
Design and Synthesis of Newly Synthesized Acrylamide Derivatives as Potential Chemotherapeutic Agents against MCF-7 Breast Cancer Cell Line Lodged on PEGylated Bilosomal Nano-Vesicles for Improving Cytotoxic Activity

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biology
2.2.1. Cytotoxic Activity against Breast Cancer Cell Line MCF-7
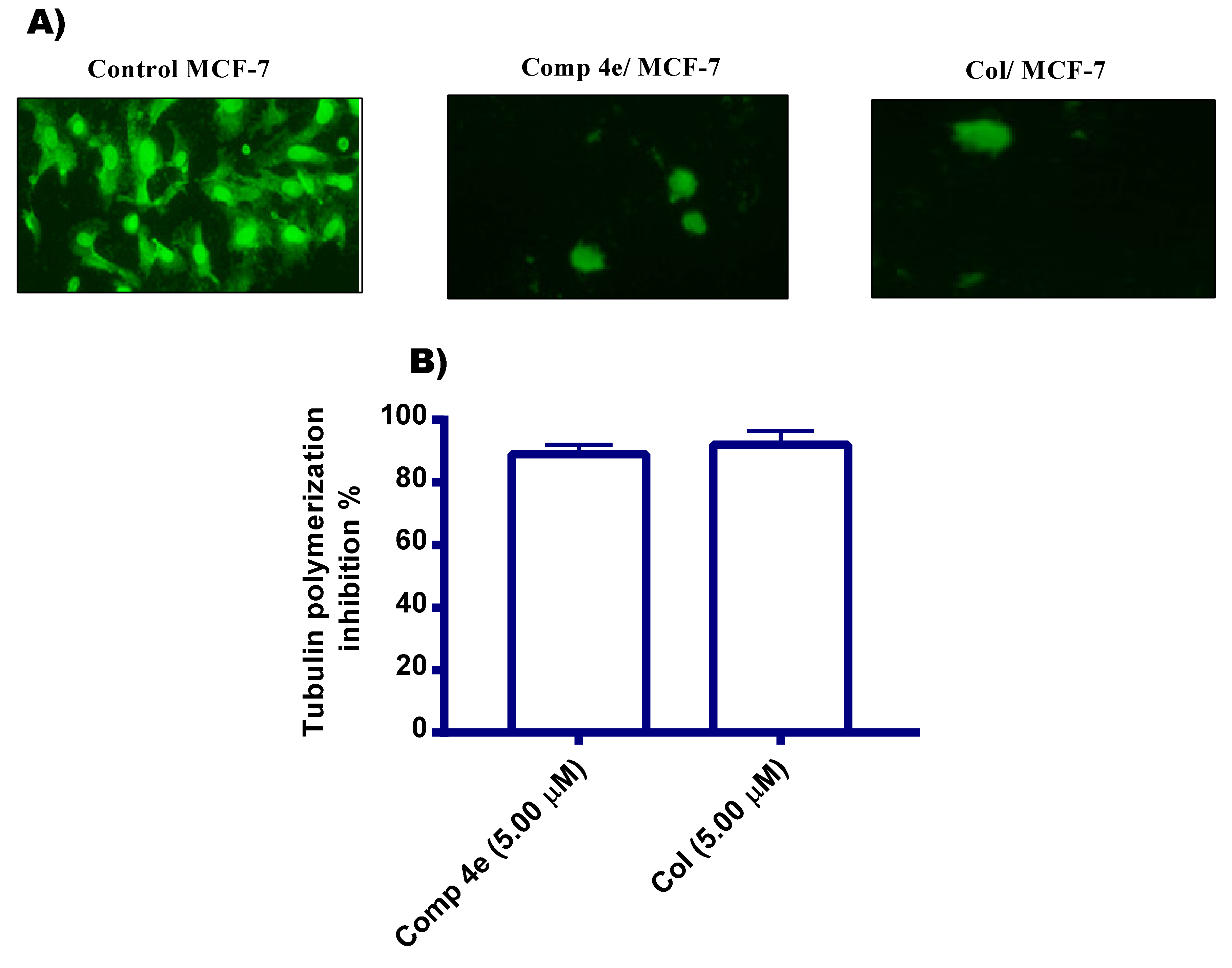
2.2.2. Tubulin Polymerization Inhibition Assays
2.2.3. DNA Flow Cytometry Analysis
Cell Cycle Analysis
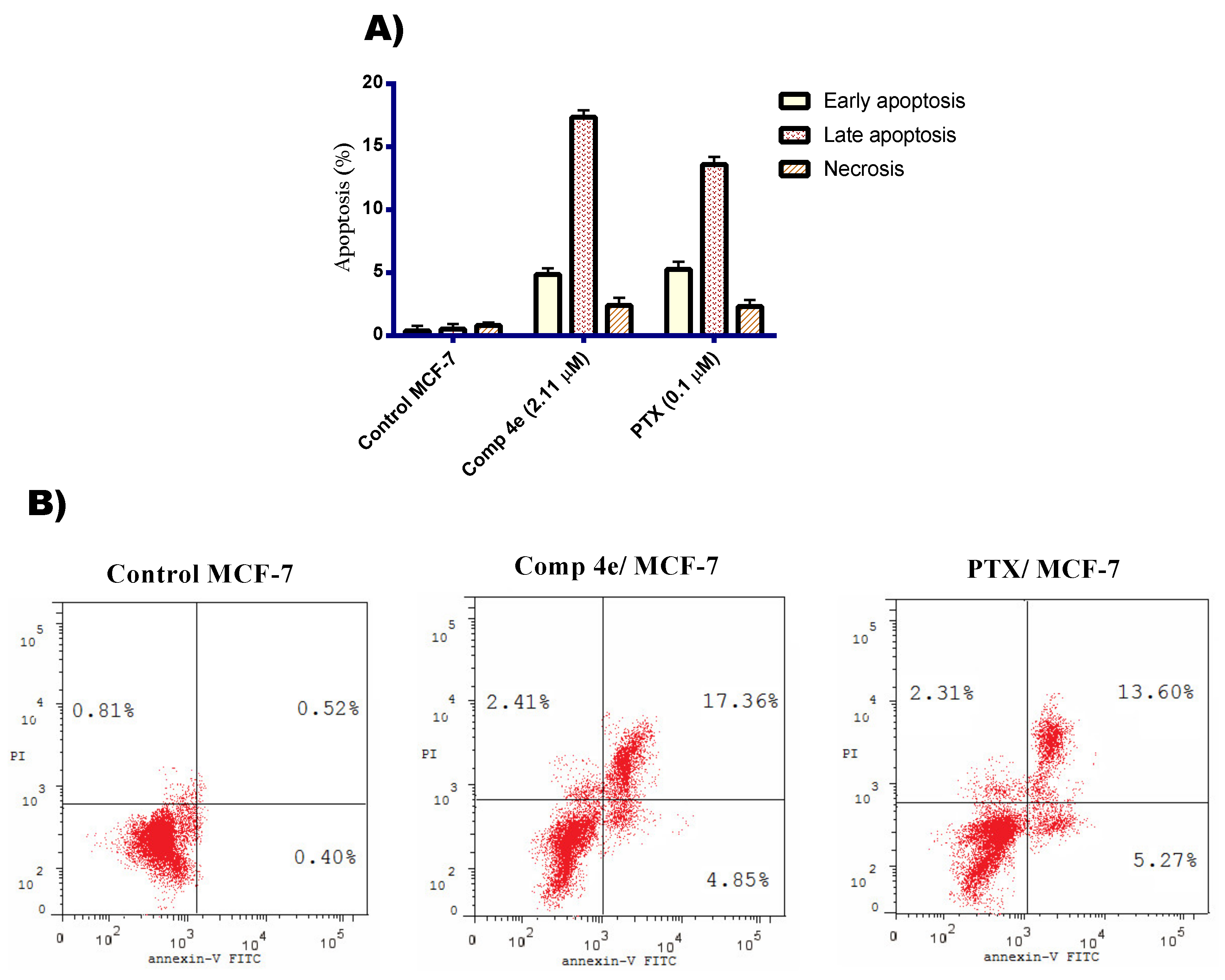
Annexin V/ FITC Apoptosis Staining Assay
2.2.4. Caspase 3/7 Assay of Compound 4e
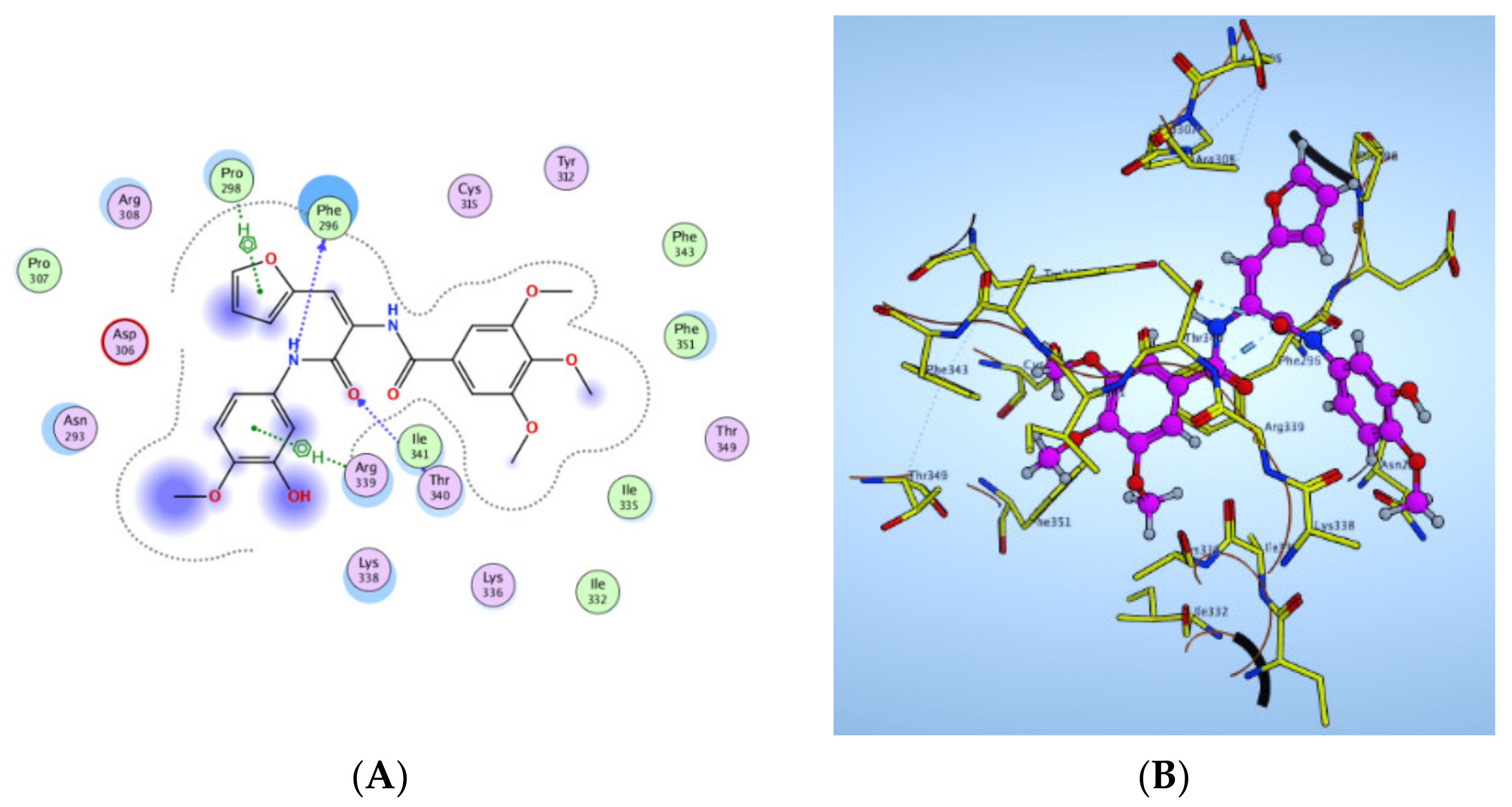
2.2.5. Molecular Docking Study
2.3. Evolvement of PEGylated Bilosomal Nano-Vesicles
2.3.1. In Silico Predictive ADME Study for Targeted Compound (4e)
2.3.2. Experimental Design, Fabrication and Statistical Evaluation of 4e Pegylated Bilosomes
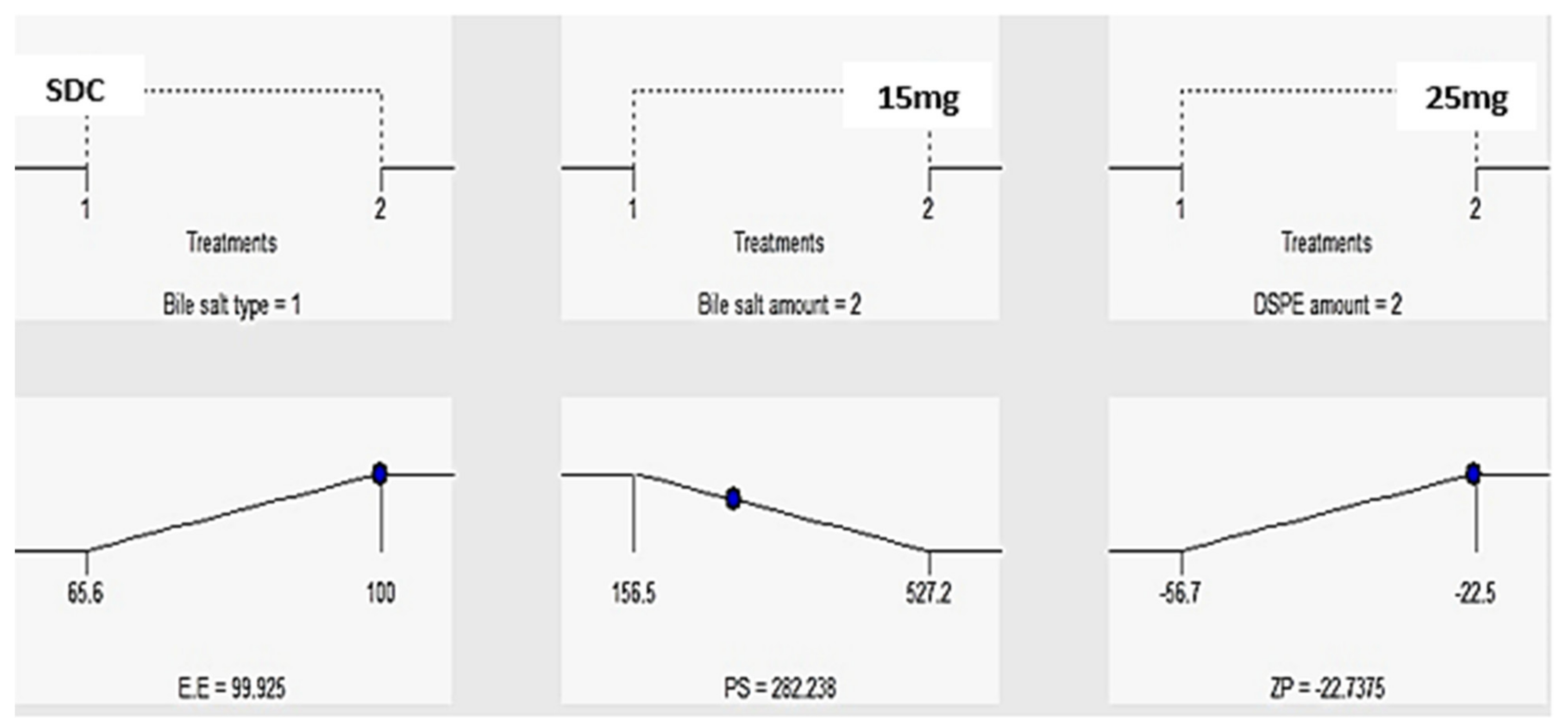
Influence of the Fabrication Variables on E.E%
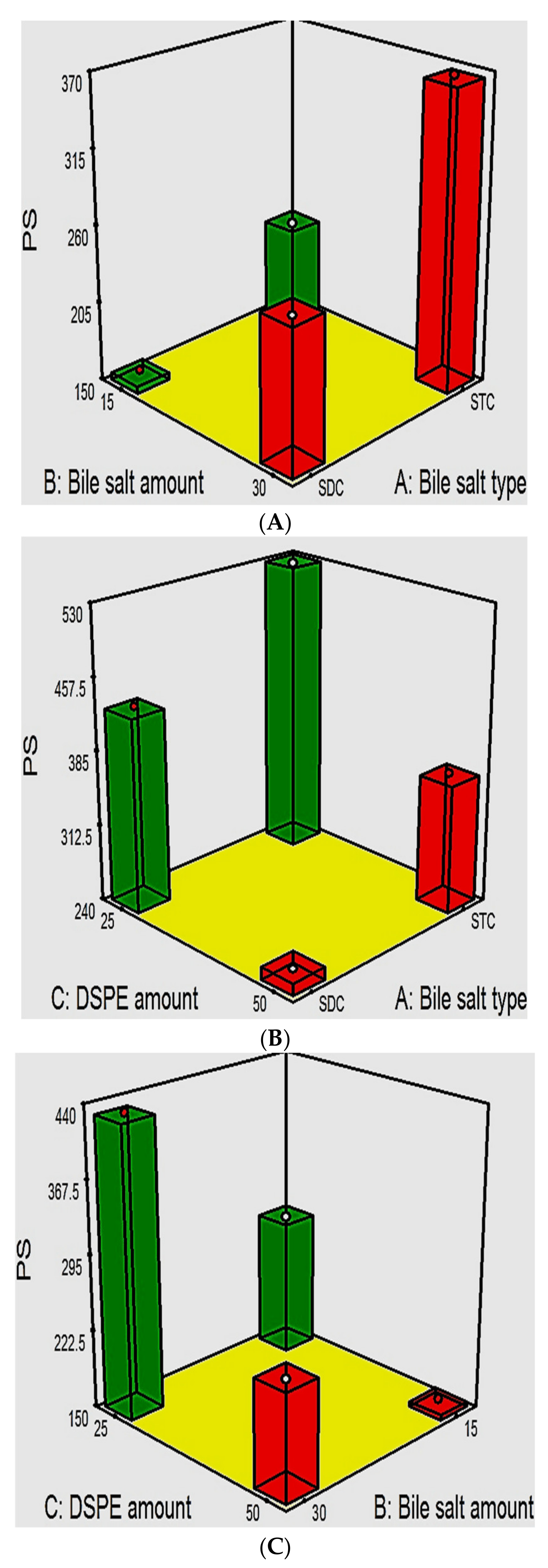
PDI and the Influence of the Fabrication Variables on PS
The Influence of the Compounding Variables on ZP
2.3.3. Statistical Optimization and Validation of the Optimal 4e-Loaded PEGylated Bilosomes
2.3.4. In Vitro Investigation of the Optimized 4e-Loaded PEGylated Bilosome
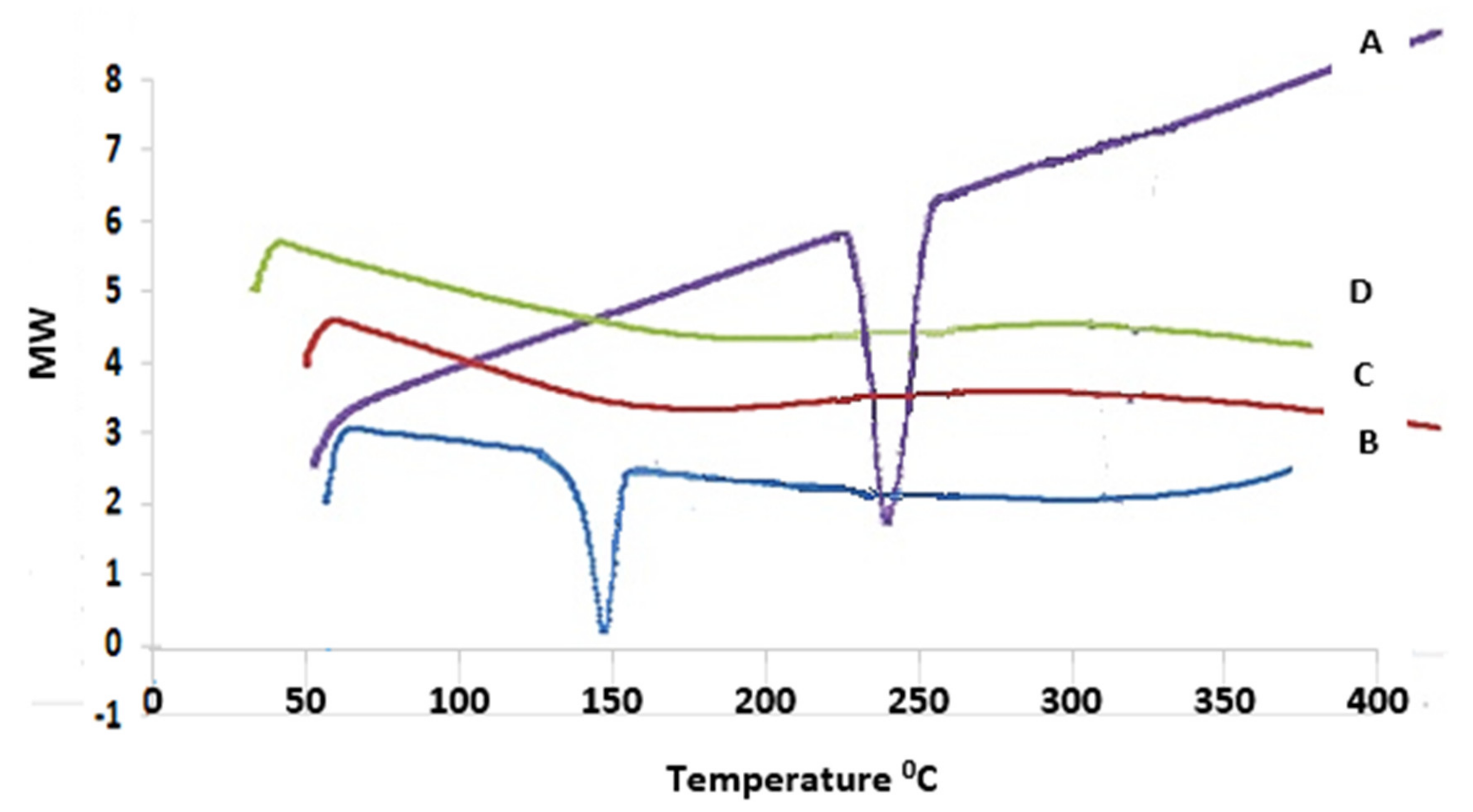
Differential Scanning Calorimetry (DSC)
Transmission Electron Microscope TEM
Cytotoxic Activity of the Optimized Formula (F7) Compared to Target Compound 4e
3. Material and Methods
3.1. Material
3.2. Chemistry
3.2.1. General Procedure for the Synthesis of 4-(Furan-2-ylmethylene)-2-(3,4,5-trimethoxyphenyl)oxazol-5(4H)-one (1)
3.2.2. General Procedure for the Synthesis of Ethyl 3-(Furan-2-yl)-2-(3,4,5-trimethoxybenzamido)acrylate (2)
3.2.3. General Procedure for the Synthesis of N-[1-(Furan-2-yl)-3-(isopropylamino)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide (3)
3.2.4. General Procedure for the Synthesis of N-[1-(Furan-2-yl)-3-oxo-3-(arylamino)prop-1-en-2-yl]-3,4,5-trimethoxybenzamide 4a–e
N-[1-(Furan-2-yl)-3-oxo-3-(o-tolylamino)prop-1-en-2-yl]-3,4,5-trimethoxybenzamide (4a)
N-[1-(Furan-2-yl)-3-oxo-3-(m-tolylamino)prop-1-en-2-yl]-3,4,5-trimethoxybenzamide (4b)
N-[1-(Furan-2-yl)-3-(4-methoxyphenylamino)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide (4c)
N-[1-(Furan-2-yl)-3-(4-hydroxyphenylamino)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide (4d)
N-[1-(Furan-2-yl)-3-(3-hydroxy-4-methoxyphenylamino)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide (4e)
