
Association between Olaparib Exposure and Early Toxicity in BRCA-Mutated Ovarian Cancer Patients: Results from a Retrospective Multicenter Study

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Results
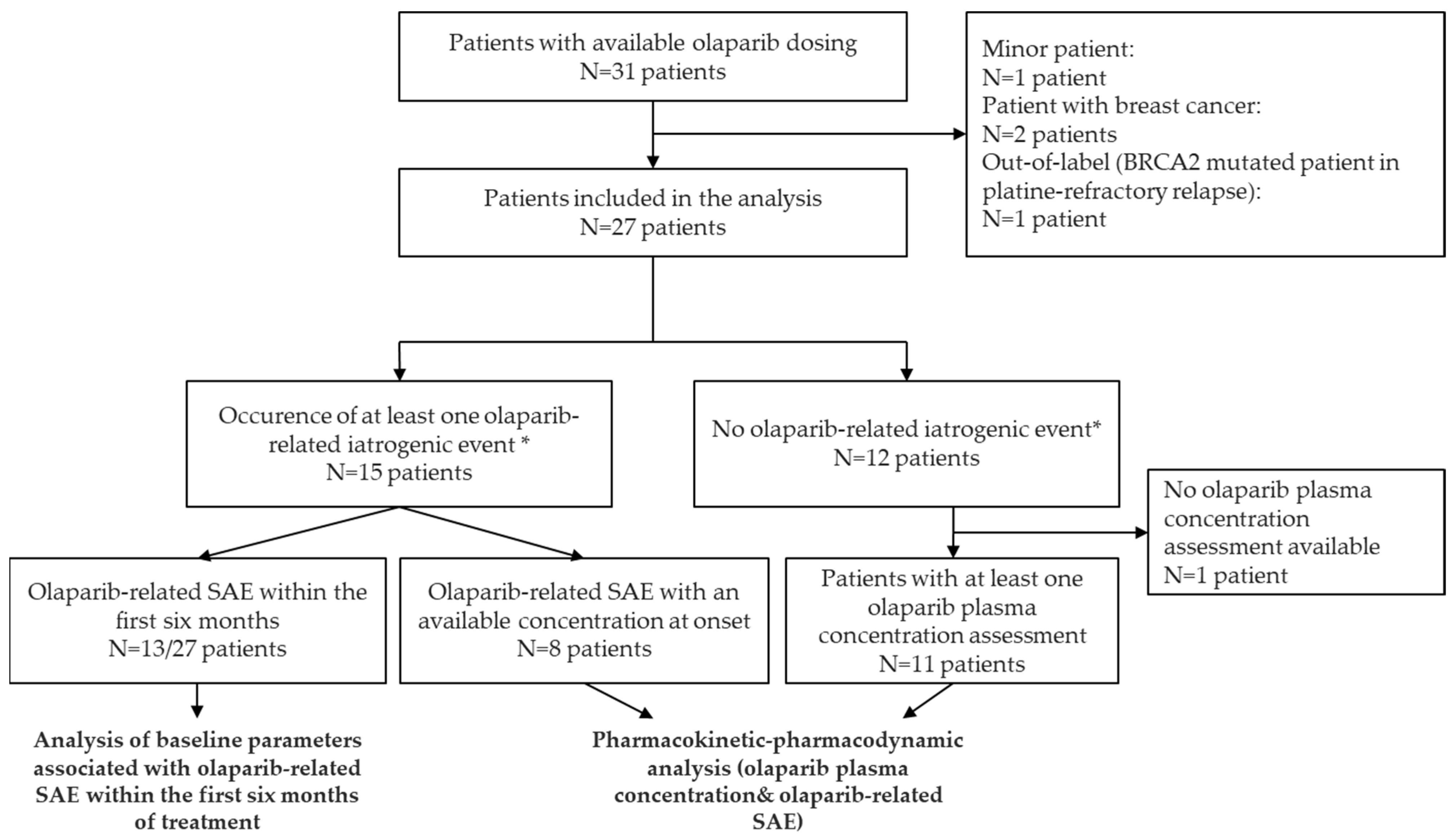
2.1. Patient Characteristics
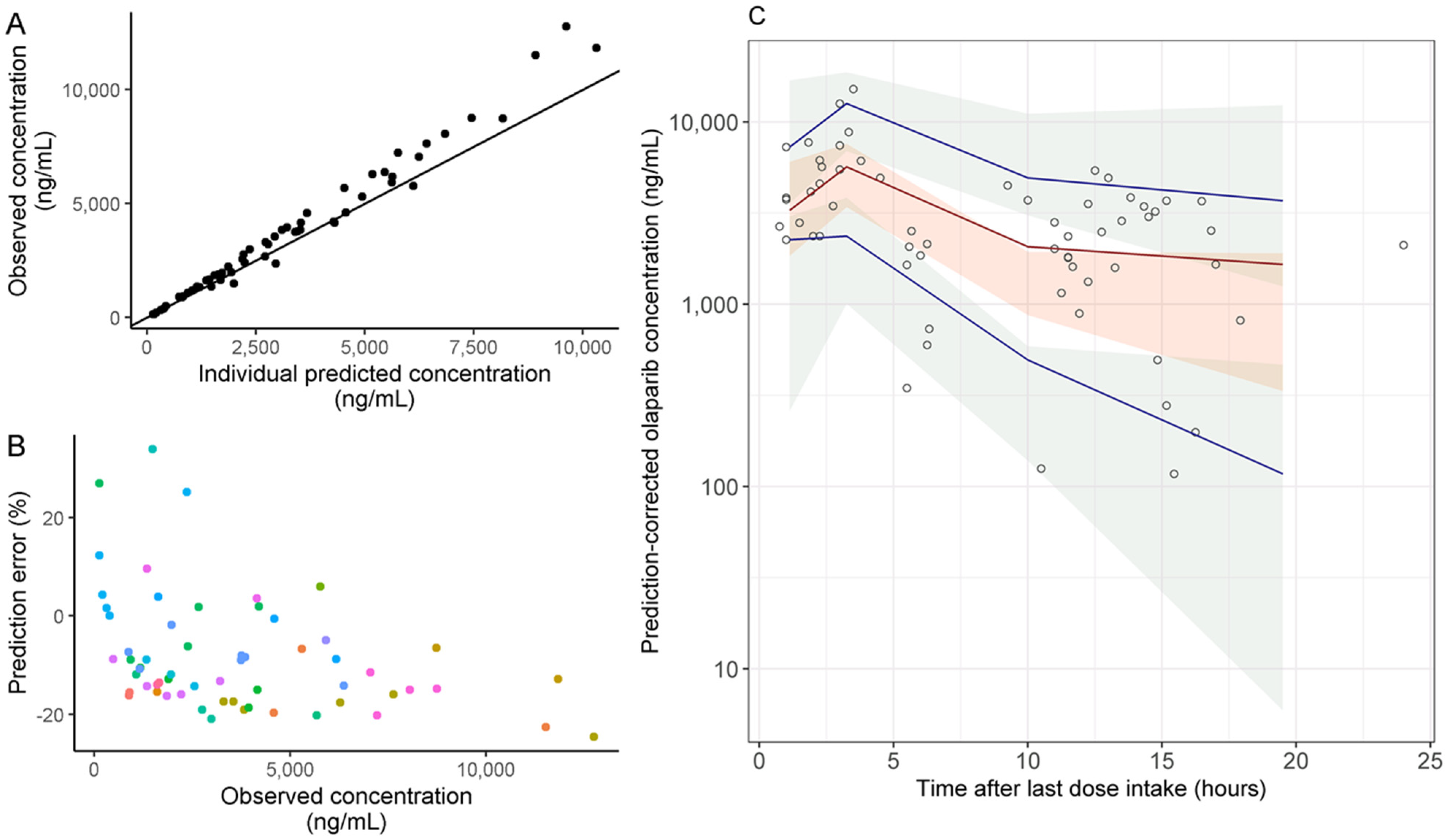
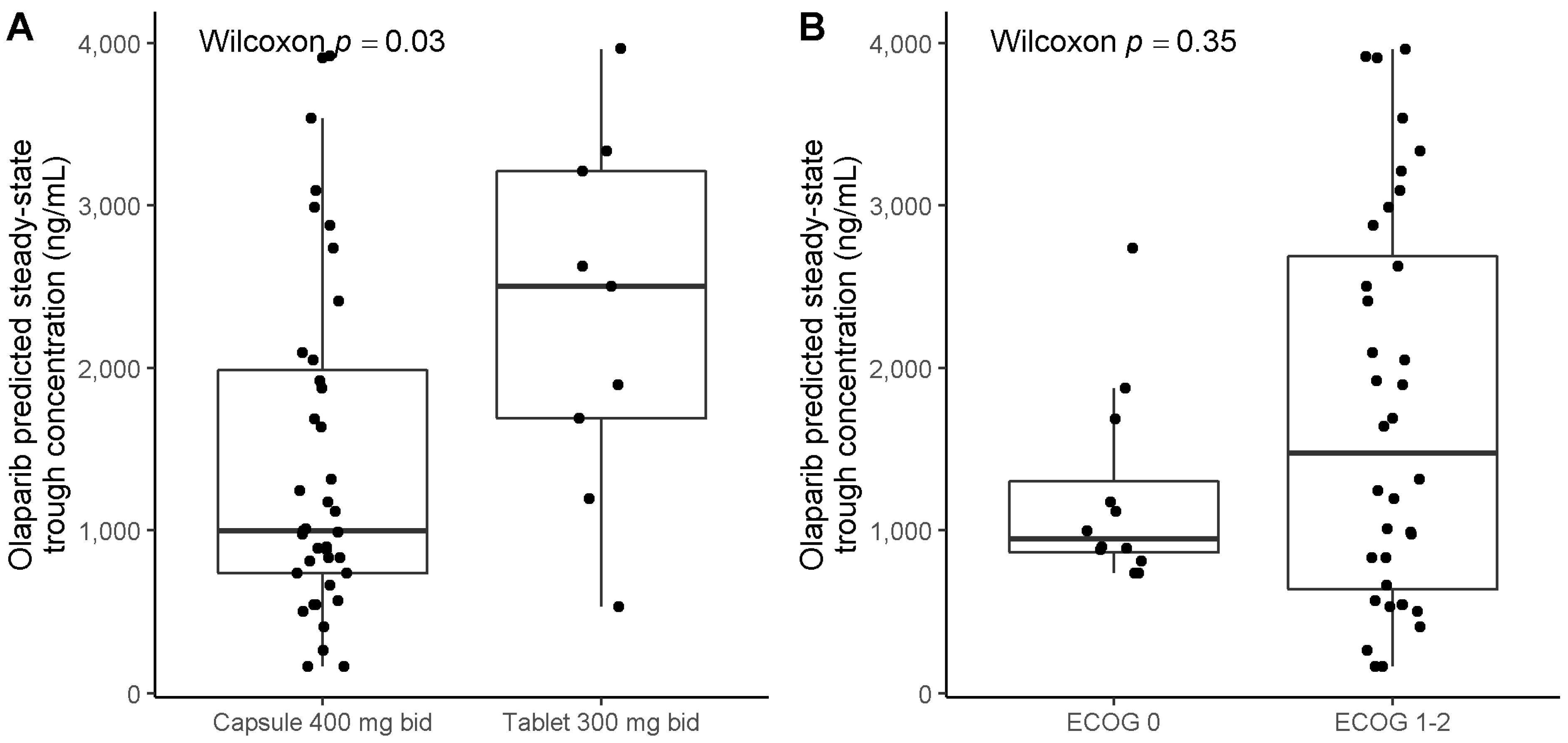
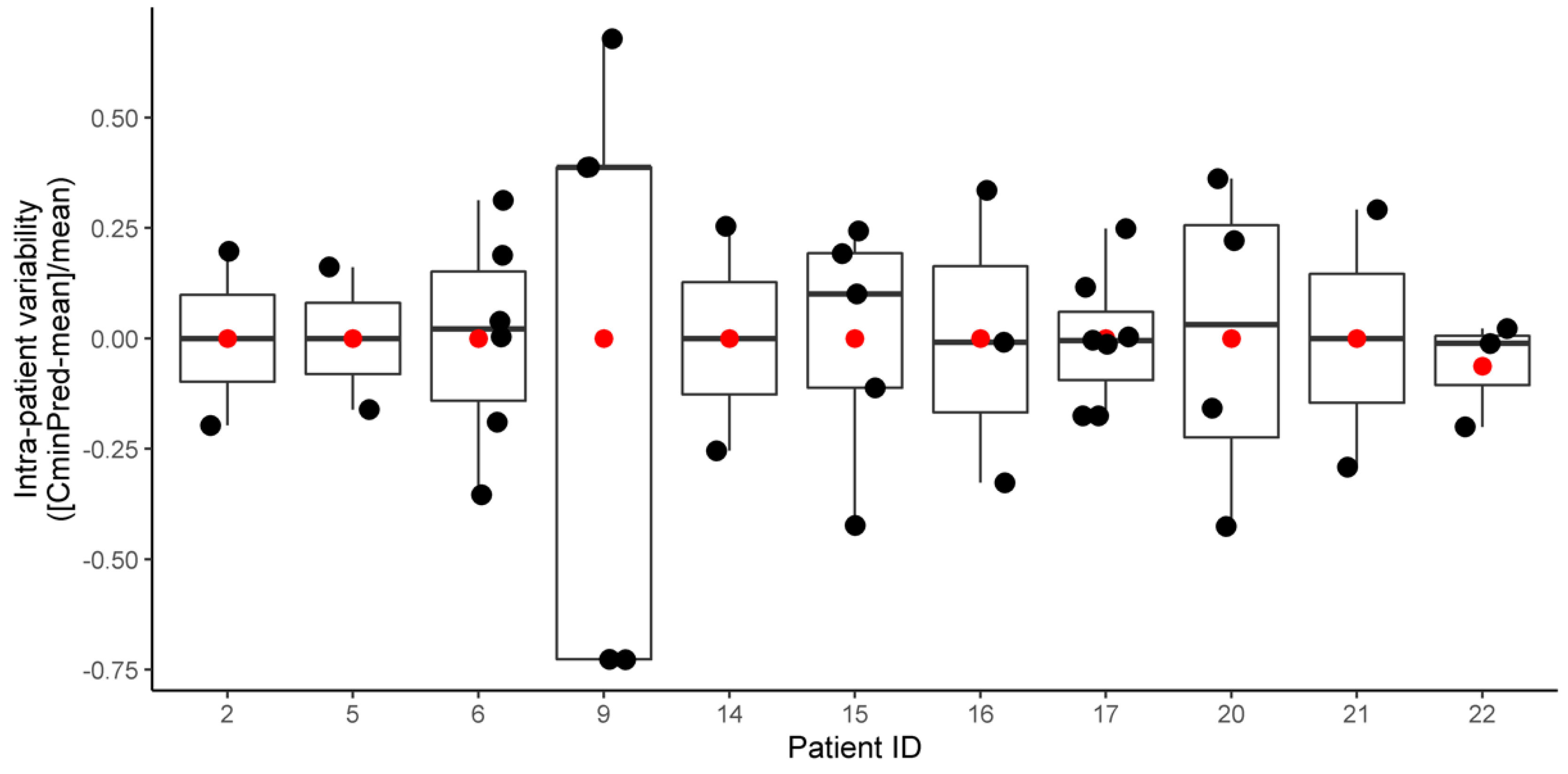
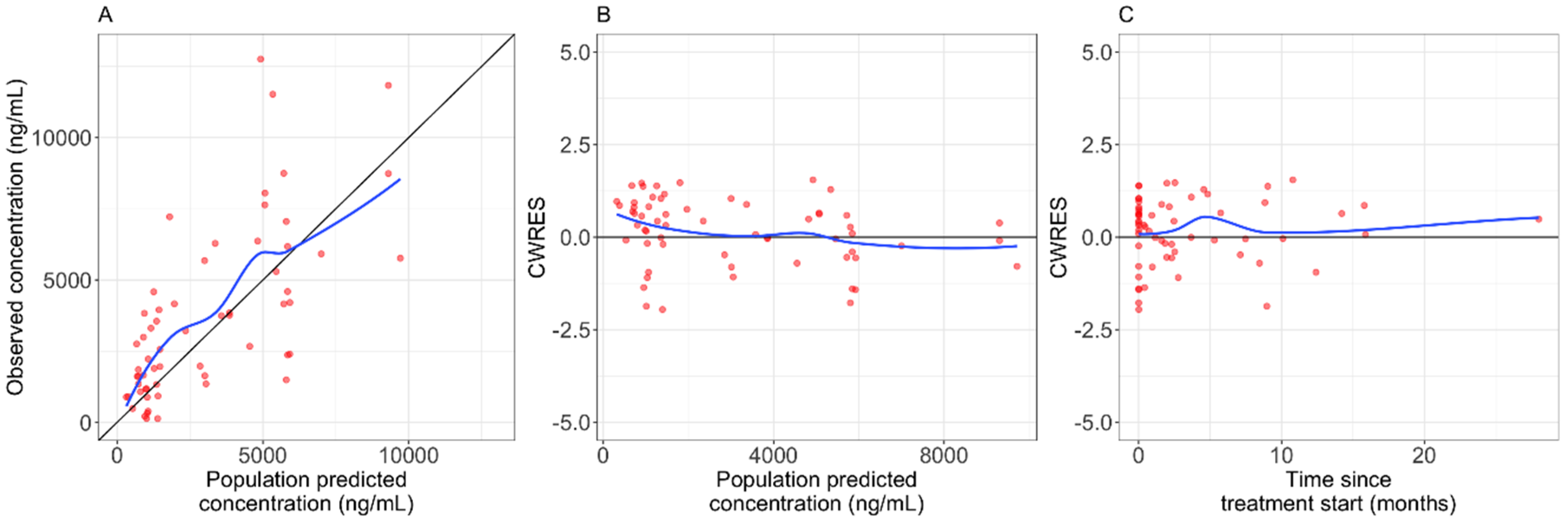
2.2. Pharmacokinetic Analysis
2.3. Association between Patients’ Baseline Characteristics and Onset of Clinically Significant Adverse Events (SAE)
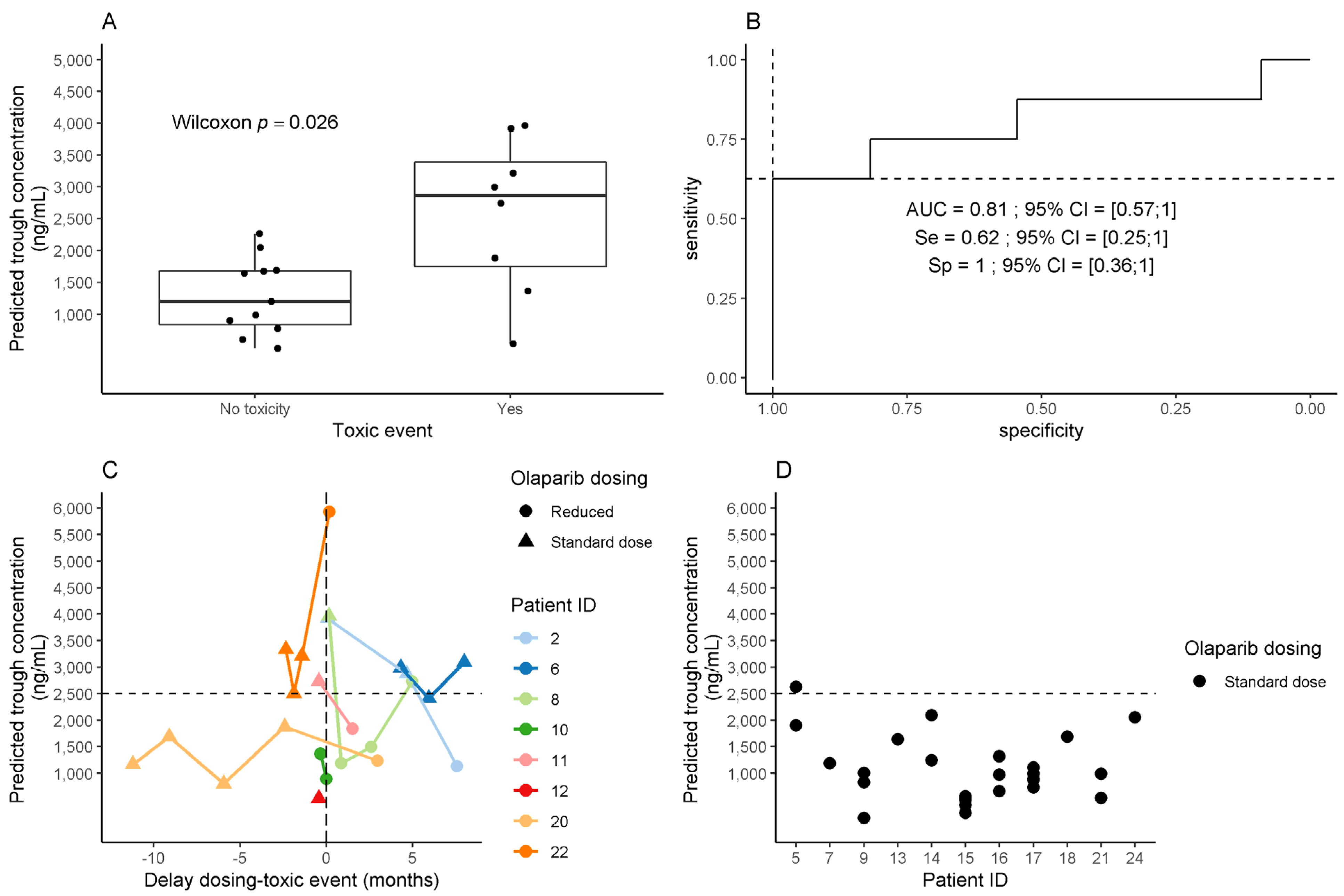
2.4. Association between Olaparib Exposure and Toxicity
2.5. Pharmacokinetic Drug–Drug Interactions
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Data Collection
4.3. Pharmacokinetic Analysis
4.4. Pharmacodynamic Analysis
4.4.1. Olaparib Toxicity
4.4.2. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Description | Value Used in the Predictions (Mean Estimate from Zhou et al.) |
