
Development and Performance Verification of the PBPK Model for Antazoline and Its Metabolite and Its Utilization for Pharmacological Hypotheses Formulating

 , , ,
, , ,
Abstract
:1. Introduction
1.1. Antazoline
1.2. Study Aims
- To perform in vitro experiments towards antazoline’s ADME characterization and present their results,
- To build and verify the antazoline PBPK model based on the in vitro ADME data with the aim of its pharmacokinetic characterization,
- To build a model of the antazoline metabolite based on the available phys-chem data and extend the previously built PBPK model.
2. Results
In Vitro Assays
3. Discussion
4. Materials and Methods
4.1. ADME Parameters Analysis
4.1.1. Binding Assays
4.1.2. Metabolic Stability
4.1.3. Clinical Studies
4.2. PBPK Modelling and Simulation—Development and Qualification
Models
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piotrowski, R.; Giebułtowicz, J.; Baran, J.; Sikorska, A.; Gralak-Łachowska, D.; Soszyńska, M.; Wroczyński, P.; Kułakowski, P. Antazoline-Insights into Drug-Induced Electrocardiographic and Hemodynamic Effects: Results of the ELEPHANT II Substudy. Ann. Noninvasive Electrocardiol. 2017, 22, e12441. [Google Scholar] [CrossRef] [PubMed]
- Farkowski, M.M.; Maciag, A.; Dabrowski, R.; Pytkowski, M.; Kowalik, I.; Szwed, H. Clinical Efficacy of Antazoline in Rapid Cardioversion of Paroxysmal Atrial Fibrillation—A Protocol of a Single Center, Randomized, Double-Blind, Placebo-Controlled Study (the AnPAF Study). Trials 2012, 13, 162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciag, A.; Farkowski, M.; Chwyczko, T.; Beckowski, M.; Syska, P.; Kowalik, I.; Pytkowski, M.; Wozniak, J.; Dabrowski, R.; Szwed, H. Efficacy and Safety of Antazoline in Rapid Cardioversion of Paroxysmal Atrial Fibrillation (the AnPAF Study). Europace 2017, 19, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Frommeyer, G.; Sterneberg, M.; Dechering, D.G.; Kaese, S.; Bögeholz, N.; Pott, C.; Fehr, M.; Bogossian, H.; Milberg, P.; Eckardt, L. Effective Suppression of Atrial Fibrillation by the Antihistaminic Agent Antazoline: First Experimental Insights into a Novel Antiarrhythmic Agent. Cardiovasc. Ther. 2017, 35, e12244. [Google Scholar] [CrossRef] [PubMed]
