1. Introduction
Recent advances in studies on the biology of extracellular vesicles (EVs) demonstrated their exceptional role in intercellular communication [
1], both in physiological and pathological conditions [
2]. Among other processes, EV-mediated cell signaling cascades are currently extensively investigated in the terms of immune regulation. EVs have also been proposed to substitute for the activity of parental immune cells; however, they seem to be less effective [
3]. At present, EVs receive special attention as physiological delivery tools, the usage of which reduces the side effects of treatment. However, the latter application is still fraught with many challenges, including enhancing their biological effectiveness and directing them towards desired target cells [
4].
Shortly after the discovery of suppressor T (Ts) cells, one of their subpopulations was shown to inhibit mouse hapten-induced contact hypersensitivity (CHS) reaction by generating so-called T suppressor factor (TsF) [
5,
6]. Our recent research uncovered that TsF consists of miRNA-150 carried by EVs, hereinafter called Ts-EVs. Those downregulate both hapten-induced CHS [
5,
7,
8], and delayed-type hypersensitivity (DTH) to protein antigens, such as ovalbumin (OVA) [
9], and casein [
10]. Both miRNA-150 and Ts-EVs are produced by CD8+ Ts cells, not expressing FoxP3, and activated through the intravenous administration of syngeneic red blood cells coupled with hapten or protein antigen [
5]. Interestingly, Ts-EVs are surface coated with antigen-specific antibody light chains derived by B1a cells activated by skin immunization [
7,
11]. This ensures the antigen specificity of immune suppression mediated by Ts-EV-delivered miRNA-150 [
12].
Our subsequent detailed studies revealed that miRNA-150-carrying Ts-EVs target antigen-presenting cells (APCs), especially antigen-primed macrophages, both in hapten-induced CHS and in OVA-induced DTH reactions [
8,
9]. In turn, Ts-EV-targeted macrophages suppress DTH immune responses by inhibiting the activation and proliferation of effector T lymphocytes and by increasing their apoptosis [
8,
13]. In addition, macrophages treated with TsF were previously shown to release the macrophage suppressor factor (MSF) of barely characterized nature [
6]. Moreover, Tung et al. have recently demonstrated that regulatory T cell-derived EVs induce tolerogenic phenotype in targeted dendritic cells due to the transmission of miRNA-150 [
14]. Together with our observations, this implies a crucial role of miRNA-150 in tolerogenic interactions between regulatory/suppressor T lymphocytes and APCs. However, this speculation remained unclear, and thus the current studies aimed at investigating the exact mechanism of suppressive action of Ts-EV-targeted macrophages on effector T cells.
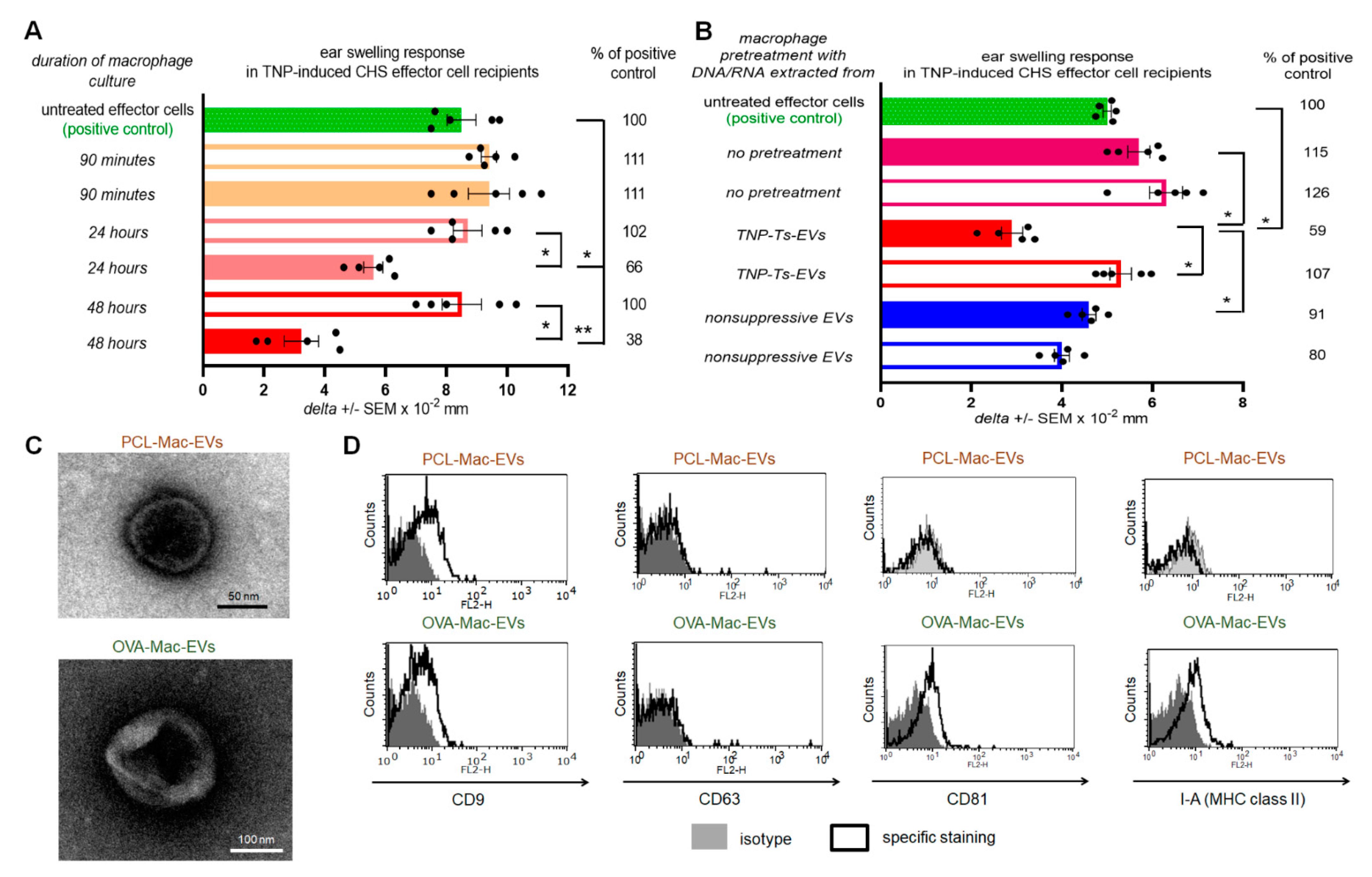
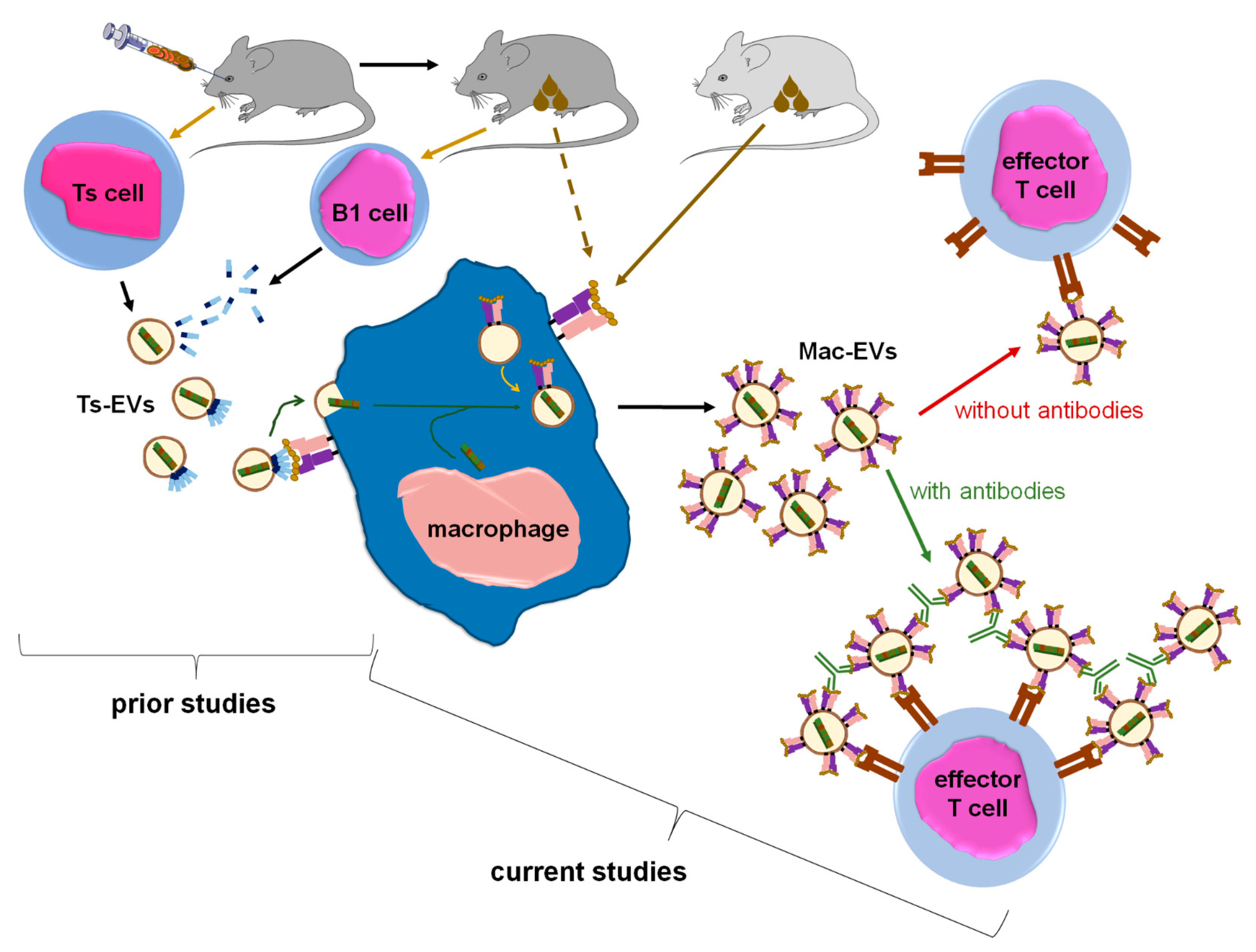
To examine how APCs treated with Ts-EVs suppress effector T lymphocytes, we cultured Ts-EV-pretreated macrophages and tested the resulting supernatant for suppressive activity in vivo, showing that the DTH suppression is mediated by macrophage-derived EVs, hereinafter called Mac-EVs. Furthermore, the suppressive action of Mac-EVs was found to be miRNA-150-dependent, triggered by immune synapse formation, and could be either abolished by pre-incubation with anti-CD9 antibodies or enhanced by pre-incubation with antigen-specific antibodies that can specifically bind to Mac-EVs. The latter finding led us to hypothesize