|---|---|---|
| CL (L h−1) | Steady-state clearance | 3.60 |
| θECOG-PS on CL | Effect of ECOG-PS on clearance for ECOG-PS ≥ 1 a | −0.240 |
| Vc (L) | Volume of distribution of the central compartment | 2.57 |
| Vp (L) | Volume of distribution of the peripheral compartment | 19.7 |
| Q (L h−1) | Intercompartmental clearance | 1.11 |
| Frel capsule b | Relative bioavailability for capsule > 100 mg dose | 0.282 |
| Frel tablet c | Relative bioavailability for tablet formulation | 0.627 |
| ka (h−1) capsule | First-order absorption rate constant for capsules | 0.247 |
| ka (h−1) tablet strength 100 mg | First-order absorption rate constant for tablets strength 100 mg | 0.374 |
| ka (h−1) tablet strength 150 mg | First-order absorption rate constant for tablets strength 150 mg | 0.267 |
| D1 (h) capsule | Duration of zero-order absorption for capsules | 0.901 |
| D1 (h) tablet | Duration of zero-order absorption for tablets | 0.467 |
| ω CL (CV%) | Inter-individual variability in CL | 0.340 (58.3) |
| ω Vc (CV%) | Inter-individual variability in Vc | 0.448 (66.9) |
| ω Vp (CV%) | Inter-individual variability in Vp | 1.44 (120) |
| ω Q (CV%) | Inter-individual variability in Q | 0.560 (77.2) |
| ω D1 (CV%) | Inter-individual variability in D1 | 0.534 (73.1) |
| ω ka (CV%) | Inter-individual variability in ka | 0.063 (25.1) |
| Proportional residual error | Residual unexplained variability | 0.354 |


References
- Evers, B.; Drost, R.; Schut, E.; de Bruin, M.; van der Burg, E.; Derksen, P.W.B.; Holstege, H.; Liu, X.; van Drunen, E.; Beverloo, H.B.; et al. Selective Inhibition of BRCA2-Deficient Mammary Tumor Cell Growth by AZD2281 and Cisplatin. Clin. Cancer Res. 2008, 14, 3916–3925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib Monotherapy as Primary Treatment in Unselected Triple Negative Breast Cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [Green Version]