- Farkowski, M.M.; Maciag, A.; Zurawska, M.; Kowalik, I.; Szwed, H.; Pytkowski, M. Clinical Effectiveness and Safety of Antazoline-Based Therapy in Patients with Stable Coronary Artery Disease Undergoing Pharmacological Cardioversion of Short-Duration Atrial Fibrillation in the Emergency Department. Cardiovasc. Ther. 2018, 36, e12469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkowski, M.M.; Maciąg, A.; Żurawska, M.; Kołakowski, K.; Gardziejczyk, P.; Kowalik, I.; Szwed, H.; Pytkowski, M. Rapid Pharmacological Cardioversion of Recent-Onset Atrial Fibrillation Using Antazoline in Elderly Patients. Pol. Arch. Intern. Med. 2021, 32, 3259–3269. [Google Scholar] [CrossRef] [PubMed]
- Palimonka, K.; Paśko, P.; Szuta, M.; Sowizdraniuk, J. Antazoline Renaissance In The Treatment Of Cardiac Arrhythmia: A Review. Acta Pol. Pharm.-Drug Res. 2020, 77, 209–219. [Google Scholar] [CrossRef]
- Farkowski, M.M.; Maciąg, A.; Żurawska, M.; Pytkowski, M.; Kowalik, I.; Woźniak, J.; Sterliński, M.; Szwed, H. Comparative Effectiveness and Safety of Antazoline-based and Propafenone-based Strategies for Pharmacological Cardioversion of Short-duration Atrial Fibrillation in the Emergency Department. Pol. Arch. Intern. Med. 2016, 126, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giebułtowicz, J.; Piotrowski, R.; Baran, J.; Kułakowski, P.; Wroczyński, P. Application of a Novel Liquid Chromatography/Tandem Mass Spectrometry Method for the Determination of Antazoline in Human Plasma: Result of ELEPHANT-I [ELEctrophysiological, Pharmacokinetic and Hemodynamic Effects of PHenazolinum (ANTazoline Mesylate)] Human Pharmacokinetic Study. J. Pharm. Biomed. Anal. 2016, 123, 113–119. [Google Scholar] [CrossRef]
- Srzednicki, M.; Sadowski, Z.; Kulikowski, A. [Evaluation of the anti-arrhythmia effectiveness of Phenazolinum Polfa in paroxysmal atrial fibrillation]. Pol. Tyg. Lek. 1990, 45, 924–927. [Google Scholar] [PubMed]
