
Virtual Screening Based on Machine Learning Explores Mangrove Natural Products as KRASG12C Inhibitors

 , and
, and
Abstract
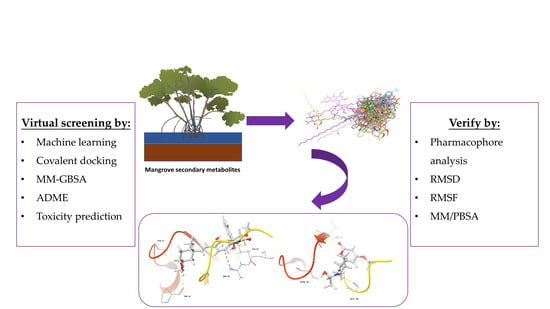
:
1. Introduction
2. Result
2.1. Candidate Compound Library Data
2.2. Machine Learning Models
2.3. Random Forest Classification Model
2.4. Chemical Space
2.5. Prediction of Prospects
2.6. Docking
2.7. MM-GBSA
2.8. ADME
2.9. Pharmacophore Analysis
2.10. Root Mean Square Deviation (RMSD) Analysis
2.11. Root Mean Square Volatility (RMSF) Analysis
2.12. MM/PBSA Analysis
3. Discussion
4. Materials and Methods
4.1. Protein Pretreatment
4.2. Machine Learning
4.2.1. Data
4.2.2. Machine Learning Models
4.2.3. QSAR Modeling
4.2.4. Principal Component Analysis
4.3. Covalent Docking
4.4. ADME
4.5. Pharmacophore Modeling and Matching Validation
4.6. Molecular Dynamics (MD) Simulation
4.7. MM-PBSA
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ruhil, S.; Balhara, M.; Dhankhar, S.; Chhillar, A.K. Aegle marmelos (Linn.) Correa: A potential source of Phytomedicine. J. Med. Plants Res. 2011, 5, 1497–1507. [Google Scholar]
- Duraipandiyan, V.; Ignacimuthu, S. Antibacterial and antifungal activity of Cassia fistula L.: An ethnomedicinal plant. J. Ethnopharmacol. 2007, 112, 590–594. [Google Scholar] [CrossRef]
- Bunyapraphatsara, N.; Jutiviboonsuk, A.; Sornlek, P.; Therathanathorn, W.; Aksornkaew, S.; Fong, H.H.; Pezzuto, J.M.; Kosmeder, J. Pharmacological studies of plants in the mangrove forest. Thai J. Phytopharm. 2003, 10, 1–12. [Google Scholar]
- Bobbarala, V.; Vadlapudi, V.R.; Naidu, C.K. Antimicrobial potentialities of mangrove plant Avicennia marina. J. Pharm. Res. 2009, 2, 1019–1021. [Google Scholar]
- Chen, S.; Cai, R.; Liu, Z.; Cui, H.; She, Z. Secondary metabolites from mangrove-associated fungi: Source, chemistry and bioactivities. Nat. Prod. Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef] [PubMed]
- Kano, Y.; Gebregiworgis, T.; Marshall, C.B.; Radulovich, N.; Poon, B.P.K.; St-Germain, J.; Cook, J.D.; Valencia-Sama, I.; Grant, B.M.M.; Herrera, S.G.; et al. Tyrosyl phosphorylation of KRAS stalls GTPase cycle via alteration of switch I and II conformation. Nat. Commun. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, Y.; Li, Y.; Zhou, J.; Wu, Y.; Cui, Y.; Yang, G.; Hong, Y. Mutations of p53 and KRAS activate NF-κB to promote chemoresistance and tumorigenesis via dysregulation of cell cycle and suppression of apoptosis in lung cancer cells. Cancer Lett. 2015, 357, 520–526. [Google Scholar] [CrossRef]
- Drosten, M.; Barbacid, M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell 2020, 37, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.; Bryant, K. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef]
- Fakih, M. Targeting mechanisms of resistance to anti-EGF receptor therapy in KRAS wild-type colorectal cancer: The path to more personalized medicine. Future Oncol. 2013, 9, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Seashore-Ludlow, B.; Rees, M.G.; Cheah, J.H.; Cokol, M.; Price, E.V.; Coletti, M.E.; Jones, V.; Bodycombe, N.E.; Soule, C.K.; Gould, J.; et al. Harnessing Connectivity in a Large-Scale Small-Molecule Sensitivity Dataset. Cancer Discov. 2015, 5, 1210–1223. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Meng, Q.; Wang, C.; Li, F.; Zhu, Z.; Liu, S.; Shi, Y.; Huang, J.; Chen, S.; Li, C. Investigation of transrenal KRAS mutation in late stage NSCLC patients correlates to disease progression. Biomarkers 2016, 22, 654–660. [Google Scholar] [CrossRef]