- Ricci, A.D.; Rizzo, A.; Novelli, M.; Tavolari, S.; Palloni, A.; Tober, N.; Abbati, F.; Mollica, V.; DE Lorenzo, S.; Turchetti, D.; et al. Specific Toxicity of Maintenance Olaparib Versus Placebo in Advanced Malignancies: A Systematic Review and Meta-Analysis. Anticancer Res. 2020, 40, 597–608. [Google Scholar] [CrossRef]
- Liposits, G.; Wulff, C.N.; Otland, A.; Fokdal, L.U. Olaparib Treatment in Older Patients with Ovarian Cancer: Need for “real-World” Data beyond Clinical Trials. Ecancermedicalscience 2020, 14, 1104. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, D.; Bologna, A.; Cecere, S.C.; De Matteis, E.; Scandurra, G.; Zamagni, C.; Arcangeli, V.; Artioli, F.; Bella, M.; Blanco, G.; et al. Sharing Real-World Experiences to Optimize the Management of Olaparib Toxicities: A Practical Guidance from an Italian Expert Panel. Support. Care Cancer 2020, 28, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Steeghs, N.; Nijenhuis, C.M.; Schellens, J.H.M.; Beijnen, J.H.; Huitema, A.D.R. Practical Guidelines for Therapeutic Drug Monitoring of Anticancer Tyrosine Kinase Inhibitors: Focus on the Pharmacokinetic Targets. Clin. Pharm. 2014, 53, 305–325. [Google Scholar] [CrossRef]