3.2.5. General Procedure for the Synthesis of N-[3-(Arylamino)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide 5a–d
N-[1-(Furan-2-yl)-3-((3-methylbenzyl)amino)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide (5a)
N-[1-(Furan-2-yl)-3-((2-methoxybenzyl)amino)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide (5b)
N-[1-(Furan-2-yl)-3-(3-methoxybenzylamino)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide (5c)
N-[3-((3,4-Dimethoxybenzyl)amino)-1-(furan-2-yl)-3-oxoprop-1-en-2-yl]-3,4,5-trimethoxybenzamide (5d)
3.3. Biological Studies
3.3.1. Cytotoxic Activity against Breast MCF-7 Cancer Cell Line
3.3.2. Tubulin Assays
3.3.3. DNA Flow Cytometry Analysis
Cell Cycle Analysis Compound 4e
Annexin V FITC/PI Apoptosis Detection Staining Assay
3.3.4. Caspase 3/7 Green Flow Cytometry Assay
3.3.5. Molecular Docking Study
3.4. Tailoring of 4e-Loaded PEGylated Bilosome
3.5. HPLC Investigation
3.6. In Vitro Analysis and Optimization of 4e-Loaded PEGylated Bilosomes
3.6.1. Investigation of the Entrapment Efficiency Percentage (EE%)
3.6.2. Investigation of Zeta Potential, Vesicle Size and PDI
3.7. Conduction of Experimental Design and Selecting the Optimal 4e-Loaded PEGylated Bilosome
3.8. In Vitro Investigation of the Optimum 4e-Loaded PEGylated Bilosomal Formula
3.8.1. Lyophilization of the Optimized PEGylated Bilosomal Formula
3.8.2. Differential Scanning Calorimetry (DSC)
3.8.3. Transmission Electron Microscopy (TEM)
3.8.4. In Vitro Release Study of the Optimal Formula
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peerzada, M.N.; Hamel, E.; Bai, R.; Supuran, C.T.; Azam, A. Deciphering the key heterocyclic scaffolds in targeting microtubules, kinases and carbonic anhydrases for cancer drug development. Pharmacol. Ther. 2021, 225, 107860. [Google Scholar] [CrossRef]
- Meiring, J.; Shneyer, B.I.; Akhmanova, A. Generation and regulation of microtubule network asymmetry to drive cell polarity. Curr. Opin. Cell Biol. 2020, 62, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Dutour-Provenzano, G.; Etienne-Manneville, S. Intermediate filaments. Curr. Biol. 2021, 31, R522–R529. [Google Scholar] [CrossRef] [PubMed]
- Oláh, J.; Lehotzky, A.; Szunyogh, S.; Szénási, T.; Orosz, F.; Ovádi, J. Microtubule-Associated Proteins with Regulatory Functions by Day and Pathological Potency at Night. Cells 2020, 9, 357. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, C.; Lalli, E. Targeting the cytoskeleton against metastatic dissemination. Cancer Metastasis Rev. 2021, 40, 89–140. [Google Scholar] [CrossRef]
