The Delivery Challenge in Neurodegenerative Disorders: The Nanoparticles Role in Alzheimer’s Disease Therapeutics and Diagnostics


Abstract
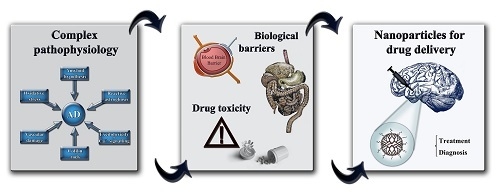
:
1. Introduction
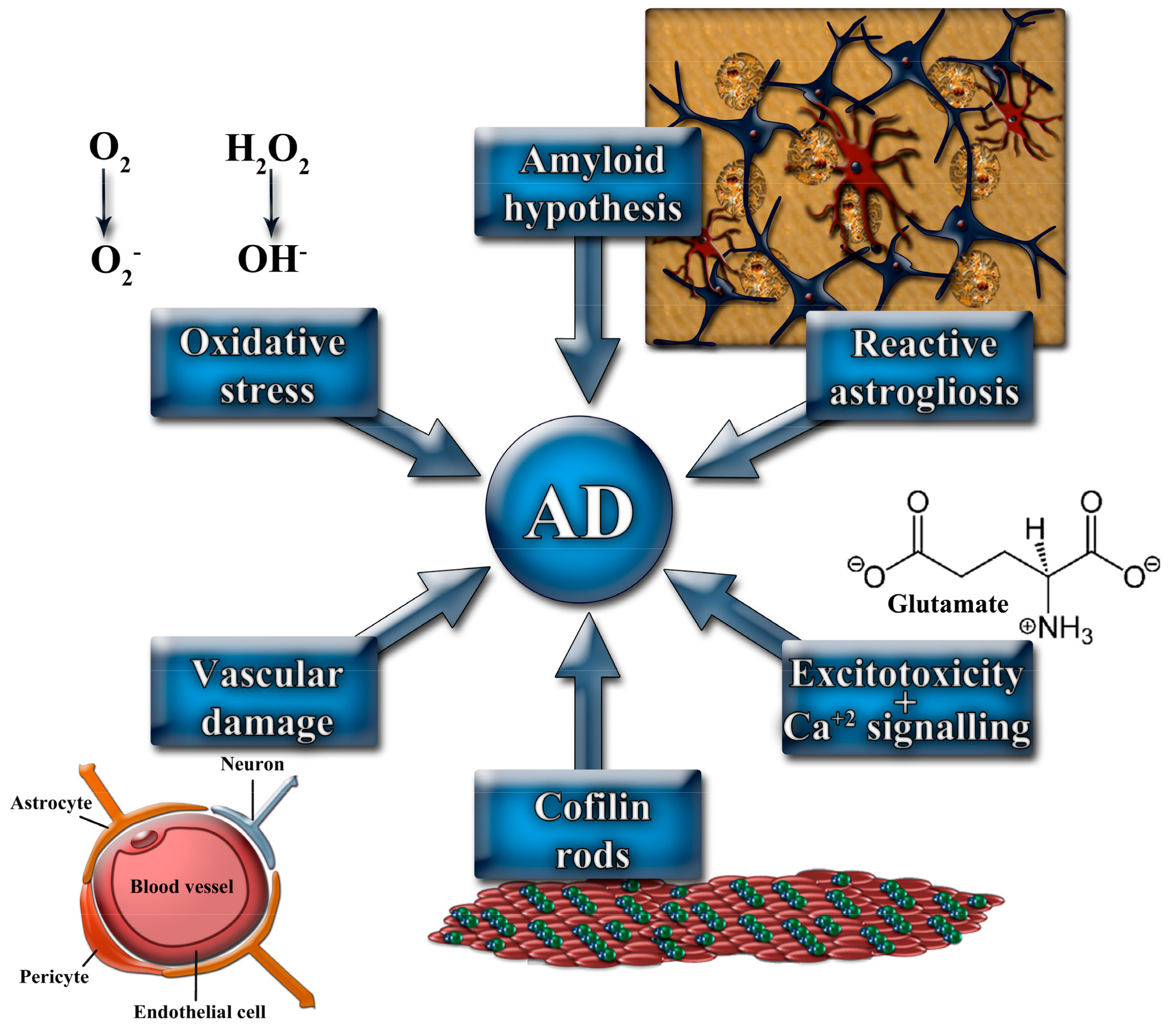
2. Main Mechanisms Involved in AD Pathophysiology
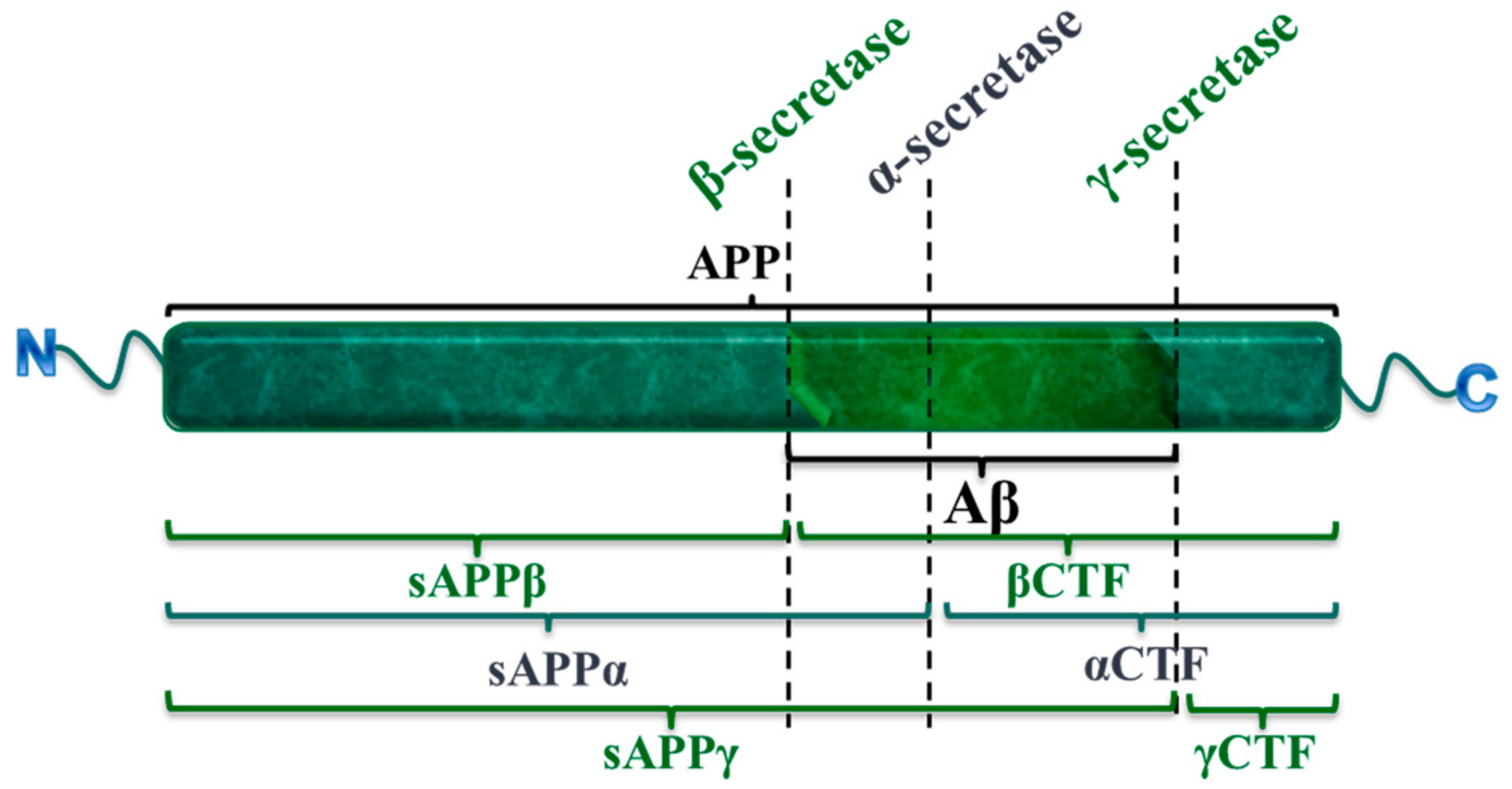
2.1. The Amyloid Cascade Hypothesis
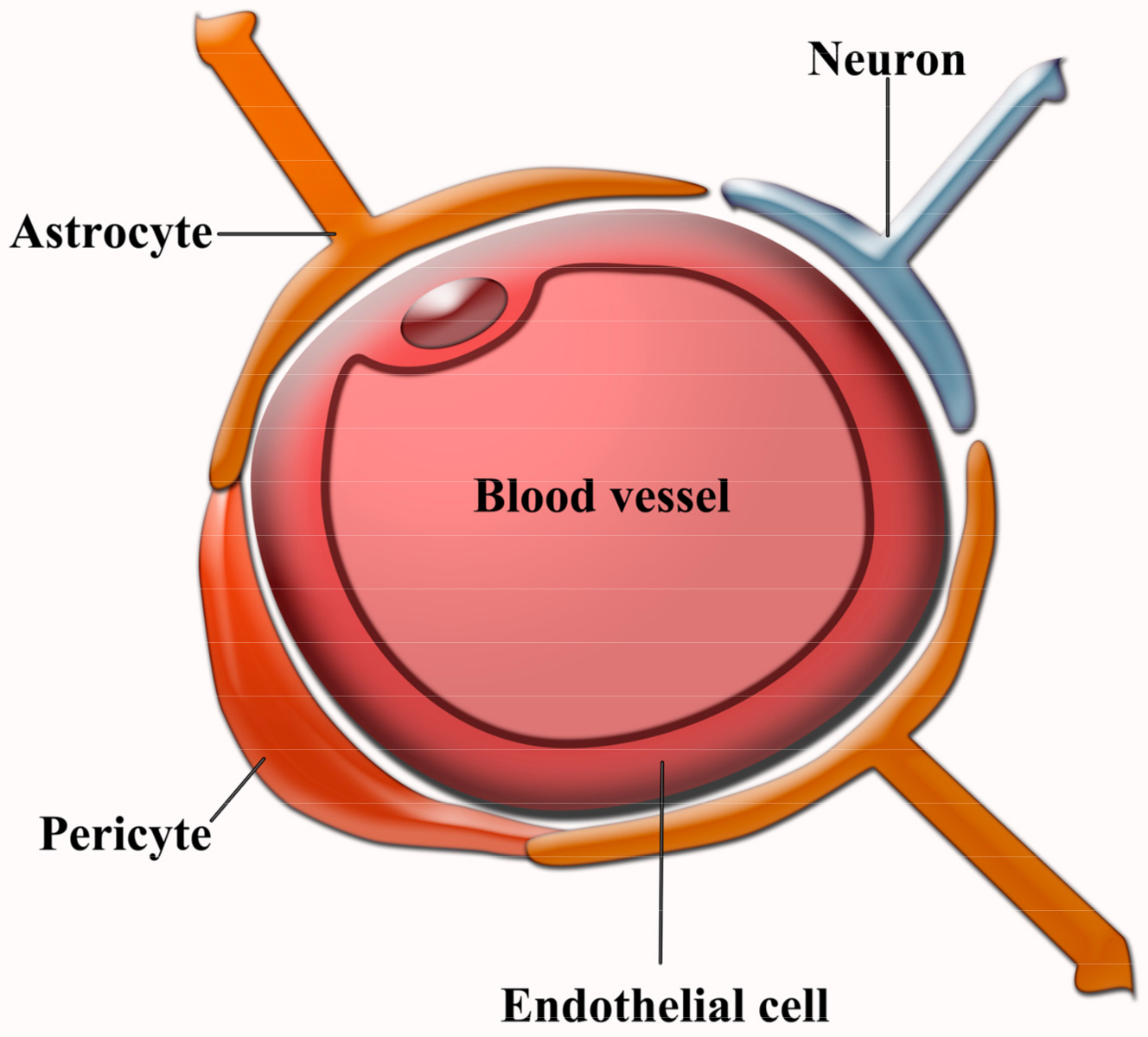
2.2. Vascular Damage
2.3. Glial Cells Involvement in AD Pathophysiology
2.4. Oxidative Stress in AD
2.5. Role of Actin Depolymerizing Factor (ADF)/Cofilin Rods in AD Pathogenesis
2.6. NMDAr Signaling and AD
3. The Delivery Challenge into the CNS
3.1. BBB Crossing in AD
3.2. NPs for the Therapeutic and Diagnostic Compounds Delivery into the CNS in AD Animal Models
3.2.1. Lipid NPs (LNPs)
3.2.2. Dendrimers (DDs)
3.2.3. Polymeric NPs (PNPs)
3.2.4. Magnetic NPs (MNPs)
3.2.5. Gold NPs (AuNPs)
3.2.6. Carbon Nanotubes (CNTs)
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferretti, M.T.; Iulita, M.F.; Cavedo, E.; Chiesa, P.A.; Dimech, A.S.; Chadha, A.S.; Baracchi, F.; Girouard, H.; Misoch, S.; Giaxobini, E.; et al. Sex differences in Alzheimer disease—the gateway to precision medicine. Nat. Rev. Neurol. 2018, 14, 457–456. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translation of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin. Anat. 1995, 8, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, S.E.; Pippin, J.J.; Barnard, N.D. Animal models of Alzheimer disease: Historical pitfalls and a path forward. ALTEX 2014, 31, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Bloom, G.S. Cytoskeletal pathologies of Alzheimer disease. Cell Motil. Cytoskeleton 2009, 66, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maloney, M.T.; Minamide, L.S.; Kinley, A.W.; Boyle, J.A.; Bamburg, J.R. Beta-secretase-cleaved amyloid precursor protein accumulates at actin inclusions induced in neurons by stress or amyloid beta: A feedforward mechanism for Alzheimer’s disease. J. Neurosci. 2005, 25, 11313–11321. [Google Scholar] [CrossRef] [PubMed]
- Dzamba, D.; Harantova, L.; Butenko, O.; Anderova, M. Glial Cells—The Key Elements of Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 894–911. [Google Scholar] [CrossRef] [PubMed]
