
Rhinacanthin-C but Not -D Extracted from Rhinacanthus nasutus (L.) Kurz Offers Neuroprotection via ERK, CHOP, and LC3B Pathways


 ,
,  and
and 
Abstract
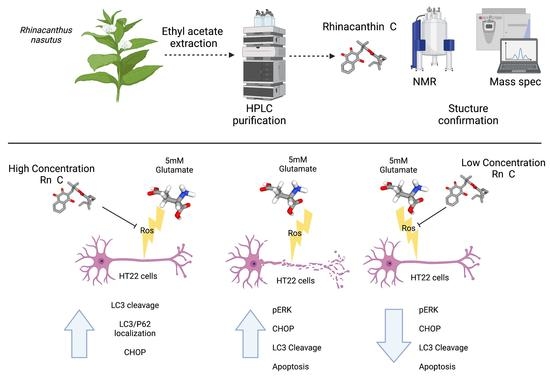
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Plant Material and Rhinacanthin Extraction
2.3. Rhinacanthin Purity Analysis—Recycling Preparative-HPLC
2.4. Nuclear Magnetic Resonance Analysis
2.5. Ultrahigh-Performance Liquid Chromatography-Mass Spectrometry
2.6. Cell Culture
2.7. Cell Viability Assay
2.8. Annexin V/Propidium Iodide Staining
2.9. Reactive Oxygen Species Analysis
2.10. Western Blotting
2.11. Immunofluorescence Colocalization of LC3B and p62 (Sequestosome-1)
2.12. Statistical Analysis
3. Results
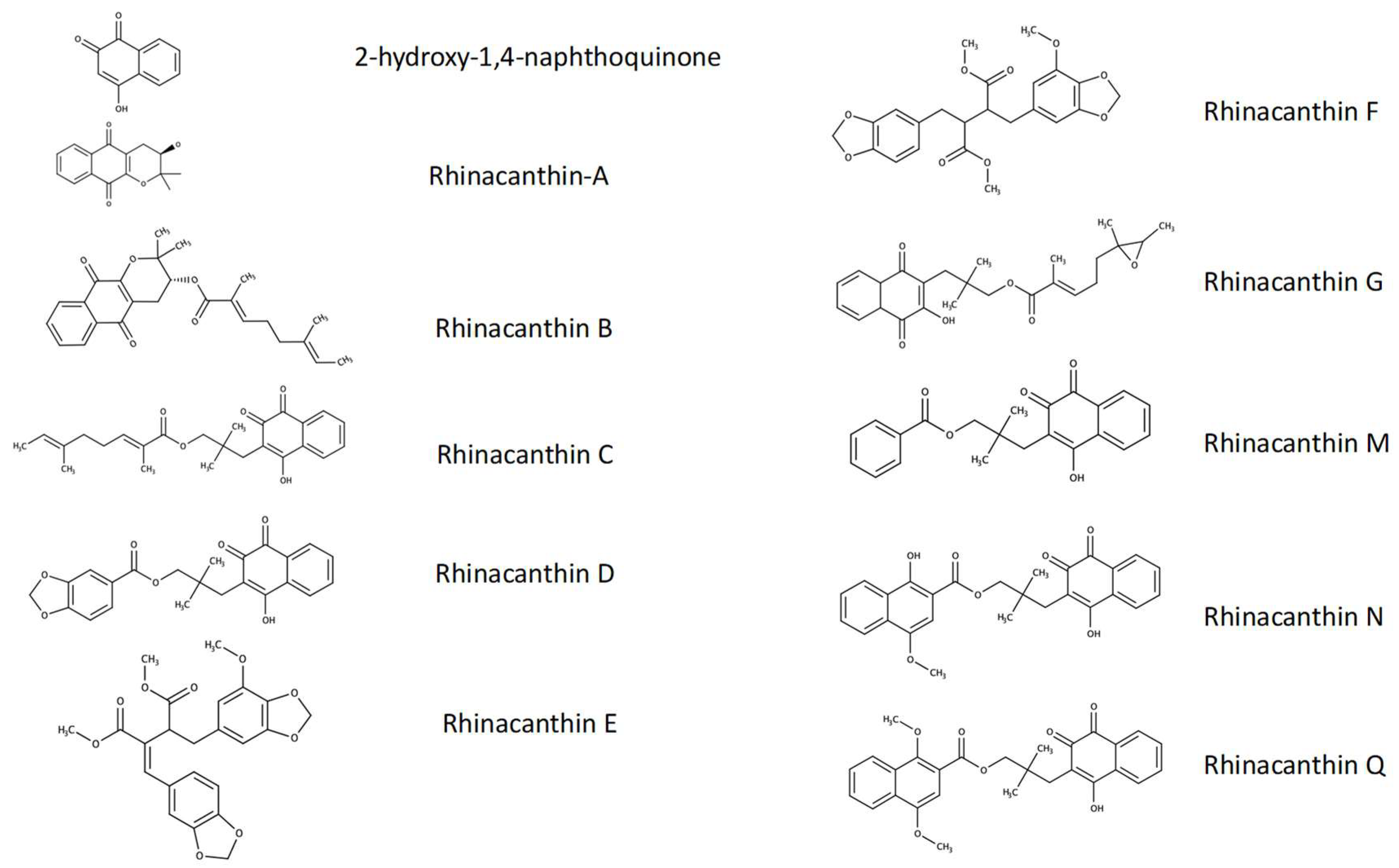
3.1. Identification, Yield, and Purity of Rn-C and Rn-D
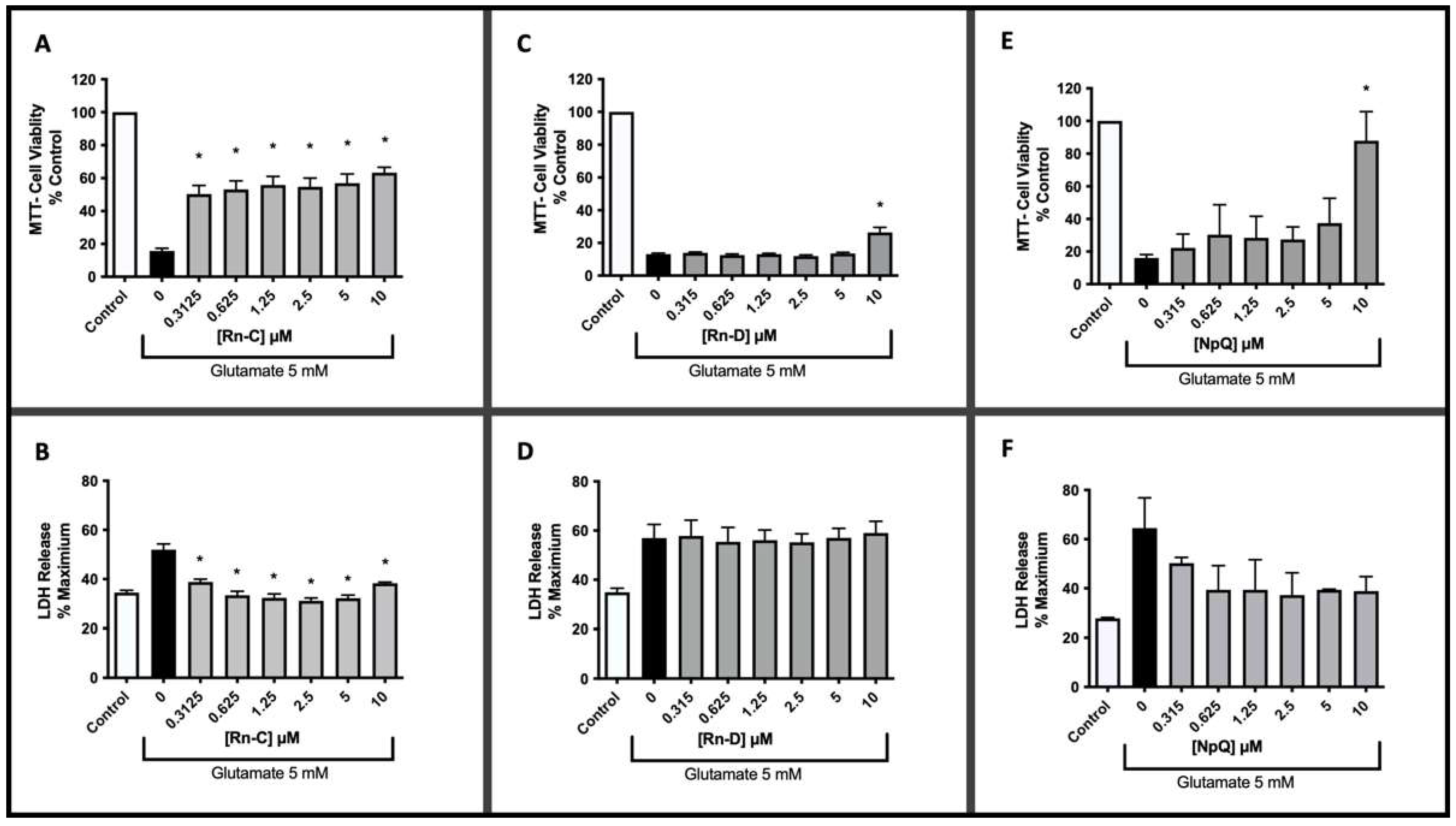
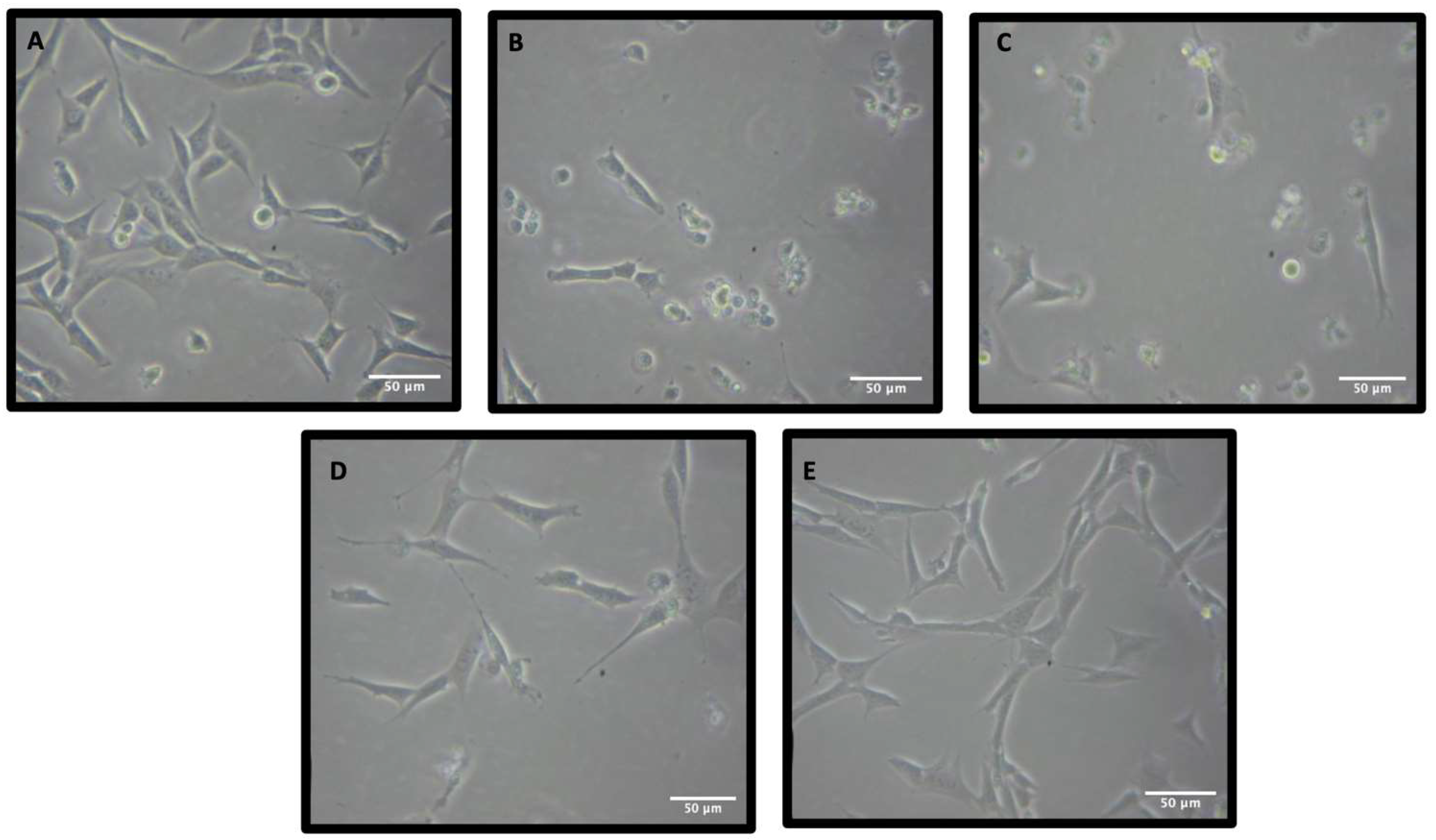
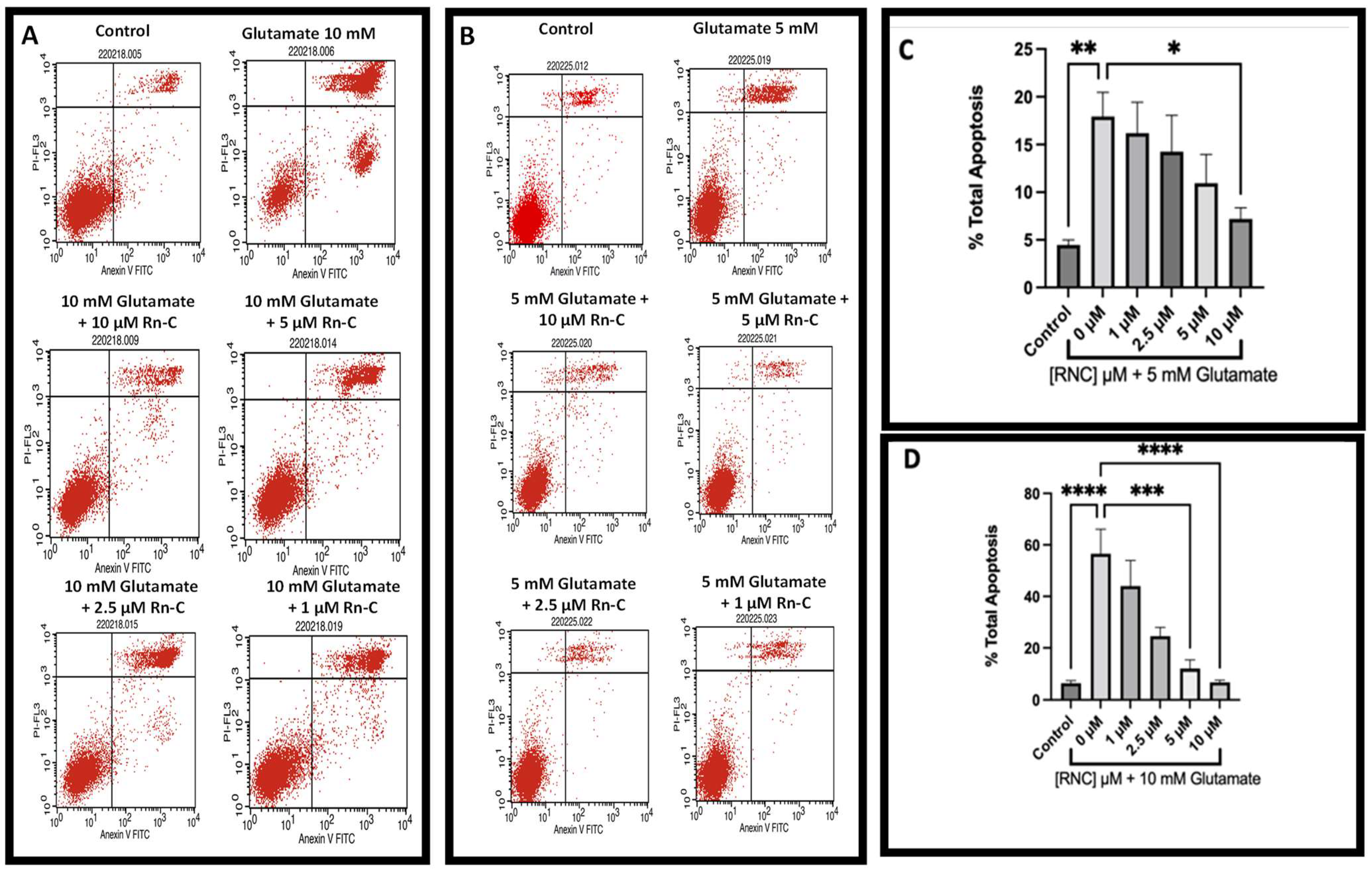
3.2. Neuroprotective Properties of Rn-C Rn-D and NpQ
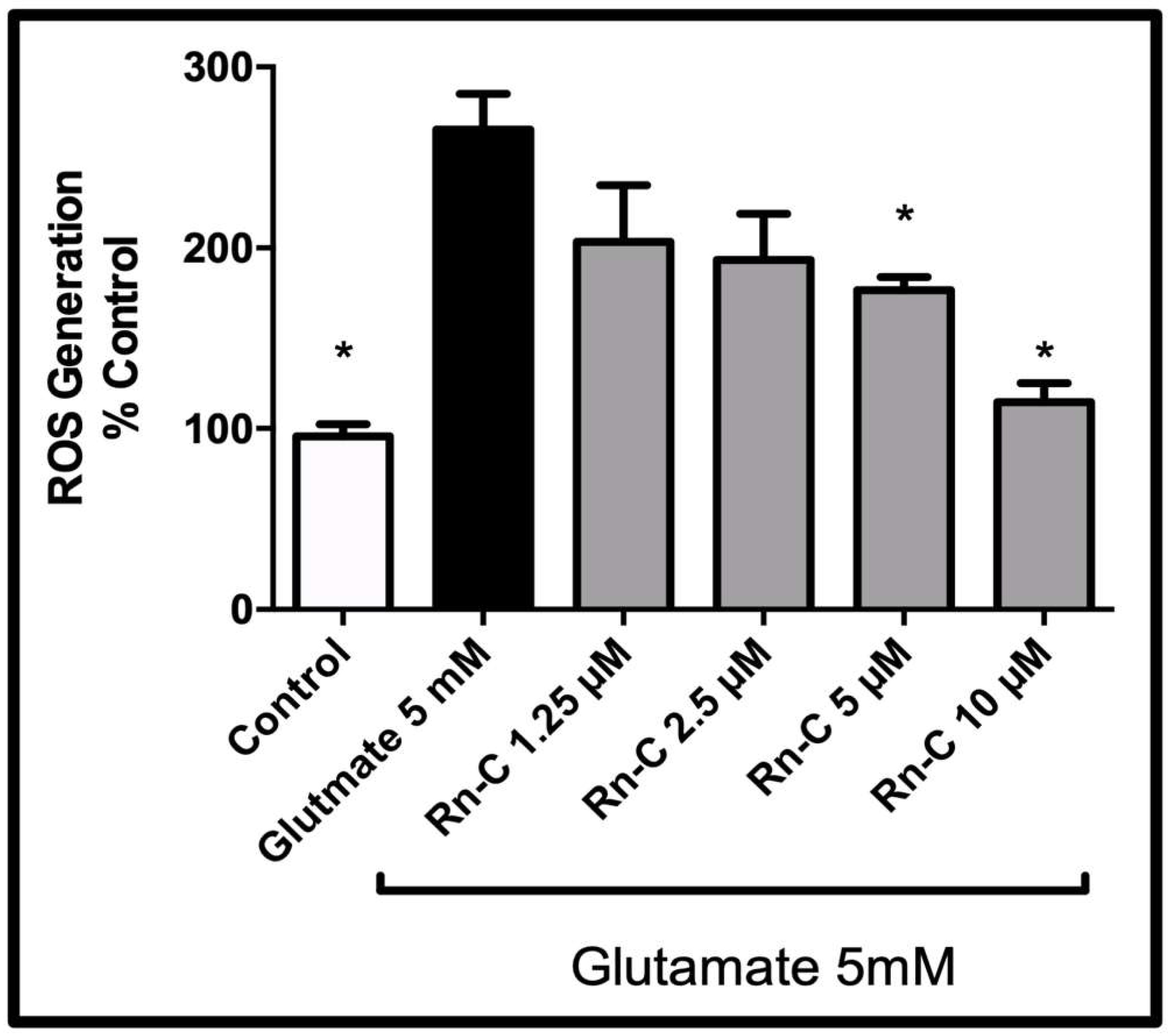
3.3. Reactive Oxygen Species Production
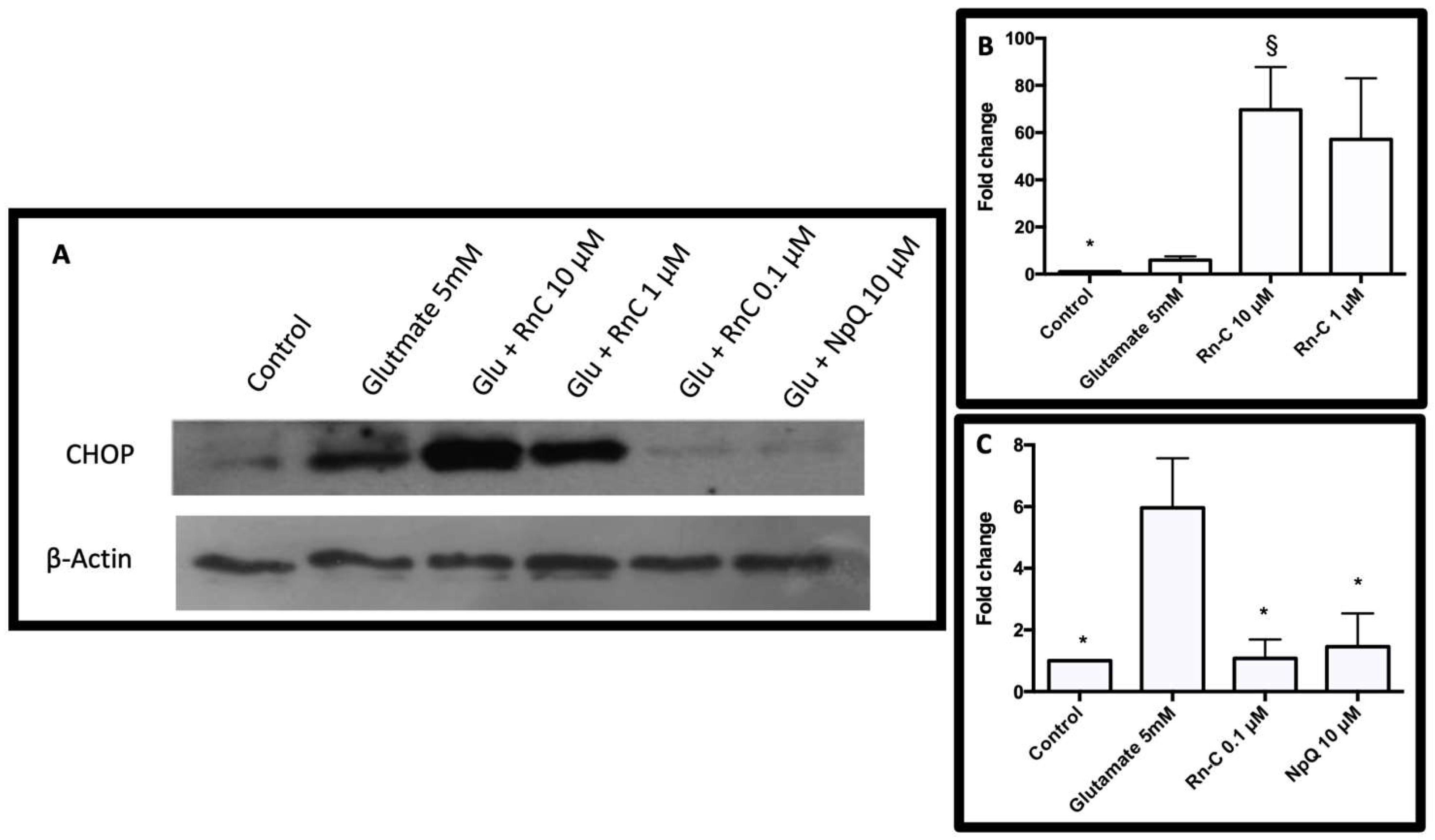
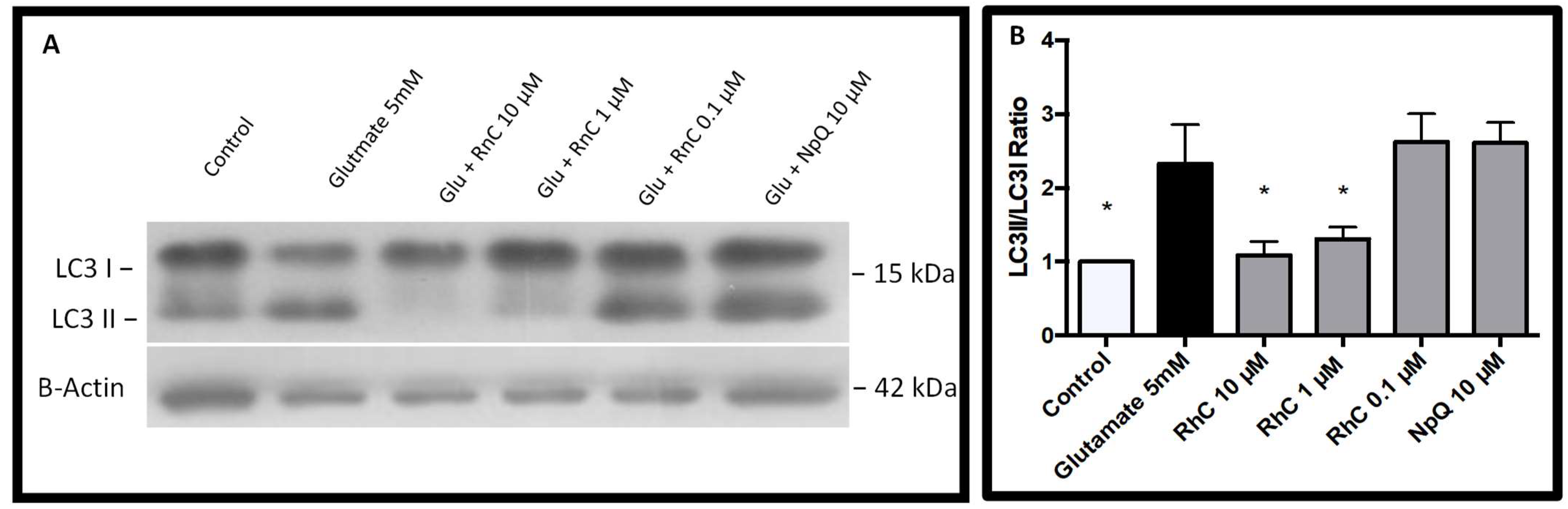
3.4. Cell Signaling in Response to Glutamate and Rn-C or NpQ
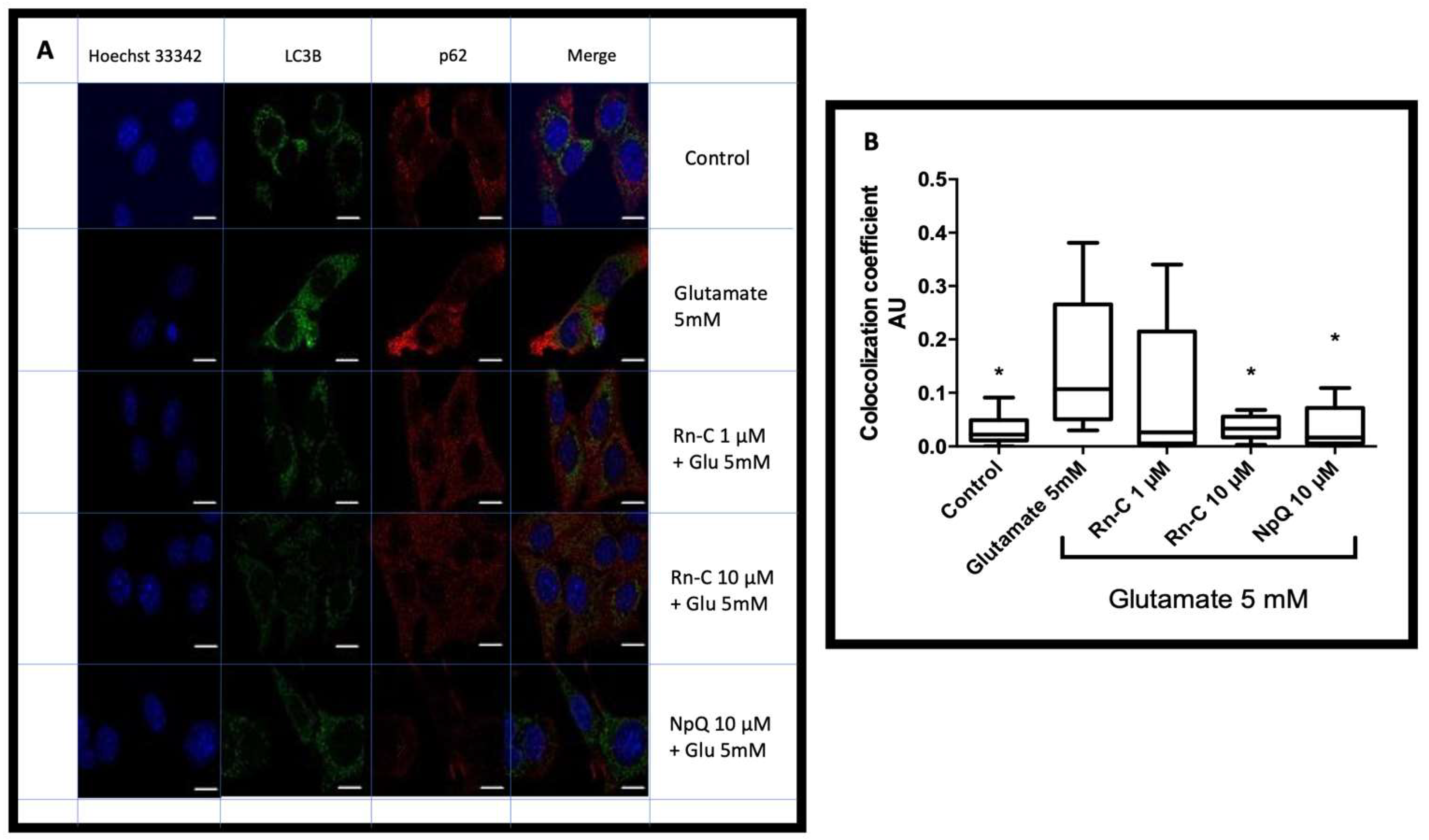
3.5. LC3 Localization with P62
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, F.; Qin, W.; Zhu, M.; Jia, J. Model-based projection of dementia prevalence in china and worldwide: 2020–2050. J. Alzheimer’s Dis. 2021, 82, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Rocca, W.A. The burden of Parkinson’s disease: A worldwide perspective. Lancet Neurol. 2018, 17, 928–929. [Google Scholar] [CrossRef]
- Dorsey, R.E.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the global burden of disease study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef]