- Takagi, M.; Shinoda, K.; Piao, J.; Mitsuiki, N.; Takagi, M.; Matsuda, K.; Muramatsu, H.; Doisaki, S.; Nagasawa, M.; Morio, T.J.B. Autoimmune lymphoproliferative syndrome–like disease with somatic KRAS mutation. J. Am. Soc. Hematol. 2011, 117, 2887–2890. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.G.; Olson, P.; Briere, T.; Wiel, C.; Bergo, M.O. Targeting Kras g12c-mutant cancer with a mutation-specific inhibitor. J. Intern. Med. 2020, 288, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Kaiser, C.E.; Frett, B.; Li, H.-Y. Targeting Mutant KRAS for Anticancer Therapeutics: A Review of Novel Small Molecule Modulators. J. Med. Chem. 2013, 56, 5219–5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Westover, K.D.; Ficarro, S.B.; Harrison, R.A.; Choi, H.G.; Pacold, M.E.; Carrasco, M.; Hunter, J.; Kim, N.D.; Xie, T.; et al. Therapeutic Targeting of Oncogenic K-Ras by a Covalent Catalytic Site Inhibitor. Angew. Chem. Int. Ed. 2014, 53, 199–204. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1998, 1, 55–68. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26, reprinted in Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Coutsias, E.A.; Wester, M.J. RMSD and Symmetry. J. Comput. Chem. 2019, 40, 1496–1508. [Google Scholar] [CrossRef]
- Anuar, N.F.S.K.; Wahab, R.A.; Huyop, F.; Halim, K.B.A.; Hamid, A.A.A. In silico mutation on a mutant lipase from Acinetobacter haemolyticus towards enhancing alkaline stability. J. Biomol. Struct. Dyn. 2019, 38, 4493–4507. [Google Scholar] [CrossRef]
- Oyewusi, H.A.; Huyop, F.; Wahab, R.A. Molecular docking and molecular dynamics simulation of Bacillus thuringiensis dehalogenase against haloacids, haloacetates and chlorpyrifos. J. Biomol. Struct. Dyn. 2020, 40, 1979–1994. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryde, U. QM/MM Calculations on Proteins. In Computational Approaches for Studying Enzyme Mechanism Part A; Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2016; pp. 119–158. [Google Scholar]
- Choi, J.; Tark, D.; Lim, Y.-S.; Hwang, S.B. Identification of African Swine Fever Virus Inhibitors through High Performance Virtual Screening Using Machine Learning. Int. J. Mol. Sci. 2021, 22, 13414. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Burgess, J.; Colclough, N.; Davies, N.L.; Lenz, E.M.; Orton, A.L.; Ward, R.A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Modeling 2017, 57, 3124–3137. [Google Scholar] [CrossRef] [PubMed]
- King, R.D.; Hirst, J.D.; Sternberg, M. New approaches to QSAR: Neural networks and machine learning. Perspect. Drug Discov. Des. 1993, 1, 279–290. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.-S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Jovic, A.; Brkic, K.; Bogunovic, N. A review of feature selection methods with applications. In Proceedings of the 2015 38th International Convention on Information and Communication Technology, Electronics and Microelectronics (MIPRO), Opatija, Croatia, 25–29 May 2015; pp. 1200–1205. [Google Scholar]