- Piotrowski, R.; Kryński, T.; Baran, J.; Futyma, P.; Stec, S.; Kułakowski, P. Antazoline for Rapid Termination of Atrial Fibrillation during Ablation of Accessory Pathways. Cardiol. J. 2014, 21, 299–303. [Google Scholar] [CrossRef] [Green Version]
- Kuch, M.; Janiszewski, M.; Dłuzniewski, M.; Mamcarz, A. Antazoline-Ineffective or Misprized in the Treatment of Paroxysmal Atrial Fibrillation? Pol. Prz. Kardiol. 2000, 2, 247–251. [Google Scholar]
- Shebley, M.; Sandhu, P.; Emami Riedmaier, A.; Jamei, M.; Narayanan, R.; Patel, A.; Peters, S.A.; Reddy, V.P.; Zheng, M.; de Zwart, L.; et al. Physiologically Based Pharmacokinetic Model Qualification and Reporting Procedures for Regulatory Submissions: A Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Edginton, A.N. Using Physiologically Based Pharmacokinetic Modeling for Mechanistic Insight: Cases of Reverse Translation. Clin. Transl. Sci. 2018, 11, 109–111. [Google Scholar] [CrossRef]
- Maharaj, A.R.; Edginton, A.N. Physiologically Based Pharmacokinetic Modeling and Simulation in Pediatric Drug Development. CPT Pharmacomet. Syst. Pharm. 2014, 3, e150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.D. Factorial Sampling Plans for Preliminary Computational Experiments. Technometrics 1991, 33, 161–174. [Google Scholar] [CrossRef]
- Giebułtowicz, J.; Korytowska, N.; Piotrowski, R.; Kułakowski, P.; Latacz, G.; Szymańska, E.; Wiśniowska, B.; Polak, S. Characterization of In Vitro and In Vivo Metabolism of Antazoline Using Liquid Chromatography-Tandem Mass Spectrometry. Int. J. Mol. Sci. 2020, 21, 9693. [Google Scholar] [CrossRef] [PubMed]
- Bērziņš, K.; Grante, I.; Nakurte, I.; Actiņš, A. The Influence of PH on the Stability of Antazoline: Kinetic Analysis. RSC Adv. 2015, 5, 68179–68186. [Google Scholar] [CrossRef]
- Marshall, P.B. Some Chemical and Physical Properties Associated with Histamine Antagonism. Br. J. Pharmacol. Chemother. 