that antigen-specific antibodies aggregate Mac-EVs expressing major histocompatibility complex (MHC) class II molecules. The final validation of this assumption with nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM), and in vivo assays, confirmed the significantly enhanced suppressive activity of aggregated Mac-EVs against DTH effector T cells. To the best of our knowledge, this is the first demonstration that antigen-specific antibodies could be easily used for increasing the biological activity of MHC class II-positive EVs, which appears to have a great therapeutic potential, both in enhancing EVs’ effectiveness and directed cell targeting.
3. Discussion
EVs are considered key players in intercellular signaling pathways that are able to specifically deliver the enclosed cargo to the desired target cells to modify their biological activities. The last two decades have brought about numerous discoveries that have greatly increased the knowledge of the mechanisms underlying these exceptional functions of EVs. Accordingly, our current research findings propose a novel approach to enhance EVs’ therapeutic potential, especially that EVs seem to have numerous advantages over synthetic liposomes and nanoparticles [
19]. However, rapid clearance from circulation along with insufficient targeting of desired cells could limit their biological efficacy [
3], and thus clinical usefulness [
20]. Our current research findings suggest that these disadvantages could be overcome by aggregating homogenous EVs with antigen-specific antibodies (
Figure 7).
Along these lines, after release by the parental cell, EVs seem to disperse quickly in body fluids where they mix with their counterparts from other cellular sources [
21], and form a colloidal suspension with a natural tendency to aggregate [
22]. On the one hand, EVs’ aggregation is considered an unwanted side effect that makes reliable single-vesicle profiling very difficult [
23,
24]. On the other hand, one can speculate that aggregation is a physiological mechanism, which evolved to increase EVs’ half-life in body fluids. However, such “colloid aggregates” are likely formed by morphologically and functionally variable EVs due to a very high heterogeneity of EV population that circulates within body fluids. In contrast, herein we observed that a cross-linking of EVs that express the same antigenic determinant with specific antibodies seems to produce homogeneous aggregates, characterized not only by increased half-life, but especially by augmented ability to target desired cells. This in turn greatly enhances their biological effectiveness in vivo, as observed in the current circumstances.
Our observations are in line with another report demonstrating that MHC class II-expressing EVs activated T cells less efficiently than their parental dendritic cells, unless EVs had been cross-linked with latex beads [
25]. The antibody-mediated aggregation of EVs proposed herein seems to take an advantage over the latter approach, since the antibodies could be physiologically degraded in the targeted cells, in contrast to latex beads. Furthermore, EVs released into 48-h culture supernatant by OVA-loaded, lipopolysaccharide (LPS)-stimulated dendritic cells have been shown to bind antibodies present in sera of OVA-immunized mice, and to induce OVA-specific T-cell proliferation in vivo [
26]. Thus, these observations support the hypothesis that EVs could bind antigen-specific antibodies, which potently enhances their activity.