- Verheijen, R.B.; Yu, H.; Schellens, J.H.M.; Beijnen, J.H.; Steeghs, N.; Huitema, A.D.R. Practical Recommendations for Therapeutic Drug Monitoring of Kinase Inhibitors in Oncology. Clin. Pharmacol. Ther 2017, 102, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Swaisland, H.; Leunen, K.; van Herpen, C.M.L.; Jerusalem, G.; De Grève, J.; Lolkema, M.P.; Soetekouw, P.; Mau-Sørensen, M.; Nielsen, D.; et al. Olaparib Tablet Formulation: Effect of Food on the Pharmacokinetics after Oral Dosing in Patients with Advanced Solid Tumours. Cancer Chemother. Pharmacol 2015, 76, 723–729. [Google Scholar] [CrossRef]
- Pilla Reddy, V.; Bui, K.; Scarfe, G.; Zhou, D.; Learoyd, M. Physiologically Based Pharmacokinetic Modeling for Olaparib Dosing Recommendations: Bridging Formulations, Drug Interactions, and Patient Populations. Clin. Pharmacol. Ther. 2019, 105, 229–241. [Google Scholar] [CrossRef]
- Rolfo, C.; de Vos-Geelen, J.; Isambert, N.; Molife, L.R.; Schellens, J.H.M.; De Grève, J.; Dirix, L.; Grundtvig-Sørensen, P.; Jerusalem, G.; Leunen, K.; et al. Pharmacokinetics and Safety of Olaparib in Patients with Advanced Solid Tumours and Renal Impairment. Clin. Pharm. 2019, 58, 1165–1174. [Google Scholar] [CrossRef]
- Zhou, D.; Li, J.; Bui, K.; Learoyd, M.; Berges, A.; Milenkova, T.; Al-Huniti, N.; Tomkinson, H.; Xu, H. Bridging Olaparib Capsule and Tablet Formulations Using Population Pharmacokinetic Meta-Analysis in Oncology Patients. Clin. Pharm. 2019, 58, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.D.; Somberg, J.C. Pharmacology and Pharmacokinetics of Amiodarone. J. Clin. Pharmacol. 1991, 31, 1061–1069. [Google Scholar] [CrossRef]
- NDA/BLA Multi-Disciplinary Review and Evaluation NDA 208558 LynparzaTM (Olaparib). Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwiO3u64zLXwAhUMsxQKHc3MCbsQFjAAegQIAxAD&url=https%3A%2F%2Fwww.accessdata.fda.gov%2Fdrugsatfda_docs%2Fnda%2F2017%2F208558Orig1s000MultidisciplineR.pdf&usg=AOvVaw0sCeqI6xDGleC3aRwY5l3v (accessed on 12 August 2021).