- Spiliotis, E.T.; Kesisova, I.A. Spatial regulation of microtubule-dependent transport by septin GTPases. Trends Cell Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Voloshin, T.; Schneiderman, R.S.; Volodin, A.; Shamir, R.R.; Kaynan, N.; Zeevi, E.; Koren, L.; Klein-Goldberg, A.; Paz, R.; Giladi, M.; et al. Tumor Treating Fields (TTFields) Hinder Cancer Cell Motility through Regulation of Microtubule and Acting Dynamics. Cancers 2020, 12, 3016. [Google Scholar] [CrossRef] [PubMed]
- Thanaussavadate, B.; Ngiwsara, L.; Lirdprapamongkol, K.; Svasti, J.; Chuawong, P. A synthetic 2,3-diarylindole induces microtubule destabilization and G2/M cell cycle arrest in lung cancer cells. Bioorg. Med. Chem. Lett. 2019, 30, 126777. [Google Scholar] [CrossRef]
- Zhang, J.; Li, A.; Sun, H.; Xiong, X.; Qin, S.; Wang, P.; Dai, L.; Zhang, Z.; Li, X.; Liu, Z. Amentoflavone triggers cell cycle G2/M arrest by interfering with microtubule dynamics and inducing DNA damage in SKOV3 cells. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Malarz, K.; Mularski, J.; Kuczak, M.; Mrozek-Wilczkiewicz, A.; Musiol, R. Novel Benzenesulfonate Scaffolds with a High Anticancer Activity and G2/M Cell Cycle Arrest. Cancers 2021, 13, 1790. [Google Scholar] [CrossRef]
- Chen, A.; Wen, S.; Liu, F.; Zhang, Z.; Liu, M.; Wu, Y.; He, B.; Yan, M.; Kang, T.; Lam, E.W.; et al. CRISPR/Cas9 screening identifies a kinetochore-microtubule dependent mechanism for Aurora-A inhibitor resistance in breast cancer. Cancer Commun. 2021, 41, 121–139. [Google Scholar] [CrossRef]
- Ana, G.; Kelly, P.; Malebari, A.; Noorani, S.; Nathwani, S.; Twamley, B.; Fayne, D.; O’Boyle, N.; Zisterer, D.; Pimentel, E.; et al. Synthesis and Biological Evaluation of 1-(Diarylmethyl)-1H-1,2,4-triazoles and 1-(Diarylmethyl)-1H-imidazoles as a Novel Class of Anti-Mitotic Agent for Activity in Breast Cancer. Pharmaceuticals 2021, 14, 169. [Google Scholar] [CrossRef]
- Ko, P.; Choi, J.-H.; Song, S.; Keum, S.; Jeong, J.; Hwang, Y.; Kim, J.; Rhee, S. Microtubule Acetylation Controls MDA-MB-231 Breast Cancer Cell Invasion through the Modulation of Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2021, 22, 6018. [Google Scholar] [CrossRef]
- Xia, L.-Y.; Zhang, Y.-L.; Yang, R.; Wang, Z.-C.; Lu, Y.-D.; Wang, B.-Z.; Zhu, H.-L. Tubulin Inhibitors Binding to Colchicine-Site: A Review from 2015 to 2019. Curr. Med. Chem. 2020, 27, 6787–6814. [Google Scholar] [CrossRef] [PubMed]