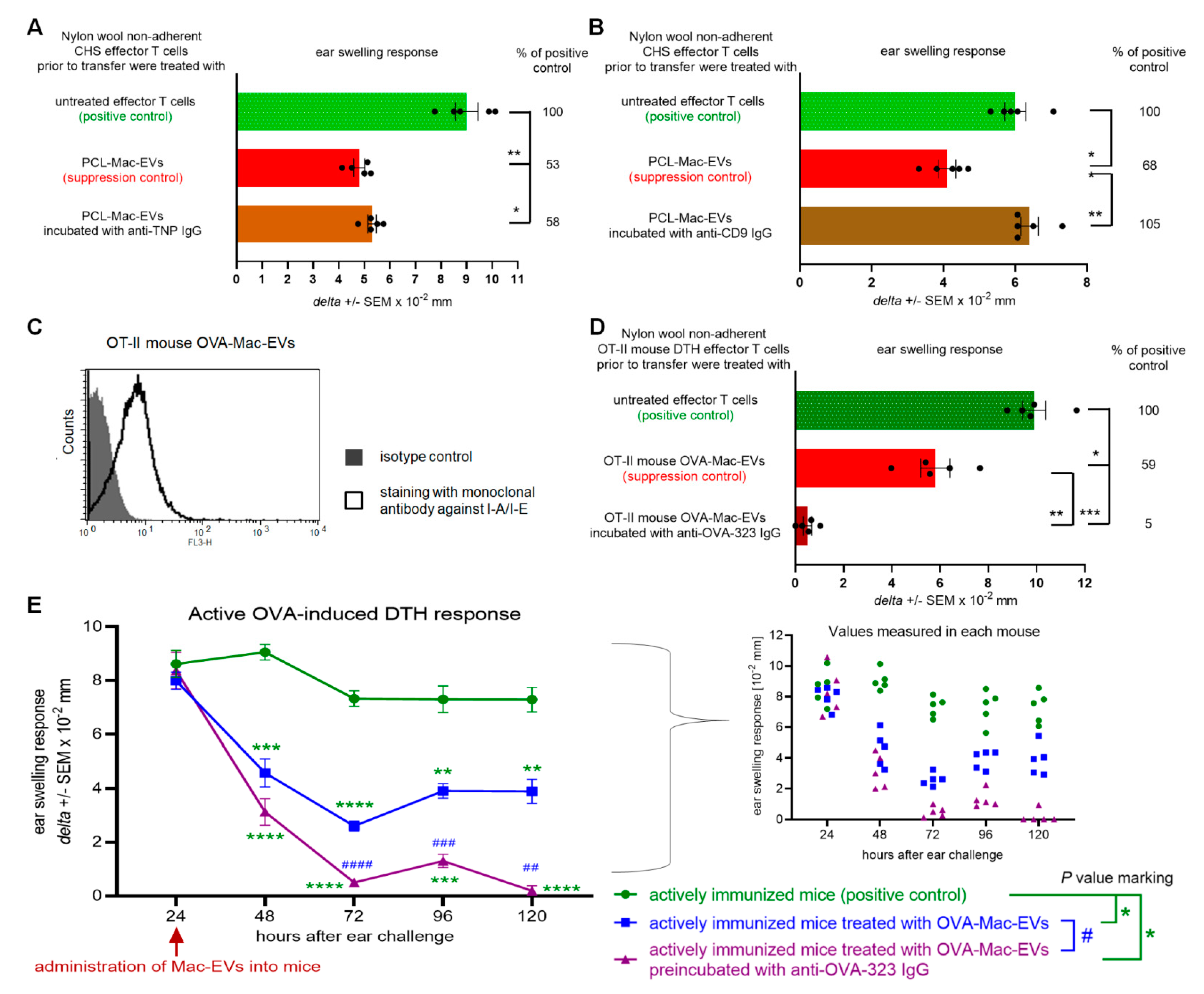
From another point of view, the pattern of expressed markers differs between PCL-Mac-EVs and OVA-Mac-EVs, especially given that only the latter were found to be MHC class II-positive. This discrepancy may result from subtle, but meaningful, differences in the activation of APCs by hapten and protein antigen. The successful presentation of antigenic peptides derived from the latter requires its endocytosis and processing by APCs, which in turn become activated. In contrast, reactive hapten derivatives could covalently bind to APC membrane proteins [
27]. As a consequence, such directly haptenated APCs [
28] may not become primed enough [
29] to be capable of translocating hapten/MHC class II complexes onto EVs’ membrane [
30]. This seems to apply to current experimental conditions, in which OVA-Ts-EV-treated macrophages from OVA-immunized OT-II mice produced MHC class II-positive OVA-Mac-EVs, while TNP-Ts-EV-treated macrophages from PCL-sensitized CBA mice released PCL-Mac-EVs that failed to express MHC class II molecules. This assumption is additionally supported by the fact that, in contrast to OVA-Mac-EVs, the suppressive activity of PCL-Mac-EVs was not affected by incubation with anti-TNP IgG antibodies. Furthermore, one can speculate that an analogous mechanism may explain the lack of CD81 expression on PCL-Mac-EVs, since this tetraspanin was reported to be involved in both the activation of antigen-primed macrophages [
31], and the trafficking of MHC class II molecules to EVs during their intracellular biogenesis [
32].
On the other hand, both PCL- and OVA-Mac-EVs expressed a CD9 marker involved in sorting of MHC class II molecules into EVs [
30], and likely playing a pivotal role in Evs’ trafficking and uptake by targeted cells [
32]. Thus, in current conditions, we assumed that anti-CD9 monoclonal antibodies abolished the suppressive activity of PCL-Mac-Evs by impairing their docking to the membrane and further uptake by CHS effector T cells. It has been postulated that the fusogenic activity of CD9 depends on its association with different adhesion molecules [
33], including lymphocyte function-associated antigen-1 (LFA-1) [
34], on acceptor cells. Accordingly, the internalization of Evs by tumor cells has been recently found to be either impaired by treatment with antigen-binding fragment (Fab) of anti-CD9 antibody or slightly enhanced by incubation with anti-CD9 antibody [
18]. The latter observation is contradictory to our findings. However, in those circumstances, Evs and tumor cells were incubated together in the presence of anti-CD9 antibodies or their derived Fab portions. This allowed simultaneous binding of CD9 on both Evs and acceptor cells, each by one of the antideterminants of the whole IgG antibody, which has been proposed to be responsible for the enhancement of Evs’ endocytosis [
18,
35]. In contrast, in our experiments, PCL-Mac-Evs were firstly incubated with anti-CD9 monoclonal antibodies and then ultracentrifuged prior to the treatment of the CHS effector cells, which favors the binding of the EV-membrane expressed CD9 by the majority of antibody antideterminants. Similarly, the incubation of OVA-Mac-Evs with anti-OVA-323 antibodies led to their aggregation. However, some of the antideterminants might remain unoccupied, and thus could bind to the OVA-323 peptides that were stuck to the TCR of the targeted effector T cells, thereby allowing DTH suppression.
Furthermore, the binding of anti-OVA-323 antibodies and the expression of I-A/I-E molecules by OVA-Mac-Evs from OT-II mice implies that they display OVA-323 peptides complexed with MHC class II. Thus, in turn, OVA-Mac-EVs can be considered capable of the specific targeting of DTH effector T cells by interacting with their TCR. This hypothesis is supported by the observation that OVA-induced DTH effector T cells from OT-II mice cannot be suppressed by OVA-Mac-EVs when pre-incubated with OVA-323 peptide. Hence, we suggest that the expression of antigenic peptides complexed with MHC allows addressing EVs to T cells with antigen-specific TCR (
Figure 7). This in turn enables the highly specific and selective delivery of EV-transferred cargo, which has great importance in attempts to therapeutically suppress antigen-specific T cells.
On the other hand, MHC-expressing EVs are considered capable of antigen presentation [
36,
37]. However, previoys studies reported that EVs displaying peptides complexed with either MHC class I or MHC class II could not directly stimulate CD8+ cytotoxic T cells [
38] or CD4+ helper T cells [
25], respectively. Instead, for the efficient stimulation of T cells, these EVs had to be uptaken by mature dendritic cells [
25,
38]. Simultaneously, it was suggested that in such conditions EV-targeted mature dendritic cells provide costimulatory signal from CD80/CD86 molecules, which enables efficient priming of naive T lymphocytes [
39]. Conversely, artificially generated nanoparticles carrying MHC/peptide complexes but not costimulatory molecules have been recently proposed for therapeutic activation of immune tolerance owing to T-cell anergy [
40]. Accordingly, our initial analysis demonstrated that OVA-Mac-EVs do not express CD80 and CD86 costimulatory molecules (data not shown). Thus, one can speculate that OVA-Mac-EVs could induce the anergy of antigen-specific T cells by presenting OVA-323 peptide in the absence of costimulatory signals. Herein, this effect seems to reinforce the miRNA-150-mediated tolerogenic potential of OVA-Mac-EVs.