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s disease and its treatment by different approaches: A review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Hayward, J.J.; Ley, S.V.; Tranmer, G.K. Pharmaceutical strategy and innovation: An academics perspective. Chem. Med. Chem. 2007, 2, 768–788. [Google Scholar] [CrossRef] [PubMed]
- Satyanarayanajois, S.D.; Hill, R.A. Medicinal chemistry for 2020. Future Med. Chem. 2011, 3. [Google Scholar] [CrossRef]
- Farnsworth, N.R.; Akerele, O.; Bingel, A.S.; Soejarto, D.D.; Guo, Z. Medicinal plants in therapy. Bull. World Health Organ. 1985, 63, 965–981. [Google Scholar] [CrossRef]
- Jamshidi-Kia, F.; Lorigooini, Z.; Amini-Khoei, H. Medicinal plants: Past history and future perspective. J. Herbmed. Pharmacol. 2018, 7, 1–7. [Google Scholar] [CrossRef]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Andreone, B.J.; Larhammar, M.; Lewcock, J.W. Cell death and neurodegeneration. Cold Spring Harb. Perspect. Biol. 2020, 12, a036434. [Google Scholar] [CrossRef]
- Fukui, M.; Song, J.H.; Choi, J.; Choi, H.J.; Zhu, B.T. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur. J. Pharmacol. 2009, 617, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, J.; Ryu, J.; Park, S.G.; Cho, S.; Park, B.C. Activation of autophagy during glutamate-induced HT22 cell death. Biochem. Biophys. Res. Commun. 2009, 388, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Brimson, J.M.; Prasanth, M.I.; Malar, D.S.; Brimson, S.; Tencomnao, T. Rhinacanthus nasutus “tea” infusions and the medicinal benefits of the constituent phytochemicals. Nutrients. 2020, 12, 3776. [Google Scholar] [CrossRef] [PubMed]
- Sendl, A.; Chen, J.L.; Jolad, S.D.; Stoddart, C.; Rozhon, E.; Kernan, M.; Nanakorn, W.; Balick, M. Two new naphthoquinones with antiviral activity from Rhinacanthus nasutus. J. Nat. Prod. 1996, 59, 808–811. [Google Scholar] [CrossRef] [PubMed]