- Karatoprak, G.; Akkol, E.K.; Genç, Y.; Bardakci, H.; Yücel, Ç.; Sobarzo-Sánchez, E. Combretastatins: An Overview of Structure, Probable Mechanisms of Action and Potential Applications. Molecules 2020, 25, 2560. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S.; Mackenzie, G.; Greedy, B.; Armitage, S.; Chabert, J.F.D.; Bennett, E.; Nettles, J.; Snyder, J.P.; Lawrence, N.J. Combretastatin-like chalcones as inhibitors of microtubule polymerisation. Part 2: Structure-based discovery of alpha-aryl chalcones. Bioorg. Med. Chem. 2009, 17, 7711–7722. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Reddy, M.K.; Viswanath, A. The design and development of imidazothiazole–chalcone derivatives as potential anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 289–304. [Google Scholar] [CrossRef]
- Kamal, A.; Kumar, G.B.; Vishnuvardhan, M.V.P.S.; Shaik, A.B.; Reddy, V.S.; Mahesh, R.; Bin Sayeeda, I.; Kapure, J.S. Synthesis of phenstatin/isocombretastatin–chalcone conjugates as potent tubulin polymerization inhibitors and mitochondrial apoptotic inducers. Org. Biomol. Chem. 2015, 13, 3963–3981. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, K.O.; Zaki, I.; El-Deen, I.M.; Abdelhameid, M.K. A new class of diamide scaffold: Design, synthesis and biological evaluation as potent antimitotic agents, tubulin polymerization inhibition and apoptosis inducing activity studies. Bioorg. Chem. 2018, 84, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Alemi, A.; Reza, J.Z.; Haghiralsadat, F.; Jaliani, H.Z.; Karamallah, M.H.; Hosseini, S.A.; Karamallah, S.H. Paclitaxel and curcumin coadministration in novel cationic PEGylated niosomal formulations exhibit enhanced synergistic antitumor efficacy. J. Nanobiotechnol. 2018, 16, 1–20. [Google Scholar] [CrossRef]
- Muthu, M.S.; Kulkarni, S.A.; Xiong, J.; Feng, S.-S. Vitamin E TPGS coated liposomes enhanced cellular uptake and cytotoxicity of docetaxel in brain cancer cells. Int. J. Pharm. 2011, 421, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Ammar, H.O.; Mohamed, M.I.; Tadros, M.I.; Fouly, A.A. Transdermal Delivery of Ondansetron Hydrochloride via Bilosomal Systems: In Vitro, Ex Vivo, and In Vivo Characterization Studies. AAPS PharmSciTech 2018, 19, 2276–2287. [Google Scholar] [CrossRef]