Thus far, miRNA-150 has been unequivocally proven to mediate the suppressive and self-tolerogenic potential of Ts-EVs [
5,
13]. Remarkably, the current research findings demonstrated that suppressive activity of Mac-EVs depends on miRNA-150 as well (
Figure 7). Thus, we assumed that antigen-presenting macrophages play a pivotal role in miRNA-150-induced immune suppression by transferring the multiplied suppressive signal from a few Ts cells to numerous effector T cells in an antigen-specific manner. In this regard, macrophages could be compared to a “resonance tube” (
Figure 7). Interestingly, a similar function could likely be applied to dendritic cells, which were suggested to amplify both antigen-presentation and maturation processes by transferring, respectively, peptide/MHC complex-bearing EVs [
38], and EV-contained miRNAs [
41], to neighboring dendritic cells. It is also worth noting that recent research demonstrated the significant modification of dendritic cell function, cytokine production especially, under the influence of the regulatory T cell-derived EVs. In that study, one of the miRNA molecules carried by regulatory T cell EVs and suspected to play a crucial role in the activation of the tolerogenic phenotype of dendritic cells was miRNA-150 [
14]. Thus, miRNA-150 could likely be considered a general inducer of the APC tolerogenic phenotype.
Interestingly, the reduced expression of miRNA-150 in ear skin samples was detected in mice with elicited CHS response to dinitrofluorobenzene hapten [
42], bringing another piece of evidence for the involvement of miRNA-150 in the regulation of CHS. Our previous studies showed that the suppressive activity of Ts-EV-targeted macrophages is associated with increased apoptosis as well as with impaired activation and proliferation of antigen-specific effector T cells [
8,
13]. The current observations confirmed that these effects are induced in targeted CD4+ T cells by Mac-EV-transferred miRNA-150. The downregulatory activity of miRNA-150 has already been shown in immune tolerance to transplanted antigens, and was similarly associated with an inhibited activation and proliferation as well as an increased apoptosis of CD4+ T lymphocytes [
43]. Our previous research also suggested that miRNA-150 decreases T-cell reactivity to IL-2 [
5,
7]. Other studies revealed that miRNA-150-induced and IL-2-mediated signaling cascades in T cells are mutually dependent [
44,
45,
46], and thus may underlie the Mac-EV-induced effects in DTH effector T cells. However, this aspect requires further investigation. Similarly, further studies are needed to specify the molecular targets of miRNA-150 in both macrophages and DTH effector T cells.
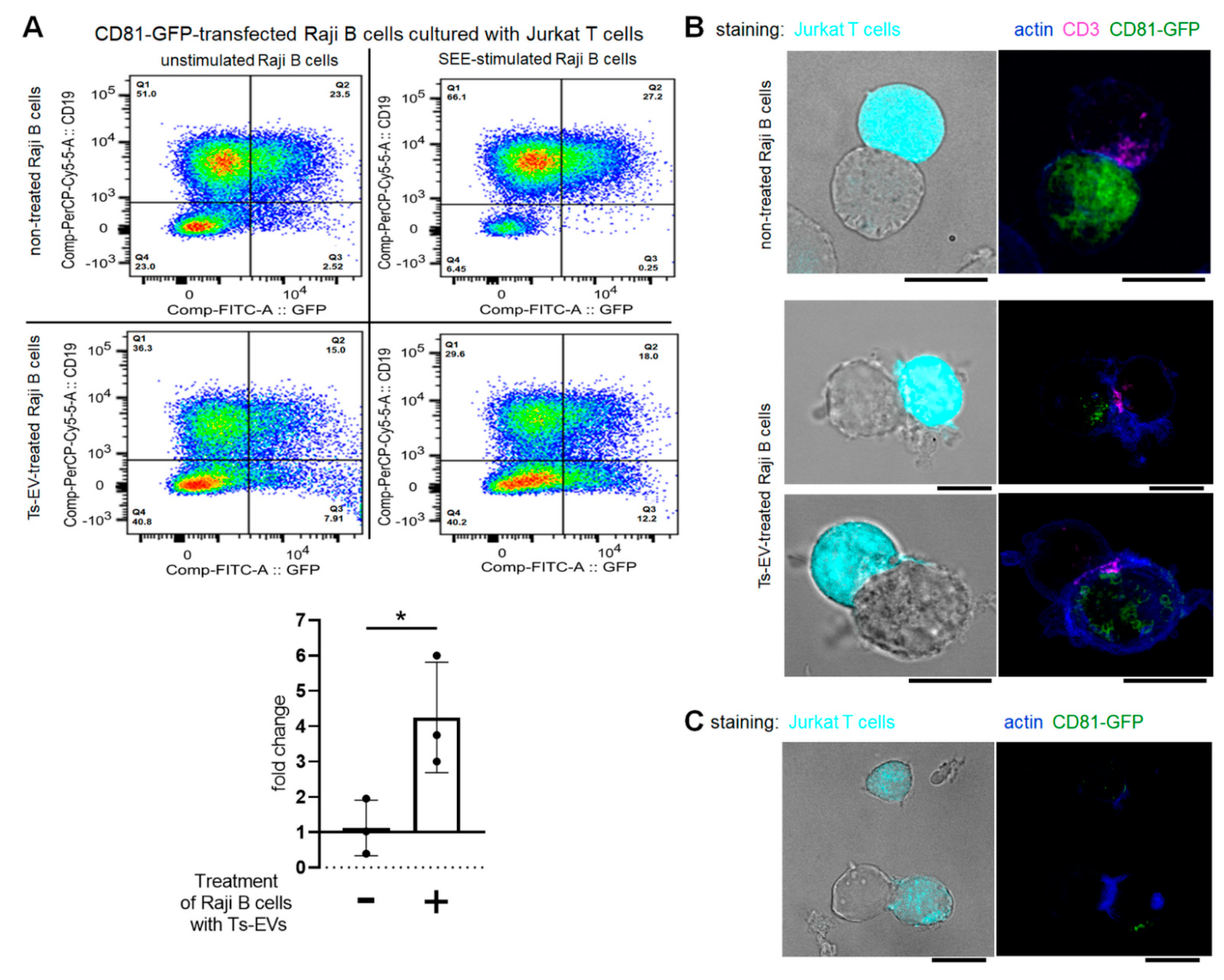
The homology of the sequences of mouse and human miRNA-150 allowed us to subject OVA-Ts-EVs to the standardized model for testing of EV-mediated intercellular interactions at the immune synapse. With this model, the unidirectional transfer of miRNA-loaded, CD63
pos EVs from intact Jurkat T cells to SEE-pulsed Raji B cells was formerly reported [
17]. Similarly to that report, CD81-GFP-transfected Raji B cells displayed almost no vesicle transfer to the Jurkat T cells after SEE-stimulation in current conditions. However, and surprisingly, the treatment of CD81-GFP-transfected Raji B cells with OVA-Ts-EVs induced the SEE-promoted transmission of CD81-GFP
pos vesicles to Jurkat T cells. This implies that Ts-EV-treated APCs may suppress effector T cells by transferring EV-enclosed regulatory miRNA-150 at the immune synapse. Additionally, we observed the accumulation of CD81 in Ts-EV-treated, SEE-stimulated Raji B cells close to the site of CD3 accumulation in Jurkat T cells. Other studies demonstrated that CD81 in APCs physiologically co-localize with T cell-expressed CD3 at the cell–cell contact area to support immune synapse formation during antigen presentation [
47]. However, after 24 h of co-culture, we found some Jurkat T cells that emitted CD81-GFP-dependent fluorescence, which seems to confirm that T cells could be targeted by CD81
pos EVs from Ts-EV-treated APCs at the immune synapse. However, these interactions remain a subject of further investigation.
Further evidence for EV-mediated signaling at the immune synapse was provided by a previous study demonstrating an alternative pathway for MHC class II sorting in antigen-loaded dendritic cells. Namely, interaction with antigen-specific CD4+ T cells induces in dendritic cells the generation of multivesicular bodies containing luminal vesicles displaying MHC class II and CD9, which are then released as EVs and transferred to these T cells [
30]. Our current results show that such EV-mediated signaling at the immune synapse could be used for the transfer of regulatory miRNA-150, and thus may ensure the antigen-specificity of the induced immune tolerance.