1955, 10, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görög, S. Ultraviolet-Visible Spectrophotometry in Pharmaceutical Analysis, 1st ed.; CRC Press: Boca Raton, FL, USA, 2017; ISBN 9781315898322. [Google Scholar]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Giebułtowicz, J.; Korytowska, N.; Sobiech, M.; Polak, S.; Wiśniowska, B.; Piotrowski, R.; Kułakowski, P.; Luliński, P. Magnetic Core–Shell Molecularly Imprinted Nano-Conjugates for Extraction of Antazoline and Hydroxyantazoline from Human Plasma—Material Characterization, Theoretical Analysis and Pharmacokinetics. Int. J. Mol. Sci. 2021, 22, 3665. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, T.; Rowland, M. Mechanistic Approaches to Volume of Distribution Predictions: Understanding the Processes. Pharm. Res. 2007, 24, 918–933. [Google Scholar] [CrossRef] [PubMed]
- Crewe, H.K.; Barter, Z.E.; Yeo, K.R.; Rostami-Hodjegan, A. Are There Differences in the Catalytic Activity per Unit Enzyme of Recombinantly Expressed and Human Liver Microsomal Cytochrome P450 2C9? A Systematic Investigation into Inter-System Extrapolation Factors. Biopharm. Drug Dispos. 2011, 32, 303–318. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
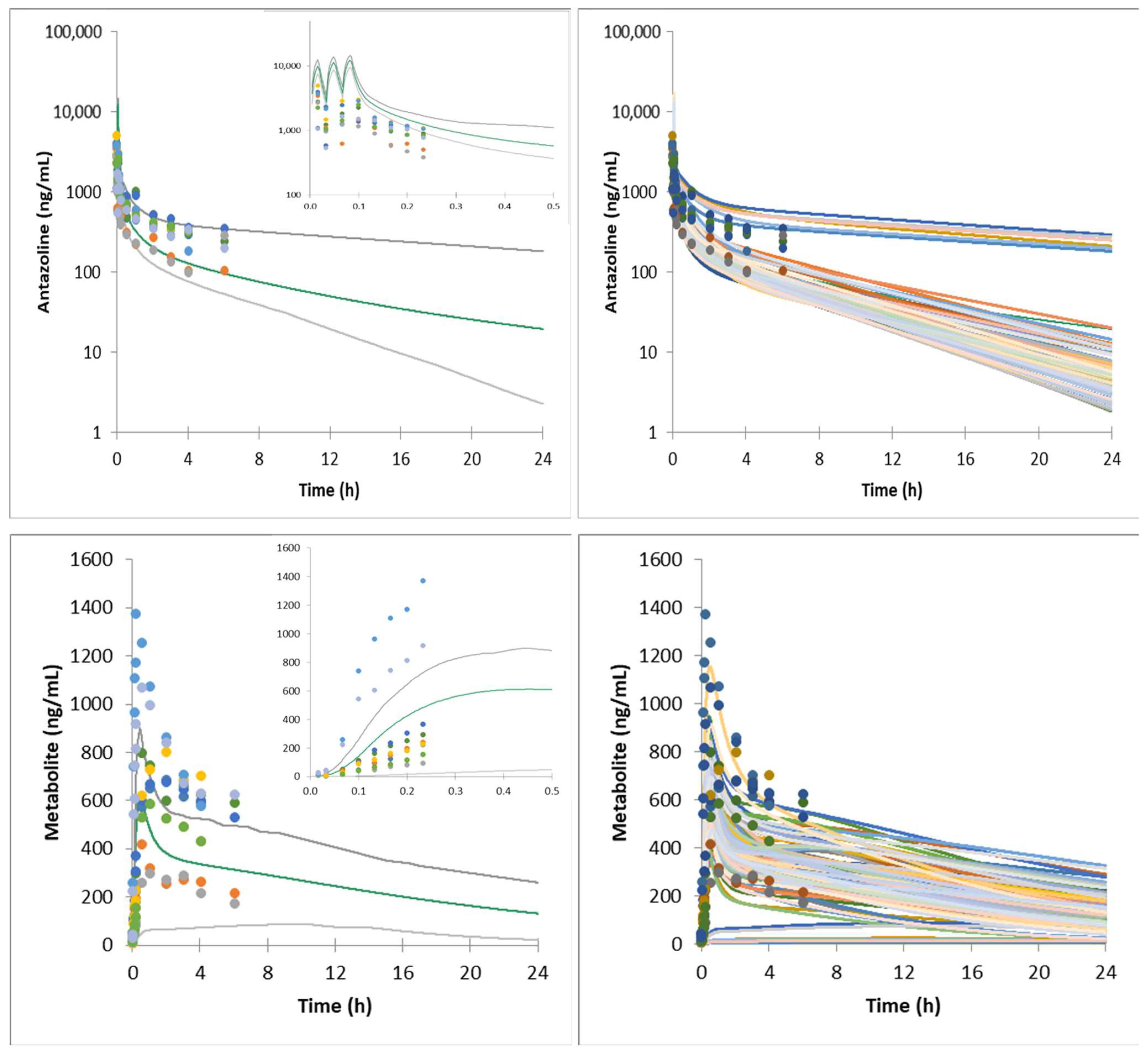{kind=link}
{kind=link}
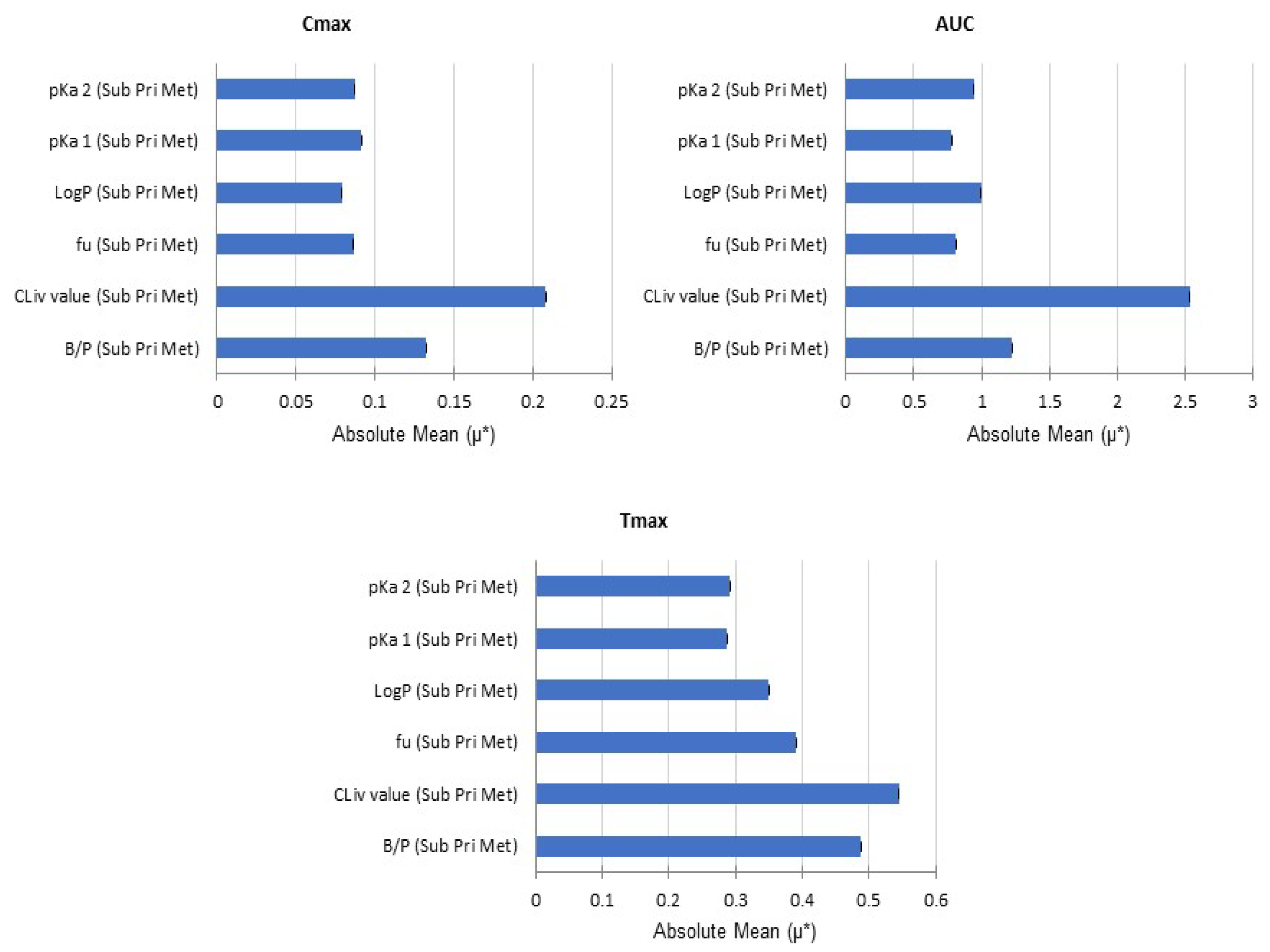{kind=link}
{kind=link}
| Binding Assays | ||||
| Parameter | Compound | Mean | SD | |
| Blood to Plasma Ratio (B/P) | Antazoline | 1.18 | 0.0161 | |
| Methazolamide (C) | 10.9 | 3.81 | ||
| Fraction Unbound (fu) | Antazoline | 0.586 | 0.0109 | |
| Warfarin (C) | 0.016 | 0.0002 | ||
| Microsomal Binding (fu) | Antazoline | 0.88 | 0.00563 | |
| Amitriptyline (C) | 0.47 | 0.00381 | ||
| Test System | Compound | CLint [µL/min/106 cells] | SE CLint | t1/2 [min] |
| Hepatocytes | Antazoline | 21.1 | 2.21 | 65.8 |
| Verapamil (C) | 78.6 | 3.85 | 17.6 | |
| HLM | Antazoline | 97.4 | 8.63 | 14.2 |
| Verapamil (C) | 373 | 23.7 | 3.72 | |
| Recombinant CYP Isoform | Compound | t1/2 [min] | SE t1/2 | Bactosomes™ Final P450 Concentration [pmol/L] |
| 2D6 | Antazoline | 0.56 | 0 | 50 |
| Dextromethorphan (C) | 1.53 | 1.17 | 50 | |
| 2C9 | Antazoline | - | - | 25 |
| Diclofenac (C) | 1.91 | 0.0800 | 25 | |
| 2C19 | Antazoline | 51 | 2.84 | 100 |
| Omeprazole (C) | 1.53 | 0.420 | 100 | |