- Zakaria, M.Y.; Fayad, E.; Althobaiti, F.; Zaki, I.; Abu Almaaty, A.H. Statistical optimization of bile salt deployed nanovesicles as a potential platform for oral delivery of piperine: Accentuated antiviral and anti-inflammatory activity in MERS-CoV challenged mice. Drug Deliv. 2021, 28, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Aburahma, M.H. Bile salts-containing vesicles: Promising pharmaceutical carriers for oral delivery of poorly water-soluble drugs and peptide/protein-based therapeutics or vaccines. Drug Deliv. 2014, 23, 1–21. [Google Scholar] [CrossRef]
- Zaki, I.; Abdelhameid, M.K.; El-Deen, I.M.; Wahab, A.H.A.A.; Ashmawy, A.M.; Mohamed, K.O. Design, synthesis and screening of 1, 2, 4-triazinone derivatives as potential antitumor agents with apoptosis inducing activity on MCF-7 breast cancer cell line. Eur. J. Med. Chem. 2018, 156, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Abdelhameid, M.K.; Zaki, I.; Mohammed, M.R.; Mohamed, K.O. Design, synthesis, and cytotoxic screening of novel azole derivatives on hepatocellular carcinoma (HepG2 Cells). Bioorg. Chem. 2020, 101, 103995. [Google Scholar] [CrossRef] [PubMed]
- Zaki, I.; Ramadan, H.M.M.; El-Sayed, E.H.; El-Moneim, M.A. Design, synthesis, and cytotoxicity screening of new synthesized imidazolidine-2-thiones as VEGFR-2 enzyme inhibitors. Arch. Pharm. 2020, 353. [Google Scholar] [CrossRef] [PubMed]
- Abu Almaaty, A.; Toson, E.; El-Sayed, E.-S.; Tantawy, M.; Fayad, E.; Abu Ali, O.; Zaki, I. 5-Aryl-1-Arylideneamino-1H-Imidazole-2(3H)-Thiones: Synthesis and In Vitro Anticancer Evaluation. Molecules 2021, 26, 1706. [Google Scholar] [CrossRef] [PubMed]
- El-Aziz, R.M.A.; Zaki, I.; El-Deen, I.M.; Abd-Rahman, M.S.; Mohammed, F.Z. In Vitro Anticancer Evaluation of Some Synthesized 2H-Quinolinone and Halogenated 2H-Quinolinone Derivatives as Therapeutic Agents. Anti-Cancer Agents Med. Chem. 2020, 20, 2304–2315. [Google Scholar] [CrossRef]
- Jiang, X.; Tsona, N.T.; Tang, S.; Du, L. Hydrogen bond docking preference in furans: O H ··· π vs. O H ··· O. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 191, 155–164. [Google Scholar] [CrossRef]
- Al-Mahallawi, A.M.; Abdelbary, A.A.; Aburahma, M.H. Investigating the potential of employing bilosomes as a novel vesicular carrier for transdermal delivery of tenoxicam. Int. J. Pharm. 2015, 485, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Saifi, Z.; Rizwanullah, M.; Mir, S.R.; Amin, S. Bilosomes nanocarriers for improved oral bioavailability of acyclovir: A complete characterization through in vitro, ex-vivo and in vivo assessment. J. Drug Deliv. Sci. Technol. 2020, 57, 101634. [Google Scholar] [CrossRef]