Along these lines, Ts-EVs are surface coated with antigen-specific antibody light chains that confer the specificity of Ts-EVs’ suppressive activity [
12]. Recent findings allowed us to conclude that OVA-Ts-EV-coating antibody light chains bind antigenic peptides complexed with MHC on APCs [
9,
21]. This enables the antigen-specific delivery of miRNA-150 to APCs, which in turn become tolerogenic. The current research showed that OVA-Ts-EV-targeted APCs release miRNA-150-carrying OVA-Mac-EVs that could be equipped with antigenic determinant complexed with MHC class II, which allows OVA-Mac-EVs to target OVA-specific T cells, likely by binding to their TCR. Thus, such a circuit allows us to maintain the antigen-specificity of EV-miRNA-150-mediated immune suppression at each of its steps. Altogether, one can conclude that EVs are naturally predisposed to specifically target desired cells to deliver selected cargo [
48].
To summarize, after discovering of the role of miRNA-150 in an antigen-specific suppression of mouse CHS and DTH responses [
5,
9], we demonstrated that Ts-EVs target antigen-presenting macrophages [
8]. Our current results show that Ts-EV-treated macrophages release Mac-EV-enclosed miRNA-150 to suppress effector T cells. Immune synapse formation can trigger the release of Mac-EVs by miRNA-150-targeted APCs. PCL-Mac-EVs target CHS effector T cells in a CD9-dependent manner, while DTH effector T cell-suppressing OVA-Mac-EVs express MHC class II and thus can bind OVA-specific antibodies, which increases their suppressive activity in vivo. The latter observation led to a unique discovery of the antibody-induced aggregation of MHC class II-positive EVs that enhances their therapeutic potential in the treatment of the active DTH response. To the best of our knowledge, this is the first demonstration of the enhancement of EVs’ biological activity due to their aggregation with antigen-specific antibodies.
4. Materials and Methods
4.1. Antigens, Antibodies, Reagents and Culture Media
Following antigens, haptens and antibodies were used: ovalbumin (OVA), OVA 323-339 peptide (OVA-323, Sigma, St Louis, MO, USA); purified and biotinylated rabbit polyclonal anti-OVA-323-339 IgG antibodies, fluorescein isothiocyanate (FITC)-conjugated rabbit polyclonal IgG isotype antibodies (Innovagen, Lund, Sweden); purified mouse anti-trinitrophenol (TNP) IgG1 monoclonal antibody of A111-3 clone, purified and phycoerythrin (PE)-conjugated rat anti-mouse CD9 monoclonal antibody (clone KMC8), PE-conjugated rat anti-mouse CD63 monoclonal antibody (clone NVG-2), PE-conjugated hamster anti-mouse CD81 monoclonal antibody (clone Eat2), PerCP-Cy- or PE-conjugated rat anti-mouse I-A/I-E monoclonal antibody of M5/114.15.2 clone, FITC- or horseradish peroxidase (HRP)-conjugated streptavidin (all from BD Biosciences, San Diego, CA, USA); PCL (TNP-Cl, Chemtronix, Swannanoa, NC, USA); trinitrobenzene sulphonic acid (TNBSA, Eastman Chemicals, Rochester, NY, USA).
Following reagents and media were used: aldehyde/sulfate latex beads 4% w/v, 4 µm (cat. No A37304, Life Technologies, ThermoFisher Scientific, Carlsbad, CA, USA); mineral oil heavy fraction, protein-free Mishell–Dutton medium, RPMI 1640, minimal essential medium with amino acids, HEPES, cacodylic buffer, 2-mercaptoethanol, phenol-chloroform mixture (Sigma, St Louis, MO, USA); Dulbecco’s phosphate-buffered saline (DPBS), penicillin/streptomycin, sodium pyruvate, L-glutamine (Gibco Life Technologies, Grand Island, NY, USA); acetone, ethanol, glucose (P.O.Ch., Gliwice, Poland); 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, Pierce, ThermoFisher Scientific, Waltham, MA, USA); ethylenediaminetetraacetic acid (EDTA, BDH, Poole, UK); extra virgin olive oil (Basso Fedelee Figli, San Michele di Serino, Italy); 3,3′,5,5′-tetramethylbenzidine (TMB) liquid substrate for enzyme-linked immunosorbent assays (ELISA, BD Biosciences, San Diego, CA, USA).
4.2. Mice
Ten- to fourteen-week old mice of the C57BL/6 and miRNA-150−/− knock-out inbred strains were from Jackson Laboratories (Bar Harbor, ME) and CBA mice were either from Jackson Laboratories or, together with OT-II T-cell receptor (TCR) transgenic mice and some of C57BL/6 mice, from the 2nd Breeding Unit of the Jagiellonian University Medical College, Faculty of Medicine (Krakow, Poland). Mice were fed autoclaved food and water ad libitum.
4.3. Induction of Tolerance and Harvest of Suppressor T Cell-Derived EVs (Ts-EVs)
Freshly collected, pelleted mouse erythrocytes were conjugated with TNP hapten by mixing with TNBSA solution in cacodylic buffer (5.7 mg/mL) in a ratio of 7 mL of TNBSA solution per 1 mL of erythrocytes, and incubating 20 min at room temperature, in darkness on hematological roller. Otherwise, mouse erythrocytes in 50% DPBS suspension were labeled with OVA protein by incubation with 1% OVA solution in DPBS (in a ratio 1:5
v/
v) for 1 h at room temperature on hematological roller in the presence of EDC used as a coupling activating agent [
9,
12]. Ts cell-mediated tolerance was induced in mice as described previously [
5,
8]. In brief, CBA, miRNA-150
−/− or C57BL/6 mice were intravenously injected with 0.2 mL of 10% DPBS suspension of either TNP-coupled or OVA-coupled syngeneic erythrocytes on days 0 and 4, which, on day 9, was followed by, respectively, contact sensitization on shaved abdominal skin with 0.15 mL of 5% PCL solution in ethanol:acetone (3:1
v/
v) [
5,
8], or by double intradermal immunization with 0.2 mL of 0.5 mg/mL OVA-saline solution on day 8 and 9 [
9,
49]. On day 11 spleens and lymph nodes containing activated Ts cells were collected from tolerized mice and single cell suspensions were cultured in protein-free Mishell–Dutton medium at a concentration of 2 × 10
7 cells/mL for 48 h [
5,
9,
10]. The resulting supernatant was subsequently centrifuged at 300
g and 3000
g for 10 min, filtered through 0.45 µm and 0.22 µm molecular filters and then ultracentrifuged twice at 100,000
g for 70 min at 4 °C [
5,
8,
9]. Resulting pellet was resuspended in DPBS [
5], and used as either TNP-Ts-EVs or OVA-Ts-EVs, respectively [
5,
9].
Pellets of non-suppressive, control vesicles, so-called negative factor [
5,
7,
8], were obtained by ultracentrifugation of supernatants of 48-h culture of lymph node and spleen cells collected from naive mice, and processed as above.
In some instances, mixtures of DNA/RNA were extracted with phenol-chloroform from culture supernatants containing either Ts-EVs or control, non-suppressive vesicles (negative factor) by modified Chomczynski method [
50], as described previously [
7]. Where indicated, DNA/RNA extract was further treated with either DNase (D7231) or RNase A (R4375, both from Sigma, St Louis, MO, USA), or incubated with anti-miR-150 (Dharmacon RNAi Technologies, Lafayette, CO, USA), as described previously [
7]. OVA-Ts-EVs were similarly incubated with anti-miR-150 (Dharmacon RNAi Technologies, Lafayette, CO, USA) [
9].