- Cervellati, C.; Wood, P.L.; Romani, A.; Valacchi, G.; Squerzanti, M.; Sanz, J.M.; Ortolani, B.; Zuliani, G. Oxidative challenge in Alzheimer’s disease: State of knowledge and future needs. J. Investig. Med. 2016, 64, 21–32. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Morales, I.; Guzman-Martinez, L.; Cerda-Troncoso, C.; Farias, G.A.; Maccioni, R.B. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front. Cell. Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Lu, P.H.; Mintz, J. Human brain myelination and amyloid beta deposition in Alzheimer’s disease. Alzheimers Dement. 2007, 3, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.P.; Zhao, J.; Li, S. Roles of NG2 glial cells in diseases of the central nervous system. Neurosci. Bull. 2011, 27, 413–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Tang, Y.; Fan, Z.; Meng, Y.; Yang, G.; Luo, J.; Ke, Z.-J. Autophagy is involved in oligodendroglial precursor-mediated clearance of amyloid peptide. Mol. Neurodegener. 2013, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Shen, Y. Interruption of beta-catenin signaling reduces neurogenesis in Alzheimer’s disease. J. Neurosci. 2009, 29, 6545–6557. [Google Scholar] [CrossRef] [PubMed]
- Finsterwald, C.; Magistretti, P.J.; Lengacher, S. Astrocytes: New Targets for the Treatment of Neurodegenerative Diseases. Curr. Pharm. Des. 2015, 21, 3570–3581. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iram, T.; Trudler, D.; Kain, D.; Kanner, S.; Galron, R.; Vassar, R.; Barzilai, A.; Binder, P.; Fishelson, Z.; Frenkel, D. Astrocytes from old Alzheimer’s disease mice are impaired in Abeta uptake and in neuroprotection. Neurobiol. Dis. 2016, 96, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.E.; Bamburg, J.R. Peptide regulation of cofilin activity in the CNS: A novel therapeutic approach for treatment of multiple neurological disorders. Pharmacol. Ther. 2017, 175, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Henriques, A.G.; Oliveira, J.M.; Carvalho, L.P.; da Cruz e Silva, O.A. Abeta Influences Cytoskeletal Signaling Cascades with Consequences to Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 1391–1407. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Boggess, T.; Uhlar, C.; Wang, X.; Khan, H.; Cappos, G.; Joly-Amado, A.; De Narvaez, D.; Majid, S.; Minamide, L.S. RanBP9 at the intersection between cofilin and Abeta pathologies: Rescue of neurodegenerative changes by RanBP9 reduction. Cell Death Dis. 2015, 6, 1676. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Koleske, A.J. Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 2013, 14, 536–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronicke, R.; Mikhaylova, M.; Ronicke, S.; Meinhardt, J.; Schroder, U.H.; Fandrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid beta oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.; Amaral, M.H.; Lobo, J.M.; Silva, A.C. Therapeutic Strategies for Alzheimer’s and Parkinson’s Diseases by Means of Drug Delivery Systems. Curr. Med. Chem. 2016, 23, 3618–3631. [Google Scholar] [CrossRef] [PubMed]
- Wen, M.M.; El-Salamouni, N.S.; El-Refaie, W.M.; Hazzah, H.A.; Ali, M.M.; Tosi, G.; Farid, R.M.; Blanco-Prieto, M.J.; Billa, N.; Hanafy, A.S. Nanotechnology-based drug delivery systems for Alzheimer’s disease management: Technical, industrial, and clinical challenges. J. Control. Release 2017, 245, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.Z.; Lazarevic-Pasti, T.D.; Bondzic, A.M.; Vasic, V.M. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Cena, V.; Jativa, P. Nanoparticle crossing of blood-brain barrier: A road to new therapeutic approaches to central nervous system diseases. Nanomedicine 2018, 12, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Li, X.; Guan, M.; Li, X.; Hai, L.; Wu, Y. Design, synthesis and biological evaluation of multivalent glucosides with high affinity as ligands for brain targeting liposomes. Eur. J. Med. Chem. 2014, 72, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Shao, K.; Zhang, Y.; Ding, N.; Huang, S.; Wu, J.; Li, J.; Yang, C.; Leng, Q.; Ye, L.; Lou, J.; et al. Functionalized nanoscale micelles with brain targeting ability and intercellular microenvironment biosensitivity for anti-intracranial infection applications. Adv. Healthc. Mater. 2015, 4, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.S.; Jung, H.J.; Oh, J.S.; Song, D.Y. Use of PEGylated Immunoliposomes to Deliver Dopamine Across the Blood-Brain Barrier in a Rat Model of Parkinson’s Disease. CNS Neurosci. Ther. 2016, 22, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.Z.; Cheng, Y.; Cai, R.Q.; Wang, W.W.; Cui, H.; Liu, M.; Zhang, B.L.; Mei, Q.B.; Zhou, S.Y. The enhancement of siPLK1 penetration across BBB and its anti glioblastoma activity in vivo by magnet and transferrin co-modified nanoparticle. Nanomedicine 2018, 14, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Frauen, H.D.; Zhang, B.; Nordhues, B.A.; Bijan, S.; Lin, Y.C.; Zamudio, F.; Sabbagh, J.J.; Selenica, M.L.; Dickey, C.A. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Noble, G.T.; Stefanick, J.F.; Ashley, J.D.; Kiziltepe, T.; Bilgicer, B. Ligand-targeted liposome design: Challenges and fundamental considerations. Trends Biotechnol. 2014, 32, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.B.; Gamarra, L.F. Getting into the brain: Liposome-based strategies for effective drug delivery across the blood-brain barrier. Int. J. Nanomed. 2016, 11, 5381–5414. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.A.; Jung, A.R.; Lakshmana, M.K.; Bedrossian, A.; Lim, Y.; Bu, J.H.; Park, S.A.; Koo, E.H.; Mook-Jung, I.; Kang, D.E. Pivotal role of the RanBP9-cofilin pathway in Abeta-induced apoptosis and neurodegeneration. Cell Death Differ. 2012, 19, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Roh, S.E.; Woo, J.A.; Lakshmana, M.K.; Uhlar, C.; Ankala, V.; Boggess, T.; Liu, T.; Hong, Y.H.; Mook-Jung, I.; Kim, S.J.; et al. Mitochondrial dysfunction and calcium deregulation by the RanBP9-cofilin pathway. FASEB J. 2013, 27, 4776–4789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, C.M.; Magnaudeix, A.; Naves, T.; Vincent, F.; Lalloue, F.; Jauberteau, M.O. The Ins and Outs of Nanoparticle Technology in Neurodegenerative Diseases and Cancer. Curr. Drug Metab. 2015, 16, 609–632. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Solinis, M.A.; Rodriguez-Gascon, A.; Almeida, A.J.; Preat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomedicine 2016, 12, 143–161. [Google Scholar] [CrossRef] [PubMed]
- Perez-Martinez, F.C.; Guerra, J.; Posadas, I.; Cena, V. Barriers to non-viral vector-mediated gene delivery in the nervous system. Pharm. Res. 2011, 28, 1843–1858. [Google Scholar] [CrossRef] [PubMed]
- Klementieva, O.; Aso, E.; Filippini, D.; Benseny-Cases, N.; Carmona, M.; Juves, S.; Appelhans, D.; Cladera, J.; Ferrer, I. Effect of poly(propylene imine) glycodendrimers on beta-amyloid aggregation in vitro and in APP/PS1 transgenic mice, as a model of brain amyloid deposition and Alzheimer’s disease. Biomacromolecules 2013, 14, 3570–3580. [Google Scholar] [CrossRef] [PubMed]
- Sheikhpour, M.; Barani, L.; Kasaeian, A. Biomimetics in drug delivery systems: A critical review. J. Control. Release 2017, 253, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Tomalia, D.A.; Christensen, J.B.; Boas, U. Dendrimers, Dendrons, and Dendritic Polymers. Discovery, Applications and the Future; Cambridge University Press: Cambridge, UK, 2012. [Google Scholar] [CrossRef]