- Panichayupakaranant, P.; Charoonratana, T.; Sirikatitham, A. Rp-hplc analysis of rhinacanthins in rhinacanthus nasutus: Validation and application for the preparation of rhinacanthin high-yielding extract. J. Chromatogr. Sci. 2009, 47, 705–708. [Google Scholar] [CrossRef]
- Brimson, J.M.; Brimson, S.J.; Brimson, C.A.; Rakkhitawatthana, V.; Tencomnao, T. Rhinacanthus nasutus extracts prevent glutamate and amyloid-β neurotoxicity in ht-22 mouse hippocampal cells: Possible active compounds include lupeol, stigmasterol and β-sitosterol. Int. J. Mol. Sci. 2012, 13, 5074–5097. [Google Scholar] [CrossRef]
- Brimson, J.M.; Tencomnao, T. Rhinacanthus nasutus protects cultured neuronal cells against hypoxia induced cell death. Molecules 2011, 16, 6322–6338. [Google Scholar] [CrossRef]
- Chuang, K.A.; Li, M.H.; Lin, N.H.; Chang, C.H.; Lu, I.; Pan, I.; Takahashi, T.; Perng, M.D.; Wen, S.F. Rhinacanthin c alleviates amyloid-β fibrils’ toxicity on neurons and attenuates neuroinflammation triggered by lps, amyloid-β, and interferon-γ in glial cells. Oxid Med. Cell Longev. 2017, 2017, 5414297. [Google Scholar] [CrossRef]
- Chang, C.Z.; Wu, S.C.; Kwan, A.L.; Lin, C.L. Rhinacanthin-C, a fat-soluble extract from Rhinacanthus nasutus, modulates high-mobility group box 1-related neuro-inflammation and subarachnoid hemorrhage-induced brain apoptosis in a rat model. World Neurosurg. 2016, 86, 349–360. [Google Scholar] [CrossRef]
- Brimson, J.M.; Prasanth, M.I.; Plaingam, W.; Tencomnao, T. Bacopa monnieri (L.) wettst. Extract protects against glutamate toxicity and increases the longevity of Caenorhabditis elegans. J. Tradit. Complement. Med. 2020, 10, 460–470. [Google Scholar] [CrossRef]