4.4. Harvest of Macrophages and Their Treatment to Obtain Macrophage-Derived EVs (Mac-EVs)
Macrophage-enriched peritoneal exudate was induced by intraperitoneal injection of 1 mL of mineral oil into either naive, PCL-sensitized or OVA-immunized mice of CBA, C57BL/6, OT-II or miRNA-150
−/− strains [
8]. Five days later, macrophage-containing exudate was harvested by washing the peritoneal cavity with ice-cold DPBS with heparin (5 U/mL). The percentage of non-specific esterase positive macrophages in exudates in each case exceeded 95% [
51]. After washing, macrophages were treated for 30 min in 37 °C water-bath with DPBS-suspension of Ts-EVs, in a dose of approximately 1 × 10
9 Ts-EVs per 1 × 10
6 macrophages, as estimated by NTA [
5]. In some instances, macrophages were similarly treated with phenol-chloroform extracts (PCE) of Ts-EVs or negative factor vesicles in a dose of about 3 µg of PCE nucleic acid mixture per 1 × 10
6 cells [
7]. After washing out of excessive vesicles or nucleic acids at 300
g, Ts-EV/PCE-treated macrophages were cultured at 37 °C and 5% CO
2 in protein-free Mishell–Dutton medium at a concentration of 3 × 10
6 cells/mL for 48 h, unless otherwise indicated in particular experiment. The resulting supernatant was subsequently centrifuged at 300
g and 3000
g for 10 min, filtered through 0.45 µm and 0.22 µm molecular filters and then ultracentrifuged twice at 100,000
g for 70 min at 4 °C. The resulting pellet containing Mac-EVs was resuspended in DPBS for experimental usage. In some experiments supernatants above the pellets, remaining after ultracentrifugation of macrophage culture supernatants, were also used for treatment of adoptively transferred effector cells. Mac-EVs pelleted by ultracentrifugation of supernatant from the culture of TNP-Ts-EV-pretreated macrophages from mice epicutaneously sensitized with PCL were termed PCL-Mac-EVs. Otherwise, Mac-EVs pelleted by ultracentrifugation of supernatant from the culture of OVA-Ts-EV-pretreated macrophages from mice intradermally immunized with OVA were termed OVA-Mac-EVs.
Where indicated, OT-II mouse OVA-Mac-EVs were incubated with anti-miR-150 (Dharmacon RNAi Technologies, Lafayette, CO, USA) for 2 h in 37 °C water-bath, and, after washing, were used to treat OT-II mouse DTH effector cells prior to their adoptive transfer to naive wild type C57BL/6 mouse recipients.
4.5. Treatment of Mac-EVs with Antibodies
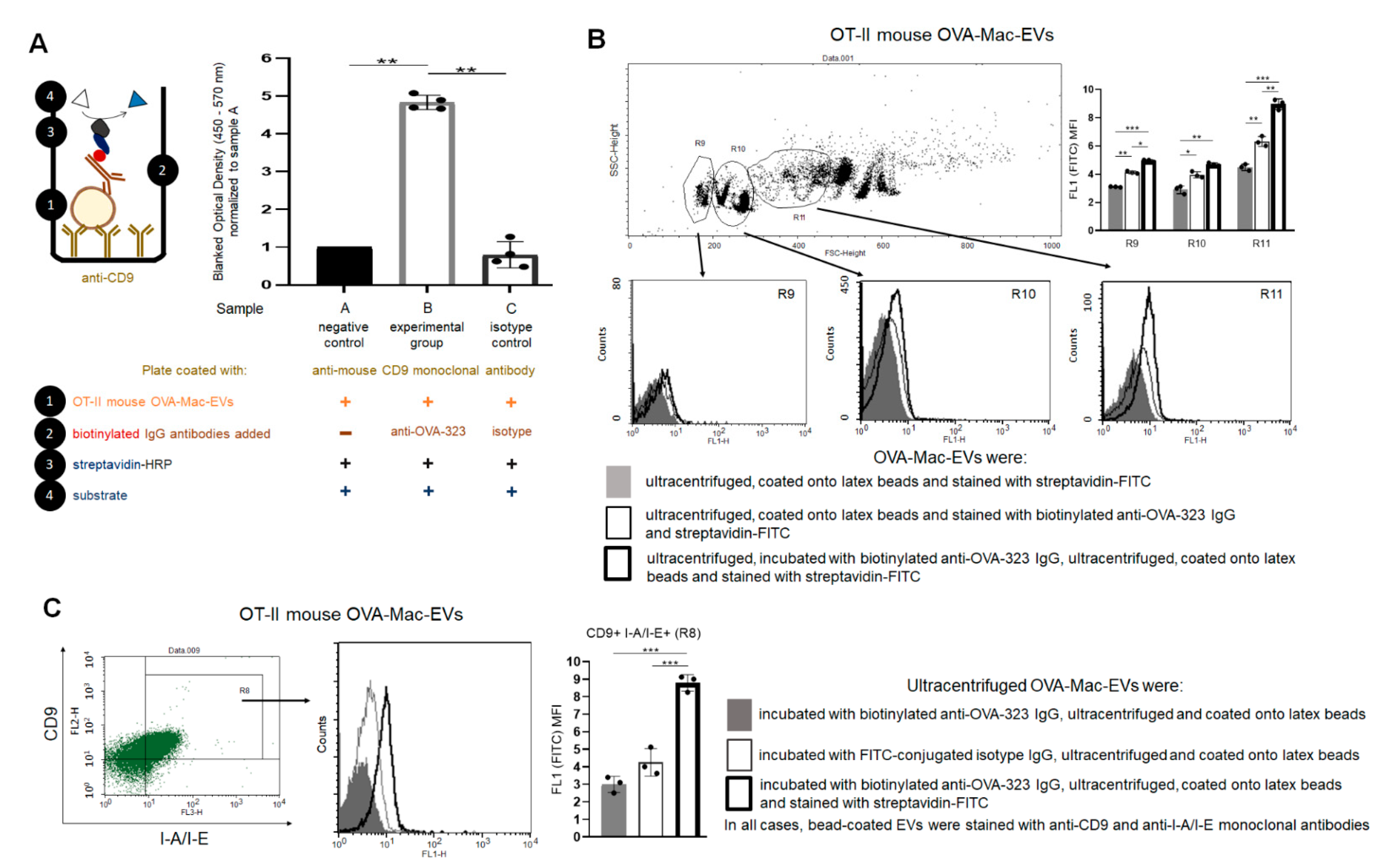
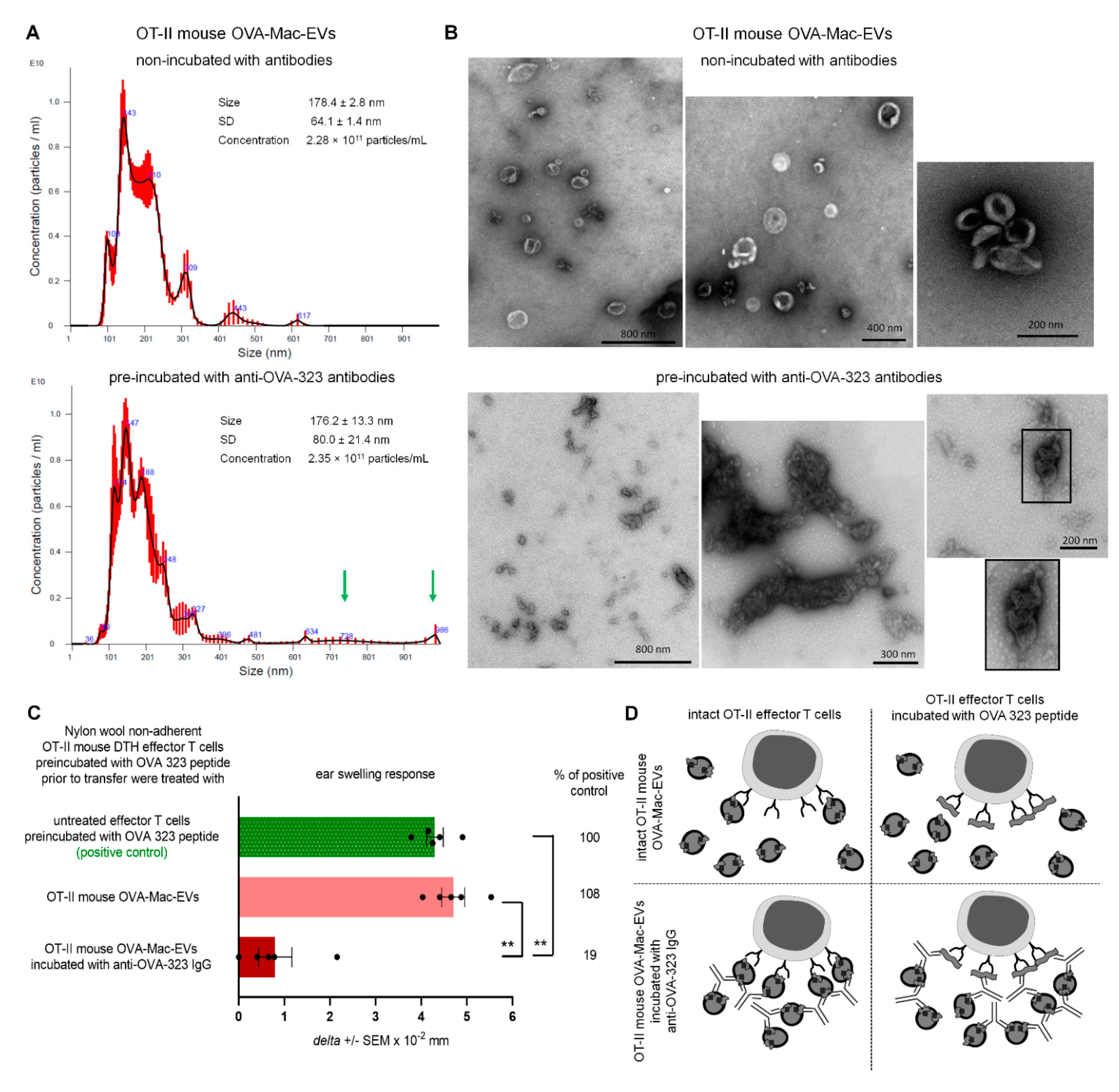
PCL-Mac-EVs were incubated overnight on ice with purified anti-TNP IgG or anti-CD9 monoclonal antibodies in a dose of 6 µg per 1 × 1010 Mac-EVs, which was followed by ultracentrifugation to remove the unbound antibodies. Pellet was then used to treat CHS effector T cells, as described below. Similarly, OVA-Mac-EVs were incubated overnight on ice with purified or biotinylated anti-OVA-323 IgG antibodies or with FITC-conjugated isotype IgG antibodies in a dose of 6 µg per 1 × 1010 Mac-EVs, which was followed by ultracentrifugation to remove the unbound antibodies. Then, pellet was either analyzed in NTA (Nanosight), visualized with TEM microscope, coupled onto latex beads and analyzed cytometrically, or used for treatment of actively immunized mice or DTH effector T cells, as described below.
4.6. Adoptive Transfer of CHS or DTH Effector Cells
CBA mice were contact sensitized with 0.15 mL of 5% PCL solution in ethanol:acetone mixture (3:1
v/
v) on shaved abdominal skin. Five days later lymph nodes and spleens were harvested from sensitized animals and proceeded for obtaining the single cell-suspension of CHS effector cells, which were then treated with PCL-Mac-EVs or control factors for 30 min in 37 °C water-bath and, after washing at 300