- Ahmed, S.; Kassem, M.A.; Sayed, S. Bilosomes as Promising Nanovesicular Carriers for Improved Transdermal Delivery: Construction, in vitro Optimization, ex vivo Permeation and in vivo Evaluation. Int. J. Nanomed. 2020, ume 15, 9783–9798. [Google Scholar] [CrossRef]
- Aziz, D.E.; Abdelbary, A.A.; Elassasy, A.I. Investigating superiority of novel bilosomes over niosomes in the transdermal delivery of diacerein: In vitro characterization, ex vivo permeation and in vivo skin deposition study. J. Liposome Res. 2018, 29, 73–85. [Google Scholar] [CrossRef]
- Albash, R.; El-Nabarawi, M.A.; Refai, H.; Abdelbary, A.A. Tailoring of PEGylated bilosomes for promoting the transdermal delivery of olmesartan medoxomil: In-vitro characterization, ex-vivo permeation and in-vivo assessment. Int. J. Nanomed. 2019, ume 14, 6555–6574. [Google Scholar] [CrossRef] [Green Version]
- Malik, N.A. Solubilization and Interaction Studies of Bile Salts with Surfactants and Drugs: A Review. Appl. Biochem. Biotechnol. 2016, 179, 179–201. [Google Scholar] [CrossRef]
- Li, N.; Fu, T.; Fei, W.; Han, T.; Gu, X.; Hou, Y.; Liu, Y.; Yang, J. Vitamin E D-alpha-tocopheryl polyethylene glycol 1000 succinate-conjugated liposomal docetaxel reverses multidrug resistance in breast cancer cells. J. Pharm. Pharmacol. 2019, 71, 1243–1254. [Google Scholar] [CrossRef]
- Stetefeld, J.; McKenna, S.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef]
- Mosallam, S.; Sheta, N.M.; Elshafeey, A.H.; Abdelbary, A.A. Fabrication of Highly Deformable Bilosomes for Enhancing the Topical Delivery of Terconazole: In Vitro Characterization, Microbiological Evaluation, and In Vivo Skin Deposition Study. AAPS PharmSciTech 2021, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Basha, M.; El-Alim, S.A.; Shamma, R.; Awad, G. Design and optimization of surfactant-based nanovesicles for ocular delivery of Clotrimazole. J. Liposome Res. 2013, 23, 203–210. [Google Scholar] [CrossRef]
- Yousry, C.; Zikry, P.M.; Salem, H.M.; Basalious, E.B.; El-Gazayerly, O.N. Integrated nanovesicular/self-nanoemulsifying system (INV/SNES) for enhanced dual ocular drug delivery: Statistical optimization, in vitro and in vivo evaluation. Drug Deliv. Transl. Res. 2020, 10, 801–814. [Google Scholar] [CrossRef]
- Abdelbary, A.A.; Abd-Elsalam, W.H.; Al-Mahallawi, A.M. Fabrication of novel ultradeformable bilosomes for enhanced ocular delivery of terconazole: In vitro characterization, ex vivo permeation and in vivo safety assessment. Int. J. Pharm. 2016, 513, 688–696. [Google Scholar] [CrossRef]