- Ko, J.; Jang, S.; Kwon, W.; Kim, S.Y.; Jang, S.; Kim, E.; Ji, Y.R.; Park, S.; Kim, M.O.; Choi, S.K. Protective effect of gip against monosodium glutamate-induced ferroptosis in mouse hippocampal ht-22 cells through the mapk signalling pathway. Antioxidants 2022, 11, 189. [Google Scholar] [CrossRef] [PubMed]
- Boonyaketgoson, S.; Rukachaisirikul, V.; Phongpaichit, S.; Trisuwan, K. Naphthoquinones from the leaves of Rhinacanthus nasutus having acetylcholinesterase inhibitory and cytotoxic activities. Fitoterapia 2018, 124, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Chatatikun, M.; Yamauchi, T.; Yamasaki, K.; Chiabchalard, A.; Aiba, S. Phyllanthus acidus (L.) skeels and Rhinacanthus nasutus (L.) kurz leaf extracts suppress melanogenesis in normal human epidermal melanocytes and reconstitutive skin culture. Asian Pac. J. Trop. Med. 2019, 12, 98–105. [Google Scholar] [CrossRef]
- Cheung, E.C.; Slack, R.S. Emerging role for erk as a key regulator of neuronal apoptosis. Sci. Signal. 2004, 2004, PE45. [Google Scholar] [CrossRef]
- Xiong, Q.; Liu, A.; Ren, Q.; Xue, Y.; Yu, X.; Ying, Y.; Gao, H.; Tan, H.; Zhang, Z.; Li, W. Cuprous oxide nanoparticles trigger reactive oxygen species-induced apoptosis through activation of Erk-dependent autophagy in bladder cancer. Cell Death Dis. 2020, 11, 366. [Google Scholar] [CrossRef]
- Ma, L.; Jiang, Y.; Dong, Y.; Gao, J.; Du, B.; Liu, D. Anti-tnf-alpha antibody attenuates subarachnoid hemorrhage-induced apoptosis in the hypothalamus by inhibiting the activation of Erk. Neuropsychiatr. Dis. Treat. 2018, 14, 525–536. [Google Scholar] [CrossRef]