g, were transferred intravenously into naive recipients (7 × 10
7 cells per mouse) that were immediately challenged to elicit CHS reaction by application of 10 µL of 0.4% PCL solution in acetone:olive oil (1:1
v/
v) on each side of both ears. Twenty four hours later CHS reaction was measured as an increase in ear thickness (ear swelling response) with engineer’s micrometer (Mitutoyo, Japan) by blinded observer [
5,
8]. This method of CHS reaction assessment is essentially non-invasive, allows for repetition of measurements and obtained results strongly correlated with other methods [
52].
CBA, OT-II or C57BL/6 mouse donors of DTH effector cells were immunized on two consecutive days (0 and 1) with OVA protein antigen (without an adjuvant) by intradermal injections into four sites of abdominal skin of 0.2 mL, in total, of a 0.5 mg/mL OVA solution in 0.9% NaCl [
9,
49]. On day 5 lymph nodes and spleens were harvested from immunized mice and obtained DTH effector cells were then treated with OVA-Mac-EVs or control factors as describe above, before intravenous transfer into naive recipients of wild type strain (7 × 10
7 cells per mouse). Twenty four hours later recipient mice were injected intradermally into both ears with 10 µL of a 0.5 mg/mL OVA solution in 0.9% NaCl (challenge) to elicit DTH reaction, measured 24 h later, as described above. Where indicated, nylon wool non-adherent CHS or DTH effector T lymphocytes were isolated by triple separation of lymph node and spleen single-cell suspensions on nylon wool column [
53], with recovery in a range of 75% for CHS effector T cells and 40% for DTH effector T cells. Then, effector T lymphocytes were treated with pelleted Mac-EVs pretreated with antibodies, as described below. DTH effector T lymphocytes from OT-II mice, prior to incubation with OVA-Mac-EVs, were incubated with OVA-323 peptide (7.5 µg/10
7 T cells) for 20 min in 37 °C water-bath.
4.7. Active Immunization
C57BL/6 were immunized on two consecutive days (0 and 1) with OVA without an adjuvant, as described above. On day 5, mice were injected intradermally in both ears with 10 µL of a 0.5 mg/mL OVA solution in 0.9% NaCl to elicit DTH reaction, measured up to 120 h after challenge. After measuring of 24-h ear swelling, mice were injected intraperitoneally with either OVA-Mac-EVs alone or OVA-Mac-EVs preincubated with anti-OVA-323 antibodies (1 × 1010 OT-II mouse OVA-Mac-EVs in 0.2 mL DPBS per mouse), or with equivalent volume of DPBS in positive control group.
4.8. In Vitro Testing of Immune Synapse Formation
The human Jurkat T-cell line (E6.1 clone) and the lymphoblastoid Raji B-cell line (Burkitt lymphoma, both acquired from the DSMZ Organization, Braunschweig, Germany, and routinely tested for mycoplasma) were cultured in RPMI 1640 medium supplemented with GlutaMAX, HEPES (Gibco Life Technologies, Grand Island, NY, USA) and 10% fetal bovine serum (FBS, Hyclone, ThermoFisher, Waltham, MA, USA). At least 24 h prior to experimental culturing, cells were moved to medium supplemented with 10% FBS obtained from stock FBS that had been previously ultracentrifuged overnight at 100,000
g to remove its own vesicles. Raji B cells (15 × 10
6 cells) resuspended in Opti-MEM medium (Gibco Life Technologies, Grand Island, NY, USA) were transiently transfected with 25 μg of CD81-GFP DNA plasmids by electroporation with the gene-pulser III system from Bio-Rad Laboratories (240 V, 975 mΩ, 27 ms) in 4 mm cuvettes (Bio-Rad, Hercules, CA, USA), as described elsewhere [
17,
47,
54]. Cells were then cultured in FBS-supplemented RPMI 1640 medium containing 2 mg/mL of G418 antibiotic (Invitrogen, Carlsbad, CA, USA) and the efficacy of electroporation and CD81-GFP plasmid incorporation was assessed cytometrically 24 h later.