- Perez-Carrion, M.D.; Perez-Martinez, F.C.; Merino, S.; Sanchez-Verdu, P.; Martinez-Hernandez, J.; Lujan, R.; Ceña, V. Dendrimer-mediated siRNA delivery knocks down Beclin 1 and potentiates NMDA-mediated toxicity in rat cortical neurons. J. Neurochem. 2012, 120, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Wasiak, T.; Ionov, M.; Nieznanski, K.; Nieznanska, H.; Klementieva, O.; Granell, M.; Caldera, J.; Majoral, J.P.; Caminade, A.M.; Klajnert, B. Phosphorus dendrimers affect Alzheimer’s (Aβ1-28) peptide and MAP-Tau protein aggregation. Mol. Pharm. 2012, 9, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Fulop, L.; Mandity, I.M.; Juhasz, G.; Szegedi, V.; Hetenyi, A.; Weber, E.; Bozso, Z.; Simon, D.; Benko, M.; Kiraly, Z.; et al. A foldamer-dendrimer conjugate neutralizes synaptotoxic beta-amyloid oligomers. PLoS ONE 2012, 7, 39485. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, J. Drug delivery to the central nervous system by polymeric nanoparticles: What do we know? Adv. Drug Deliv. Rev. 2014, 71, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Dilnawaz, F.; Sahoo, S.K. Therapeutic approaches of magnetic nanoparticles for the central nervous system. Drug Discov. Today 2015, 20, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhang, M. Multifunctional Magnetic Nanoparticles for Medical Imaging Applications. J. Mater. Chem. 2009, 19, 6258–6266. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Aguirre, C.; Morales, F.; Gallardo-Toledo, E.; Guerrero, S.; Giralt, E.; Araya, E.; Kogan, M.J. Peptides and proteins used to enhance gold nanoparticle delivery to the brain: Preclinical approaches. Int. J. Nanomed. 2015, 10, 4919–4936. [Google Scholar] [CrossRef]
- Sela, H.; Cohen, H.; Elia, P.; Zach, R.; Karpas, Z.; Zeiri, Y. Spontaneous penetration of gold nanoparticles through the blood brain barrier (BBB). J. Nanobiotechnol. 2015, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Vardharajula, S.; Ali, S.Z.; Tiwari, P.M.; Eroglu, E.; Vig, K.; Dennis, V.A.; Singh, S.R. Functionalized carbon nanotubes: Biomedical applications. Int. J. Nanomed. 2012, 7, 5361–5374. [Google Scholar] [CrossRef]