| 3A4 | Antazoline | - | - | 25 |
| Testosterone (C) | 24.6 | 4.12 | 25 | |
| 1A2 | Antazoline | 100 | ||
| Ethoxycoumarin (C) | 2.23 | 1.00 | 100 | |
| 2C8 | Antazoline | 1100 | 4210 | 50 |
| Paclitaxel (C) | 13.4 | 0.880 | 50 | |
| UGT Isoform | Test Concentration | Recombinant UGT CLint Clearance [µL/min/mg] | Recombinant UGT Half-Life t1/2 [min] | |
| 1A1 | 1 µM | <2.9 | >240 | |
| 1A3 | 1 µM | <2.9 | >240 | |
| 1A4 | 1 µM | <2.9 | >240 | |
| 1A6 | 1 µM | <5.8 | >240 | |
| 1A9 | 1 µM | <5.8 | >240 | |
| 2B7 | 1 µM | <5.8 | >240 | |
| B15 | 1 µM | <5.8 | >240 | |
| Parameter | Distribution | Lower Bound | Upper Bound |
|---|---|---|---|
| LogPo:w Value | Uniform | 1.5 | 3.5 |
| pKa 1 = acid Value (Ampholytes) | Uniform | 6 | 14 |
| pKa 2 = base Value (Ampholytes) | Uniform | 6 | 14 |
| Blood to plasma partition ratio | Uniform | 0.55 | 2 |
| i.v. Clearance value | Uniform | 2 | 80 |
| Fraction unbound in plasma | Uniform | 0.1 | 1 |
| ANTAZOLINE | |||
| Phys-chem Parameters | Compound type | Monoprotic Base | Source |
| MW | 265.35 g/mol | PubChem | |
| logP | 3.16 | Average of PubChem; ALGPS; ChemAxon reported values | |
| pKa | 9.43 | Average of PubChem; ChemAxon, Toxnet [18,19,20] | |
| Blood binding | fu plasma | 0.586 | In house data/Cyprotex |
| B/P | 1.18 | In house data | |
| Distribution | Vss | 4.97 | Simcyp Method 2 |
| Elimination | CLint HLM | 97.4 (SE 8.63) µL/min/mg protein | In house data |
| CLint hepatcyte | 21.1 (SE 2.21) µL/min/mg protein | In house data | |
| CLint CYP2D6 | 24.75 µL/min/pmol | In house data, Bactosomes | |
| CLint CYP2C19 | 0.14 µL/min/pmol | In house data, Bactosomes | |
| Additional systemic clearance | 10 L/h | Estimated * | |
| M2 METABOLITE | |||
| Phys-chem parameters | Compound type | Ampholyte | |
| MW | 281.36 g/mol | PubChem | |
| logP | 2.57 | ChemAxon | |
| pKa1 | 9.2 | ChemAxon | |
| pKa2 | 10.3 | ChemAxon | |
| Blood binding | fu plasma | 0.742 | Simcyp QSAR model |
| B/P | 1 | Assumed | |
| Distribution | Vss | 2.17 | Simcyp Method 2 |
| Elimination | CL total | 12 L/h | pkCSM [21] |