- Xiong, Y.; Zhou, D.; Zheng, K.; Bi, W.; Dong, Y. Extracellular atp binding to p2y1 receptors prevents glutamate-induced excitotoxicity: Involvement of erk1/2 signaling pathway to suppress autophagy. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Ma, Y.; Qi, Q.; He, Q.; Gilyazova, N.S.; Ibeanu, G.; Li, P.A. Neuroprotection by b355252 against glutamate-induced cytotoxicity in murine hippocampal HT-22 cells is associated with activation of ERK-3 signaling pathway. Biol. Pharm. Bull. 2021, 44, 1662–1669. [Google Scholar] [CrossRef]
- Park, J.S.; Park, J.H.; Kim, K.Y. Neuroprotective effects of myristargenol a against glutamate-induced apoptotic HT22 cell death. RSC Adv. 2019, 9, 31247–31254. [Google Scholar] [CrossRef]
- Kang, J.H.; Kim, M.H.; Lee, H.J.; Huh, J.W.; Lee, H.S.; Lee, D.S. Peroxiredoxin 4 attenuates glutamate-induced neuronal cell death through inhibition of endoplasmic reticulum stress. Free Radic. Res. 2020, 54, 207–220. [Google Scholar] [CrossRef]
- Kang, Y.; Lee, J.H.; Seo, Y.H.; Jang, J.H.; Jeong, C.H.; Lee, S.; Jeong, G.S.; Park, B. Epicatechin prevents methamphetamine-induced neuronal cell death via inhibition of ER stress. Biomol. Ther. 2019, 27, 145. [Google Scholar] [CrossRef] [PubMed]
- B’chir, W.; Chaveroux, C.; Carraro, V.; Averous, J.; Maurin, A.C.; Jousse, C.; Muranishi, Y.; Parry, L.; Fafournoux, P.; Bruhat, A. Dual role for chop in the crosstalk between autophagy and apoptosis to determine cell fate in response to amino acid deprivation. Cell. Signal. 2014, 26, 1385–1391. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Wang, S.; Ren, B.; Wang, J.; Chen, J.; Lu, J.; Zhan, S.; Fu, Y.; Huang, L.; Tan, J. CHOP favors endoplasmic reticulum stress-induced apoptosis in hepatocellular carcinoma cells via inhibition of autophagy. PLoS ONE 2017, 12, e0183680. [Google Scholar] [CrossRef] [PubMed]