- Li, W.; Zhou, Y.; Zhao, N.; Hao, B.; Wang, X.; Kong, P. Pharmacokinetic behavior and efficiency of acetylcholinesterase inhibition in rat brain after intranasal administration of galanthamine hydrobromide loaded flexible liposomes. Environ. Toxicol. Pharmacol. 2012, 34, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.Z.; Zhang, Y.Q.; Wang, Z.Z.; Wu, K.; Lou, J.N.; Qi, X.R. Enhanced brain distribution and pharmacodynamics of rivastigmine by liposomes following intranasal administration. Int. J. Pharm. 2013, 452, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Mourtas, S.; Lazar, A.N.; Markoutsa, E.; Duyckaerts, C.; Antimisiaris, S.G. Multifunctional nanoliposomes with curcumin-lipid derivative and brain targeting functionality with potential applications for Alzheimer disease. Eur. J. Med. Chem. 2014, 80, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ordonez-Gutierrez, L.; Re, F.; Bereczki, E.; Ioja, E.; Gregori, M.; Andersen, A.J.; Anton, M.; Moghimi, S.M.; Pei, J.J.; Masserini, M.; et al. Repeated intraperitoneal injections of liposomes containing phosphatidic acid and cardiolipin reduce amyloid-beta levels in APP/PS1 transgenic mice. Nanomedicine 2015, 11, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Mancini, S.; Minniti, S.; Gregori, M.; Sancini, G.; Cagnotto, A.; Couraud, P.O.; Ordoñez-Gutierrez, L.; Wandosell, F.; Salmona, M.; Re, F. The hunt for brain Abeta oligomers by peripherally circulating multi-functional nanoparticles: Potential therapeutic approach for Alzheimer disease. Nanomedicine 2016, 12, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Gregori, M.; Taylor, M.; Salvati, E.; Re, F.; Mancini, S.; Balducci, C.; Forloni, G.; Zambelli, V.; Sesana, S.; Michael, M.; et al. Retro-inverso peptide inhibitor nanoparticles as potent inhibitors of aggregation of the Alzheimer’s Abeta peptide. Nanomedicine 2017, 13, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, J.A.; Gomes, B.; Fricker, G.; Cardoso, I.; Ribeiro, C.A.; Gaiteiro, C.; Coelho, M.A.; do Carmo Pereira, M.; Rocha, S. Dual ligand immunoliposomes for drug delivery to the brain. Colloids Surf. B Biointerfaces 2015, 134, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, M.; Khan, M.; Khan, R.A.; Ahmed, B. Preparation, characterization, in vivo and biochemical evaluation of brain targeted Piperine solid lipid nanoparticles in an experimentally induced Alzheimer’s disease model. J. Drug Target. 