| N = 18 | Mean | SD |
|---|---|---|
| Age [Years] | 59 | 14 |
| Gender (M/F) | 12/6 | |
| BMI [kg (m2)−1] | 28.8 | 4.25 |
| Echocardiographic Parameters LVEF (%) | 59 | 4 |
| Laboratory Tests | ||
| WBC level [K/µL] | 6.8 | 1.9 |
| HGB level [g L−1] | 14.6 | 1.4 |
| HCT % | 42.7 | 3.7 |
| Platelet Count (PLT) [K/µL] | 220 | 51 |
| Creatinine level [mg L−1] | 1.04 | 0.38 |
| Sodium level [mmol L−1] | 141 | 2 |
| Potassium level [mmol L−1] | 4.3 | 0.2 |
| Concomitant Disorders | N (%) | |
| Atrial fibrillation | 18 (100%) | |
| Hypertension | 14 (78%) | |
| Hyperlipidemia | 6 (33%) | |
| Coronary arterial disease | 1 (5.5%) | |
| Chronic heart failure | 1 (5.5%) | |
| Chronic obstructive pulmonary disease | 1 (5.5%) | |
| Diabetes mellitus | 2 (11%) | |
| Obstructive sleep apnea | 2 (11%) | |
| Hypothyroidism | 4 (22%) | |
| Renal failure | 1 (5.5%) | |
| Antiarrhythmic Drugs | ||
| Beta-blocker | 11 (61%) | |
| Propafenon | 9 (50%) | |
| Sotalol | 3 (17%) | |
| Amiodaron | 1 (5.5%) | |
| None | 1 (5.5%) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiśniowska, B.; Giebułtowicz, J.; Piotrowski, R.; Kułakowski, P.; Polak, S. Development and Performance Verification of the PBPK Model for Antazoline and Its Metabolite and Its Utilization for Pharmacological Hypotheses Formulating. Pharmaceuticals 2022, 15, 379. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15030379
Wiśniowska B, Giebułtowicz J, Piotrowski R, Kułakowski P, Polak S. Development and Performance Verification of the PBPK Model for Antazoline and Its Metabolite and Its Utilization for Pharmacological Hypotheses Formulating. Pharmaceuticals. 2022; 15(3):379. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15030379
Chicago/Turabian StyleWiśniowska, Barbara, Joanna Giebułtowicz, Roman Piotrowski, Piotr Kułakowski, and Sebastian Polak. 2022. "Development and Performance Verification of the PBPK Model for Antazoline and Its Metabolite and Its Utilization for Pharmacological Hypotheses Formulating" Pharmaceuticals 15, no. 3: 379. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15030379