- Kasetti, R.B.; Maddineni, P.; Kiehlbauch, C.; Patil, S.; Searby, C.C.; Levine, B.; Sheffield, V.C.; Zode, G.S. Autophagy stimulation reduces ocular hypertension in a murine glaucoma model via autophagic degradation of mutant myocilin. JCI Insight 2021, 6, e143359. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Lee, S.; Park, P.H. ER stress contributes to autophagy induction by adiponectin in macrophages: Implication in cell survival and suppression of inflammatory response. Cytokine 2020, 127, 154959. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef]
- Saleem, U.; Gull, Z.; Saleem, A.; Shah, M.A.; Akhtar, M.F.; Anwar, F.; Ahmad, B.; Panichayupakaranant, P. Appraisal of anti-Parkinson activity of rhinacanthin-C in haloperidol-induced parkinsonism in mice: A mechanistic approach. J. Food Biochem. 2021, 45, e13677. [Google Scholar] [CrossRef]
- Nguyen, H.Q.; Zada, S.; Lai, T.H.; Pham, T.M.; Hwang, J.S.; Ahmed, M.; Kim, D.R. Calpain-dependent beclin1 cleavage stimulates senescence-associated cell death in HT22 hippocampal cells under the oxidative stress conditions. Neurosci. Lett. 2019, 701, 106–111. [Google Scholar] [CrossRef]
- Song, J.H.; Shin, M.S.; Hwang, G.S.; Oh, S.T.; Hwang, J.J.; Kang, K.S. Chebulinic acid attenuates glutamate-induced HT22 cell death by inhibiting oxidative stress, calcium influx and mapks phosphorylation. Bioorg. Med. Chem. Lett. 2018, 28, 249–253. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, H.N.; Kim, C.Y.; Seo, M.D.; Baek, S.H. Synergistic protection by isoquercitrin and quercetin against glutamate-induced oxidative cell death in HT22 cells via activating NRF-2 and HO-1 signaling pathway: Neuroprotective principles and mechanisms of Dendropanax morbifera leaves. Antioxidants 2021, 10, 554. [Google Scholar] [CrossRef]
