well tolerated but no therapeutic beneficial effects | [40] | |||
| Gastric cancer | Phase III | Severe musculoskeletal (MS) syndrome | [84] | |||
| Metastatic breast cancer | Phase I | Musculoskeletal pain associated with inferior survival | [85] | |||
| MMI-270 | Hydroxamate/Small molecule | Broad spectrum | Advanced solid cancer | Phase I | Rash and musculoskeletal pain | [86] |
| Prinomastat | Hydroxamate/Small molecule | Broad spectrum | Advanced cancer | Phase I | No response in tumor growth | [36] |
| Non-small cell lung cancer (stage IIIB or IV) | Phase III | Musculoskeletal syndrome | [87] | |||
| Esophagus cancer | Phase II | Unexpected thromboembolic events | [88] | |||
| Tanomastat (Bay 12-9566) | Biphenyl, thioether zinc-binding group/small molecule | MMP2, -3 and -9 | Solid tumor | Phase I | Mild toxicity, no musculoskeletal pain. No effect on tumor | [89] |
| Pancreatic cancer without prior chemotherapy | Phase III | Poorer survival | [90] | |||
| Ovarian cancer | Phase III | Well tolerated but did not impact patients’ survival | [91] | |||
| Metastat | Tetracycline derivatives/small molecule | MMP2 and -9 | Refractory solid tumors | Phase I | Subcutaneous phototoxicity | [92] |
| AIDS related Kaposi’s sarcoma | Phase I Applied with sun protection | Photosensitivity reaction | [93] | |||
| Advanced soft tissue sarcoma | Phase II Applied with sun protection | Photosensitivity reaction | [94] | |||
| Periostat®/ Doxycycline | Tetracycline derivatives/small molecule | Broad spectrum | Periodontitis | Phase III | Well tolerated; improved outcome | [41,43] |
| Asymptomatic abdominal aortic aneurysms | Phase II | Well tolerated; but no significant therapeutic effects | [95] | |||
| Multiple sclerosis | Phase II Along with IFNß-1a | Well tolerated; improved outcome | [46] | |||
| Type II diabetes | Phase III | Reduced inflammation and better insulin sensitivity | [47] | |||
| Rebimastat (BMS- 275291) | Mercaptoacyl, thiol zinc-binding group/small molecule | MMP1, -2, -8, and MT1-MMP | Advanced cancer | Phase I | Well tolerated; no tumor response | [96] |
| Early-stage breast cancer | Phase III | Study was terminated because of toxicity | [97] | |||
| Non-small cell lung cancer | Phase II along with paclitaxel and carboplatin | Well tolerated but poor therapeutic response | [38] | |||
| Non-small cell lung cancer | Phase II along with paclitaxel and carboplatin | Increased toxicity with no improved survival | [37] | |||
| S-3304 | Sulfonamide derivatives/small molecule | MMP2 and -9 | Advanced solid tumors | Phase I | Well tolerated | [98] |
| AZD1236 | MMP9 and -12 | Moderate to severe Chronic obstructive pulmonary disease (COPD) | Phase II | Well tolerated but no therapeutic efficacy | [99,100] | |
| Neovastat (AE-941) | Mixed extract from shark cartilage | Broad spectrum | Non-small cell lung cancer (stage III) | Phase III along with chemotherapy | Well tolerated but no therapeutic effect | [39] |
Table 2.
Small molecule exosite MMP inhibitors.
| Compounds | Target | Binding Site | Mechanism of Action | Assays and Models Tested on | References |
|---|---|---|---|---|---|
| N-[4 (difluoromethoxy) phenyl] 2-[(4-oxo-6-propyl 1Hpyrimidin-2yl) sulfanyl]-acetamide | MMP9 | Hemopexin (PEX) | Interfered with homodimerization; inhibition of cell migration and proliferation | Tumor growth and metastasis (xenograft mouse model) | [16] |
| NSC405020 | MT1-MMP | Hemopexin (PEX) | Interfered with homodimerization and interaction with catalytic domain | Tumor growth (xenograft mouse model) | [24] |
| JNJ0966: (N-{2-[(2-methoxyphenyl) amino]-4-methyl-4,5-bi-1,3-thiazol-2-yl} acetamide) | MMP9 | Pro-peptide domain | Inhibit activation of MMP9 without affecting MMP1, -2, -3, -9 and MT1-MMP | Autoimmune encephalomyelitis (mouse model) | [29] |
| N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio) butanamide | MMP9 | Hemopexin (PEX) | Inhibition of homodimerization; decreased cancer cell migration- blocks cancer cell invasion of basement membrane and angiogenesis | in vitro migration assay, Tumor growth (xenograft mouse model), angiogenesis (chicken chorioallantoic membrane) | [17] |
| Synthesized acrylamides (1) 3-bromo-N-(4-nitrophenyl) propenamide (2) 3-bromo-N-{4-[(pyrimidine-2-yl) sulfamoyl] phenyl} propenamide (3) 3-bromo-N-{4-[(4,6 dimethylpyrimidin-2-yl) sulfamoyl]-phenyl} propanamide | MMP9 | Hemopexin (PEX) | Inhibition of 4T1 breast cancer cell growth; inhibition of MMP9 gelatinolytic activity | in vitro migration assay and (xenograft mouse model) | [22] |
Table 3.
Regulation of MMP inhibition by antibodies.
| Name | Antibody Type | Target | Epitope/Domains | Assays and Models Tested on | References |
|---|---|---|---|---|---|
| LEM-2/5 | Monoclonal | MT1-MMP | Surface epitope; V-P loop | Migrating cancer cells; | [59] |
| lung pathology; influenza | [48] | ||||
| SD3 | Monoclonal | MMP2 and -9 | Catalytic domain | Inflammatory bowel disease (mouse model); colitis | [55] |
| REGA-3G12 | Monoclonal | MMP-9 | Catalytic domain other than Zn2+ binding | Inhibited MMP9 proteolytic activity | [56,57] |
| DX-2400 | Fab fragment | MT1-MMP | Catalytic domain | Breast cancer | [58] |
| Multiple (A4-7 Fc-ScFv, E2_C6 Fc-ScFv) | Antibody fragments | MT1-MMP | Catalytic domain outside the active site cleft, inhibiting binding to triple helical collagen | Tumor growth and proliferation (xenograft mouse model) | [101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Das, N.; Benko, C.; Gill, S.E.; Dufour, A. The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors. Pharmaceuticals 2021, 14, 31. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010031
AMA Style
Das N, Benko C, Gill SE, Dufour A. The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors. Pharmaceuticals. 2021; 14(1):31. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010031
Chicago/Turabian StyleDas, Nabangshu, Colette Benko, Sean E. Gill, and Antoine Dufour. 2021. "The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors" Pharmaceuticals 14, no. 1: 31. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14010031
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.