- Pachauri, M.; Gupta, E.D.; Ghosh, P.C. Piperine loaded PEG-PLGA nanoparticles: Preparation, characterization and targeted delivery for adjuvant breast cancer chemotherapy. J. Drug Deliv. Sci. Technol. 2015, 29, 269–282. [Google Scholar] [CrossRef]
- Kalepu, S.; Nekkanti, V. Insoluble drug delivery strategies: Review of recent advances and business prospects. Acta Pharm. Sin. B 2015, 5, 442–453. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Tan, S.-W.; Guo, Y.; Huang, J.; Chu, M.; Liu, H.; Zhang, Z. pH-Sensitive Docetaxel-Loaded d-α-Tocopheryl Polyethylene Glycol Succinate–Poly(β-amino ester) Copolymer Nanoparticles for Overcoming Multidrug Resistance. Biomacromolecules 2013, 14, 2636–2646. [Google Scholar] [CrossRef] [PubMed]
- Aldawsari, H.; Ahmed, O.; Alhakamy, N.; Neamatallah, T.; Fahmy, U.; Badr-Eldin, S. Lipidic Nano-Sized Emulsomes Potentiates the Cytotoxic and Apoptotic Effects of Raloxifene Hydrochloride in MCF-7 Human Breast Cancer Cells: Factorial Analysis and In Vitro Anti-Tumor Activity Assessment. Pharmaceutics 2021, 13, 783. [Google Scholar] [CrossRef]
- Dubey, S.; Vyas, S.P. Emulsomes for Lipophilic Anticancer Drug Delivery: Development, optimization and In Vitro drug release kinetic study. Int. J. Appl. Pharm. 2021, 114–121. [Google Scholar] [CrossRef]
- El-Halim, S.M.A.; Abdelbary, G.A.; Amin, M.M.; Zakaria, M.Y.; Shamsel-Din, H.A.; Ibrahim, A.B. Stabilized oral nanostructured lipid carriers of Adefovir Dipivoxil as a potential liver targeting: Estimation of liver function panel and uptake following intravenous injection of radioiodinated indicator. DARU J. Pharm. Sci. 2020, 28, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, M.M.; Khalil, I.; Khalil, M.A. Sertaconazole nitrate loaded nanovesicular systems for targeting skin fungal infection: In-vitro, ex-vivo and in-vivo evaluation. Int. J. Pharm. 2017, 527, 1–11. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp No. | IC50 Value (μM) | |
|---|---|---|
| MCF-7 | MCF-10A | |
| 2 | >50 | NT |
| 3 | 4.73 ± 0.27 | NT |
| 4a | 27.31 ± 1.21 | NT |
| 4b | 10.37 ± 1.09 | NT |
| 4c | 26.15 ± 1.21 | NT |
| 4d | 18.37 ± 1.14 | NT |
| 4e | 2.11 ± 0.19 | 29.27 ± 1.21 |
| 5a | 3.92 ± 0.21 | NT |
| 5b | 5.03 ± 1.01 | NT |
| 5c | 3.03 ± 0.39 | NT |
| 5d | 2.61 ± 0.32 | NT |
| Cisplatin | 1.02 ± 0.12 | 22.62 ± 0.19 |
| Formula | A (Bile Salt Type) | B (Bile Salt Amount) | C (DSPE–mPEG-2000 Amount) | A (EE%) | B (PS) | C (ZP) | PDI |