- Ismail, M.; Khan, S.; Khan, F.; Noor, S.; Sajid, H.; Yar, S.; Rasheed, I. Prevalence and Significance of Potential Drug-Drug Interactions among Cancer Patients Receiving Chemotherapy. BMC Cancer 2020, 20, 335. [Google Scholar] [CrossRef] [Green Version]
- Fabre, E.; Thomas-Schoemann, A.; Blanchet, B. Letter to the Editor Regarding the Paper by Loquai C et al. “Use of Complementary and Alternative Medicine: A Multicenter Cross-Sectional Study in 1089 Melanoma Patients”. Eur. J. Cancer 2017, 85, 158–159. [Google Scholar] [CrossRef]
- Lynparza—Summary of Product Characteristics. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwj1z7u_9fDwAhX76OAKHZyJDTMQFnoECAUQAA&url=https%3A%2F%2Fwww.ema.europa.eu%2Fen%2Fdocuments%2Fproduct-information%2Flynparza-epar-product-information_en.pdf&usg=AOvVaw3CrQwyaKRC8QKpzVqArXFi (accessed on 12 August 2021).
- Peer, C.J.; Lee, J.-M.; Roth, J.; Rodgers, L.; Nguyen, J.; Annunziata, C.M.; Minasian, L.; Kohn, E.C.; Figg, W.D. Population Pharmacokinetic Analyses of the Effect of Carboplatin Pretreatment on Olaparib in Recurrent or Refractory Women’s Cancers. Cancer Chemother. Pharmacol. 2017, 80, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Dirix, L.; Swaisland, H.; Verheul, H.M.W.; Rottey, S.; Leunen, K.; Jerusalem, G.; Rolfo, C.; Nielsen, D.; Molife, L.R.; Kristeleit, R.; et al. Effect of Itraconazole and Rifampin on the Pharmacokinetics of Olaparib in Patients With Advanced Solid Tumors: Results of Two Phase I Open-Label Studies. Clin. Ther. 2016, 38, 2286–2299. [Google Scholar] [CrossRef]
- Simon, F.; Gautier-Veyret, E.; Truffot, A.; Chenel, M.; Payen, L.; Stanke-Labesque, F.; Tod, M. Modeling Approach to Predict the Impact of Inflammation on the Pharmacokinetics of CYP2C19 and CYP3A4 Substrates. Pharm. Res. 2021, 38, 415–428. [Google Scholar] [CrossRef]
- Banerjee, S.; Gonzalez-Martin, A.; Harter, P.; Lorusso, D.; Moore, K.N.; Oaknin, A.; Ray-Coquard, I. First-Line PARP Inhibitors in Ovarian Cancer: Summary of an ESMO Open—Cancer Horizons Round-Table Discussion. ESMO Open 2020, 5, e001110. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Lim, M.C.; Kim, B.G.; Ngoi, N.Y.; Choi, C.H.; Park, S.Y.; Tan, D.S.; Go, Y.; Lee, J.Y. A Single-Arm Phase II Study of Olaparib Maintenance with Pembrolizumab and Bevacizumab in BRCA Non-Mutated Patients with Platinum-Sensitive Recurrent Ovarian Cancer (OPEB-01). J. Gynecol. Oncol. 2021, e31. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Alblihy, A.; Toss, M.S.; Algethami, M.; Al Sunni, R.; Green, A.R.; Rakha, E.A.; Madhusudan, S. XRCC1 Deficient Triple Negative Breast Cancers Are Sensitive to ATR, ATM and Wee1 Inhibitor Either Alone or in Combination with Olaparib. Ther. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef]
- Mansouri, A.; McGregor, N.; Dunn, R.; Dobbie, S.; Holmes, J.; Collins, L.; Nicum, S. Randomised Phase II Trial of Olaparib, Chemotherapy or Olaparib and Cediranib in Patients with Platinum-Resistant Ovarian Cancer (OCTOVA): A Study Protocol. BMJ Open 2021, 11. [Google Scholar] [CrossRef]
- Available online: https://cancer-druginteractions.org (accessed on 12 August 2021).
- Available online: https://www.ddi-predictor.org/ (accessed on 12 August 2021).