- Elphick, L.M.; Hawat, M.; Toms, N.J.; Meinander, A.; Mikhailov, A.; Eriksson, J.E.; Kass, G.E. Opposing roles for caspase and calpain death proteases in L-glutamate-induced oxidative neurotoxicity. Toxicol. Appl. Pharmacol. 2008, 232, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Stanciu, M.; DeFranco, D.B. Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J. Biol. Chem. 2002, 277, 4010–4017. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Chen, T.X.; Hou, M.J.; Song, J.H.; Li, P.; Liu, C.F.; Wang, P.; Zhu, B.T. Protection against glutathione depletion-associated oxidative neuronal death by neurotransmitters norepinephrine and dopamine: Protein disulfide isomerase as a mechanistic target for neuroprotection. Acta Pharmacol. Sin. 2022. [Google Scholar] [CrossRef] [PubMed]
- Van Leyen, K.; Siddiq, A.; Ratan, R.R.; Lo, E.H. Proteasome inhibition protects HT22 neuronal cells from oxidative glutamate toxicity. J. Neurochem. 2005, 92, 824–830. [Google Scholar] [CrossRef]
- Zhang, Y.; Bhavnani, B.R. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006, 7, 49. [Google Scholar] [CrossRef]
- Yoo, J.M.; Lee, B.D.; Lee, S.J.; Ma, J.Y.; Kim, M.R. Anti-apoptotic effect of n-palmitoyl serotonin on glutamate-mediated apoptosis through secretion of bdnf and activation of trkb/creb pathway in HT-22 cells. Eur. J. Lipid. Sci. Technol. 2018, 120, 1700397. [Google Scholar] [CrossRef]
- Yang, S.J.; Han, A.R.; Choi, H.R.; Hwang, K.; Kim, E.A.; Choi, S.Y.; Cho, S.W. N-adamantyl-4-methylthiazol-2-amine suppresses glutamate-induced autophagic cell death via Pi3K/AKT/mTOR signaling pathways in cortical neurons. BMB Rep. 2020, 53, 527–532. [Google Scholar] [CrossRef]
- Kumari, S.; Mehta, S.L.; Li, P.A. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: Preventive effects of selenium. PLoS ONE 2012, 7, e39382. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of ATG4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Xu, S.; Yang, P.; Qian, K.; Li, Y.; Guo, Q.; Wang, P.; Meng, R.; Wu, J.; Cao, J.; Cheng, Y. Modulating autophagic flux via ROS-responsive targeted micelles to restore neuronal proteostasis in Alzheimer’s disease. Bioact. Mater. 