For flow cytometric analysis of cells after formation of conjugates, CD81-GFP-transfected Raji B cells were pulsed with Staphylococcal enterotoxin type E (SEE) superantigen (0.5 µg/mL) for 30 min at 37 °C and, after washing, were cultured with Jurkat T cells for 24 h in the presence of OVA-Ts-EVs. Afterwards, cells were stained with fluoresceinated antibodies against CD19 (also allowing to distinguish CD19
neg T cells). A viability staining was performed as well with Ghost Dye V510 (TONBO Biosciences, San Diego, CA, USA). Cells were analyzed in a FACS Canto II (BD Bioscience, San Jose, CA, USA) and then FCS files were analyzed in FlowJo software [
55].
For immune synapse formation assay evaluated in fluorescence confocal microscopy, Jurkat T cells were loaded with the CMAC cell tracker. CD81-GFP-transfected Raji B cells were treated with OVA-Ts-EVs for 4 h at 37 °C, which was followed by pulsing with SEE (0.5 µg/mL) for 30 min at 37 °C. Afterwards, Jurkat T cells (1 × 10
5 cells) were mixed with the CD81-GFP-transfected, Ts-EV-pretreated Raji B cells (in a ratio 1:1) and plated onto Poly-L-Lys-coated slides for 1 h incubation at 37 °C, or were standardly cultured for 24 h on 96-well culture plates and then moved onto Poly-L-Lys-coated slides and incubated for 30 min at 37 °C. Then, cells were fixed with 4% paraformaldehyde and 0.12 mM sucrose in PHEM buffer and permeabilized at room temperature with 0.2% Triton X-100 in immunofluorescence solution. After blocking with immunofluorescence solution, cells were stained with selected primary and then secondary antibodies. Finally, cells were mounted on Prolong Gold and analyzed with a Leica SP5 confocal microscope (Leica, Wetzlar, Germany) fitted with a HCX PL APO×63/1.40–0.6 oil objective and images were processed and assembled using Image J software [
54].
4.9. Cytometric Analysis of Mac-EVs
Aldehyde/sulfate latex beads, washed and resuspended in DPBS, were incubated with DPBS-suspension of either PCL-Mac-EVs or OVA-Mac-EVs (in a ratio about 1 × 10
6 beads per 1 × 10
9 vesicles, as estimated by NTA) in a total volume of 1 mL of DPBS for 10 min at room temperature with gentle agitation [
56,
57,
58]. Afterwards, 1 mL of DPBS was added and samples were incubated for next 2 h at room temperature with gentle agitation. After addition of 1 mL of 100 mM glycine solution for blocking of remaining binding sites, samples were incubated for 30 min at room temperature with gentle agitation. Then, beads coated with Mac-EVs were washed thrice in DPBS with 0.1% bovine serum albumin (BSA) by centrifugation at 600
g for 10 min. After resuspending in DPBS, vesicle-coated beads were incubated with PE-conjugated anti-mouse CD9, CD63, CD81 or I-A/I-E (MHC class II) monoclonal antibodies for 40 min at room temperature in darkness, which was followed by triple washing with DPBS containing 0.1% BSA. In some instances, beads coated with OVA-Mac-EVs, pre-incubated overnight with biotinylated anti-OVA-323 IgG antibodies, were stained with streptavidin-FITC and, where indicated, with PE-conjugated anti-CD9 and PerCP-Cy-conjugated I-A/I-E monoclonal antibodies. Latex beads coated with OVA-Mac-EVs, pre-incubated overnight with FITC-conjugated isotype IgG antibodies, were only stained with PE-conjugated anti-CD9 and PerCP-Cy-conjugated I-A/I-E monoclonal antibodies. Otherwise, beads coated with OVA-Mac-EVs were stained with biotinylated anti-OVA-323 IgG antibodies for 1 h and then with streptavidin-FITC for 40 min at room temperature. After washing, DPBS-resuspended, vesicle-coated beads were acquired by a BD FACSCalibur and data were analyzed using BD CellQuest Pro software (BD Bioscience, San Jose, CA, USA).
4.10. NTA and TEM Analysis of Mac-EVs
OT-II mouse OVA-Mac-EVs that were incubated overnight on ice either alone or with anti-OVA-323 antibodies, after dilution with filtered DPBS, were subjected to NTA by an experienced observer unaware of experimental protocol and samples [
5]. Furthermore, OT-II OVA-Mac-EVs, similarly incubated with or without antibodies, were absorbed onto cupper grid and negatively stained with 3% uranyl acetate. Then, samples were visualized with TEM microscope (JEOL 2100HT, Tokyo, Japan), equipped with TVIPS camera, and analyzed with EMMENU 4 software by an experienced observer unaware of experimental protocol and samples.
4.11. Assessing of Binding of Antibodies to OVA-Mac-EVs by ELISA
Ninety six-well ELISA plates (Corning, NY, USA) were coated with purified anti-mouse CD9 antibody (4 µg/mL) diluted in sodium carbonate buffer (pH 9.5), by incubation overnight at 4 °C. After washing, blocking the plate wells with 2% BSA in PBS for 2 h at room temperature, and repeated washing, 50 µL/well of OT-II mouse OVA-Mac-EVs (approximately 1 × 108 EVs/well) in PBS was pipetted to selected wells, and the plate was incubated for 2 h at room temperature. Then, 50 µL/well of biotinylated anti-OVA-323 IgG or biotinylated isotype IgG diluted in PBS (1 µg/mL) was added to selected wells, and the plate was incubated overnight at 4 °C. After extensive washing, 100 µL/well of streptavidin-HRP (diluted 1:250 in PBS with 0.1% BSA) was added to each well, which was followed by incubation for 45 min at room temperature and extensive washing. Finally, 100 µL/well of TMB substrate was pipetted to each well and the reaction was stopped after 5 min by adding 50 µL/well of 1 M H3PO4. The absorbance was measured at 450 nm with a reference wavelength of 570 nm. Yielded absorbance values were blanked by subtracting the values measured in respective wells without added OVA-Mac-EVs and then normalized to the mean absorbance value detected in wells with added OVA-Mac-EVs but not biotinylated antibodies.
4.12. Statistics
In vivo experiments were carried out 2–3 times and representative results were shown in the figures, prepared with the use of GraphPad Prism 8, and Adobe Photoshop 22.4 software. All groups consisted of 5 mice and average value of nonspecific increase in ear thickness caused by chemical irritation by hapten or protein solution in challenged but not immunized mouse littermates was subtracted from average values in experimental groups to obtain net swelling value (delta), expressed as average delta ± standard error of the estimate of mean value (SEM). Statistical significance of the data was estimated (after control of meeting of test assumptions) in one-way Analysis of Variance (ANOVA) with post hoc RIR Tukey test with the use of STATISTICA.10 or GraphPad Prism 8 software, and p < 0.05 was considered statistically significant. In vitro experiments were performed at least twice and data were analyzed either with one-way or two-way ANOVA or with two-tailed Student t-test, while miRNA deep-sequencing was performed once, and heatmaps were drawn with GraphPad Prism 8.
4.13. Study Approval
All animal experiments were performed in accordance with the principles of the Basel Declaration and ARRIVE guidelines, and were approved by Ethics Committees of both Yale (approval number 07381) and Jagiellonian (approvals number 39/2011, 154/2013, 51/2017 and 433/2020) Universities.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