| Variable | Value |
|---|---|
| Patients and cancer characteristics at diagnostic | |
| Age, median [Q1–Q3] (years), (27 ‡) | 59 [53–66] |
| Histological subtypes, N (%), (27 ‡) | |
| High grade ovarian serous carcinoma | 25 (93%) |
| Others | 2 (7%) |
| FIGO stage at diagnosis, N (%), (27 ‡) | |
| I | 2 (8%) |
| III | 22 (81%) |
| IVB | 3 (11%) |
| BRCA mutations §, N (%), (27 ‡) | |
| BRCA1 | 19 (70%) |
| BRCA2 | 8 (30%) |
| Initial therapeutic management, N (%), (27 ‡) | |
| Induction/neoadjuvant platinum-based chemotherapy | 14 (52%) |
| Cytoreductive surgery | 25 (93%) |
| Complete resection achieved | 23 (85%) |
| Adjuvant platinum-based chemotherapy | 25 (93%) |
| Patient characteristics at olaparib initiation | |
| Age, median [Q1–Q3] (years), (27 ‡) | 63 [57–72] |
| ECOG-PS, N (%), (27 ‡) | |
| 0 | 5 (19%) |
| 1 | 16 (59%) |
| 2 | 6 (22%) |
| Body mass index (kg/m2), median [Q1–Q3], (25 ‡) | 23 [20–26] |
| Hemoglobin (g/dL), median [Q1–Q3], (26 ‡) | 11.8 [11.1–12.4] |
| White blood cells count (G/L), median [Q1–Q3], (25 ‡) | 5.1 [3.6–6.2] |
| Platelet count (G/L), median [Q1–Q3], (26 ‡) | 233 [187–288] |
| Serum albumin (g/L), median [Q1–Q3], (20 ‡) | 42 [39–44] |
| Estimated creatinine clearance (Cockcroft–Gault formula), median [Q1–Q3] (mL/min) (26 ‡) | 78 [54–97] |
| Cancer characteristics at olaparib initiation | |
| Olaparib introduction setting, N (%), (27 ‡) | |
| Maintenance after adjuvant chemotherapy | 7 (26%) |
| First relapse | 12 (44%) |
| Beyond first relapse | 8 (30%) |
| Number of metastatic sites, N (%), (27 ‡) | |
| Complete remission at olaparib initiation | 10 (37%) |
| 1 | 11 (40%) |
| 2 | 5 (19%) |
| 3 | 1 (4%) |
| Metastatic sites, N (%), (27 ‡) | |
| Peritoneal metastases | 15 (55%) |
| Node metastases | 4 (15%) |
| Visceral abdominal metastases | 3 (11%) |
| Extra abdominal metastases | 2 (7%) |
| Olaparib formulation and dosing, N (%), (27 ‡) | |
| Capsule 400 mg bid | 16 (59%) |
| Capsule 200 mg bid | 1 (4%) |
| Tablet 300 mg bid | 10 (37%) |
| Total | 27 (100%) |
| Categories | SAE within 6 Months * Odds Ratio † [95% CI] | p-Value † |
|---|---|---|
| Age at olaparib initiation (years), (27 ‡), for each additional year | 1.01 [0.99; 1.03] | 0.13 |
| ECOG-PS > 1, (27 ‡) | 2.66 [0.42; 22.5] | 0.31 |
| Body mass index (kg/m2), (25 ‡), for each additional unit | 1.03 [0.99; 1.07] | 0.10 |
| Serum albumin (g/L), (20 ‡), for each additional unit | 0.95 [0.91; 1.00] | 0.10 |
| Renal insufficiency, (27 ‡), Cockcroft–Gault estimated clearance < 60 mL/min | 0.77 [0.15; 3.85] | 0.75 |
| Hemoglobin (g/dL), (26 ‡), for each additional unit | 0.93 [0.72; 1.18] | 0.57 |
| Olaparib formulation (27 ‡), capsule (reference) vs. tablet | 2.14 [0.44; 11.3] | 0.34 |
| Olaparib introduction setting (27 ‡), maintenance (reference) vs. first relapse and beyond | 0.26 [0.03; 1.57] | 0.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Velev, M.; Puszkiel, A.; Blanchet, B.; de Percin, S.; Delanoy, N.; Medioni, J.; Gervais, C.; Balakirouchenane, D.; Khoudour, N.; Pautier, P.; et al. Association between Olaparib Exposure and Early Toxicity in BRCA-Mutated Ovarian Cancer Patients: Results from a Retrospective Multicenter Study. Pharmaceuticals 2021, 14, 804. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14080804
Velev M, Puszkiel A, Blanchet B, de Percin S, Delanoy N, Medioni J, Gervais C, Balakirouchenane D, Khoudour N, Pautier P, et al. Association between Olaparib Exposure and Early Toxicity in BRCA-Mutated Ovarian Cancer Patients: Results from a Retrospective Multicenter Study. Pharmaceuticals. 2021; 14(8):804. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14080804
Chicago/Turabian StyleVelev, Maud, Alicja Puszkiel, Benoit Blanchet, Sixtine de Percin, Nicolas Delanoy, Jacques Medioni, Claire Gervais, David Balakirouchenane, Nihel Khoudour, Patricia Pautier, and et al. 2021. "Association between Olaparib Exposure and Early Toxicity in BRCA-Mutated Ovarian Cancer Patients: Results from a Retrospective Multicenter Study" Pharmaceuticals 14, no. 8: 804. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14080804