2022, 11, 300–316. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, B.; Sun, J.; Song, J.; Nie, L.; Zhu, K. Fraxetin suppresses reactive oxygen species-dependent autophagy by the Pi3K/AKT pathway to inhibit isoflurane-induced neurotoxicity in hippocampal neuronal cells. J. Appl. Toxicol. 2022, 42, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Unsicker, K. Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience 2006, 138, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.L.; Park, S.Y.; Kim, Y.H.; Oh, J.I.; Lee, S.J.; Park, G. The neuroprotective effects of cordycepin inhibit glutamate-induced oxidative and ER stress-associated apoptosis in hippocampal HT22 cells. NeuroToxicology 2014, 41, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rakkhittawattana, V.; Panichayupakaranant, P.; Prasanth, M.I.; Brimson, J.M.; Tencomnao, T. Rhinacanthin-C but Not -D Extracted from Rhinacanthus nasutus (L.) Kurz Offers Neuroprotection via ERK, CHOP, and LC3B Pathways. Pharmaceuticals 2022, 15, 627. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050627
Rakkhittawattana V, Panichayupakaranant P, Prasanth MI, Brimson JM, Tencomnao T. Rhinacanthin-C but Not -D Extracted from Rhinacanthus nasutus (L.) Kurz Offers Neuroprotection via ERK, CHOP, and LC3B Pathways. Pharmaceuticals. 2022; 15(5):627. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050627
Chicago/Turabian StyleRakkhittawattana, Varaporn, Pharkphoom Panichayupakaranant, Mani I. Prasanth, James M. Brimson, and Tewin Tencomnao. 2022. "Rhinacanthin-C but Not -D Extracted from Rhinacanthus nasutus (L.) Kurz Offers Neuroprotection via ERK, CHOP, and LC3B Pathways" Pharmaceuticals 15, no. 5: 627. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050627