- Frank, E.; Hall, M.A.; Witten, I.H. The WEKA Workbench; Morgan Kaufmann: Burlington, MA, USA, 2016. [Google Scholar]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Toledo Warshaviak, D.; Golan, G.; Borrelli, K.W.; Zhu, K.; Kalid, O. Structure-Based Virtual Screening Approach for Discovery of Covalently Bound Ligands. J. Chem. Inf. Modeling 2014, 54, 1941–1950. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPEAnte-Chamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.; Carchman, R.; Long, R.; Denslow, S.A. La Crosse Viral Infection in Hospitalized Pediatric Patients in Western North Carolina. Hosp. Pediatrics 2012, 2, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Modeling 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Precision | Recall | F-Measure | MCC |
|---|---|---|---|---|
| Active | 0.946 | 0.993 | 0.969 | 0.825 |
| Inactive | 0.962 | 0.758 | 0.847 | 0.825 |
| Weighted Avg | 0.949 | 0.949 | 0.946 | 0.825 |
| Class | Precision | Recall | F-Measure | MCC |
|---|---|---|---|---|
| Active | 0.795 | 0.939 | 0.861 | 0.289 |
| Inactive | 0.600 | 0.273 | 0.375 | 0.289 |
| Weighted Avg | 0.746 | 0.773 | 0.740 | 0.289 |
| Name | 2D Structure | RMSD | Docking Score |
|---|---|---|---|
| 8 |  | 8.7699 | −6.518 |
| 14 |  | 9.1083 | −6.707 |
| 15 |  | 8.3822 | −6.432 |
| 31 |  | 10.9337 | −6.52 |
| 44 |  | 7.9196 | −6.916 |
| 75 |  | 8.4722 | −6.65 |
| 102 |  | 12.8430 | −7.618 |
| 127 |  | 12.7782 | −7.701 |
| Molecule | MW | Rotatable Bonds | H-Bond Acceptors | H-Bond Donors | ESOL Log S | TPSA | WLOGP | GI Absorption | log Kp (cm/s) |
|---|---|---|---|---|---|---|---|---|---|
| 14 | 241.28 | 5 | 5 | 2 | −1.93 | 71.7 | 0.95 | High | −7.02 |
| 31 | 323.35 | 2 | 4 | 3 | −2.69 | 85.16 | −0.38 | High | −7.58 |
| 44 | 437.53 | 4 | 5 | 4 | −4.66 | 99.02 | 3.01 | High | −6.6 |
| 127 | 315.25 | 3 | 7 | 2 | −3.04 | 120.03 | 1.54 | High | −7.11 |
| ID | Features | Rank | Direct Hit | Partial Hit | Max Fit |
|---|---|---|---|---|---|
| 1 | HHHHAAA | 52.937 | 111 | 000 | 7 |
| 2 | HHHHAAA | 52.338 | 111 | 000 | 7 |
| 3 | HHHHAAA | 51.858 | 111 | 000 | 7 |
| 4 | HHHHHAA | 51.669 | 111 | 000 | 7 |
| 5 | HHHHHAA | 51.530 | 111 | 000 | 7 |
| 6 | HHHHAAA | 51.409 | 111 | 000 | 7 |
| 7 | HHHHHAA | 51.359 | 111 | 000 | 7 |
| 8 | HHHHHAA | 51.352 | 111 | 000 | 7 |
| 9 | HHHHAAA | 51.242 | 111 | 000 | 7 |
| 10 | HHHHAAA | 51.236 | 111 | 000 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, L.; Zheng, T.; Wang, Q.; Liao, Y.; Zheng, X.; Zhong, A.; Huang, Z.; Luo, H. Virtual Screening Based on Machine Learning Explores Mangrove Natural Products as KRASG12C Inhibitors. Pharmaceuticals 2022, 15, 584. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050584
Luo L, Zheng T, Wang Q, Liao Y, Zheng X, Zhong A, Huang Z, Luo H. Virtual Screening Based on Machine Learning Explores Mangrove Natural Products as KRASG12C Inhibitors. Pharmaceuticals. 2022; 15(5):584. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050584
Chicago/Turabian StyleLuo, Lianxiang, Tongyu Zheng, Qu Wang, Yingling Liao, Xiaoqi Zheng, Ai Zhong, Zunnan Huang, and Hui Luo. 2022. "Virtual Screening Based on Machine Learning Explores Mangrove Natural Products as KRASG12C Inhibitors" Pharmaceuticals 15, no. 5: 584. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050584