|---|---|---|---|---|---|---|---|
| F1 | SDC | 30 | 50 | 85.4 ± 4.7 | 249.9 ± 21.5 | −45.9 ± 3.8 | 0.25 ± 0.02 |
| F2 | STC | 30 | 50 | 65.6 ± 2.3 | 367.6 ± 29.7 | −56.7 ± 7.4 | 0.21 ± 0.03 |
| F3 | SDC | 15 | 50 | 93.2 ± 3.6 | 156.5 ± 18.2 | −37.8 ± 2.6 | 0.31 ± 0.07 |
| F4 | STC | 15 | 50 | 72.6 ± 2.1 | 219.8 ± 13.8 | −47.7 ± 2.3 | 0.34 ± 0.05 |
| F5 | SDC | 30 | 25 | 94.4 ± 4.2 | 432.4 ± 21.3 | −38.3 ± 3.1 | 0.28 ± 0.085 |
| F6 | STC | 30 | 25 | 77.3 ± 3.3 | 527.2 ± 24.6 | −50.6 ± 5.6 | 0.56 ± 0.08 |
| F7 | SDC | 15 | 25 | 100 ± 5.6 | 280.3 ± 15.4 | −22.5 ± 3.4 | 0.23 ± 0.05 |
| F8 | STC | 15 | 25 | 81.5 ± 2.9 | 336.2 ± 18.9 | −35.8 ± 3.7 | 0.39 ± 0.074 |
| Responses | EE (%) | PS (nm) | ZP (mV) |
|---|---|---|---|
| R2 | 0.9999 | 0.9997 | 0.9994 |
| Adjusted R2 | 0.9997 | 0.9979 | 0.996 |
| Predicted R2 | 0.9970 | 0.9810 | 0.9633 |
| Adequate precision | 172.6 | 73.1 | 53.6 |
| Significant factors | A, B, C | A, B, C | A, B, C |
| Observed value of the optimal formula (F7) | 100 | 280.3 | −22.5 |
| Predicted value of the optimal formula (F7) | 99.9 | 282.2 | −22.73 |
| Absolute deviation % | 0.1 | 0.68 | 1.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaki, I.; Abou-Elkhair, R.A.I.; Abu Almaaty, A.H.; A. Abu Ali, O.; Fayad, E.; Ahmed Gaafar, A.G.; Zakaria, M.Y. Design and Synthesis of Newly Synthesized Acrylamide Derivatives as Potential Chemotherapeutic Agents against MCF-7 Breast Cancer Cell Line Lodged on PEGylated Bilosomal Nano-Vesicles for Improving Cytotoxic Activity. Pharmaceuticals 2021, 14, 1021. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14101021
Zaki I, Abou-Elkhair RAI, Abu Almaaty AH, A. Abu Ali O, Fayad E, Ahmed Gaafar AG, Zakaria MY. Design and Synthesis of Newly Synthesized Acrylamide Derivatives as Potential Chemotherapeutic Agents against MCF-7 Breast Cancer Cell Line Lodged on PEGylated Bilosomal Nano-Vesicles for Improving Cytotoxic Activity. Pharmaceuticals. 2021; 14(10):1021. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14101021
Chicago/Turabian StyleZaki, Islam, Reham A. I. Abou-Elkhair, Ali H. Abu Almaaty, Ola A. Abu Ali, Eman Fayad, Ahmed Gaafar Ahmed Gaafar, and Mohamed Y. Zakaria. 2021. "Design and Synthesis of Newly Synthesized Acrylamide Derivatives as Potential Chemotherapeutic Agents against MCF-7 Breast Cancer Cell Line Lodged on PEGylated Bilosomal Nano-Vesicles for Improving Cytotoxic Activity" Pharmaceuticals 14, no. 10: 1021. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14101021