2012, 21, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, A.; Frozza, R.L.; Meneghetti, A.; Hoppe, J.B.; Battastini, A.M.; Pohlmann, A.R.; Guterres, S.S.; Salbego, C.G. Indomethacin-loaded lipid-core nanocapsules reduce the damage triggered by Aβ1-42 in Alzheimer’s disease models. Int. J. Nanomed. 2012, 7, 4927–4942. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Asghar, S.; Gao, S.; Su, Z.; Song, J.; Huo, M.; Meng, W.; Ping, Q.; Xiao, Y. A novel LDL-mimic nanocarrier for the targeted delivery of curcumin into the brain to treat Alzheimer’s disease. Colloids Surf. B Biointerfaces 2015, 134, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Misra, S.; Chopra, K.; Sinha, V.R.; Medhi, B. Galantamine-loaded solid-lipid nanoparticles for enhanced brain delivery: Preparation, characterization, in vitro and in vivo evaluations. Drug Deliv. 2016, 23, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Vedagiri, A.; Thangarajan, S. Mitigating effect of chrysin loaded solid lipid nanoparticles against Amyloid β25-35 induced oxidative stress in rat hippocampal region: An efficient formulation approach for Alzheimer’s disease. Neuropeptides 2016, 58, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, J.A.; Andrade, S.; Duarte, A.; Neves, A.R.; Queiroz, J.F.; Nunes, C.; Sevin, E.; Fenart, L.; Gosselet, F.; Coelho, M.A.; et al. Resveratrol and Grape Extract-loaded Solid Lipid Nanoparticles for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 277. [Google Scholar] [CrossRef] [PubMed]
- Rassu, G.; Soddu, E.; Posadino, A.M.; Pintus, G.; Sarmento, B.; Giunchedi, P.; Gavini, E. Nose-to-brain delivery of BACE1 siRNA loaded in solid lipid nanoparticles for Alzheimer’s therapy. Colloids Surf. B Biointerfaces 2017, 152, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Klajnert, B.; Wasiak, T.; Ionov, M.; Fernandez-Villamarin, M.; Sousa-Herves, A.; Correa, J.; Riguera, R.; Fernandez-Megia, E. Dendrimers reduce toxicity of Aβ 1-28 peptide during aggregation and accelerate fibril formation. Nanomedicine 2012, 8, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Kumar, K.P.; Ramasamy, M.; Suresh, B. Chitosan nanoparticles as a new delivery system for the anti-Alzheimer drug tacrine. Nanomedicine 2010, 6, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Fazil, M.; Md, S.; Haque, S.; Kumar, M.; Baboota, S.; Sahni, J.K.; Ali, J. Development and evaluation of rivastigmine loaded chitosan nanoparticles for brain targeting. Eur. J. Pharm. Sci. 2012, 47, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.K.; Agarwal, S.; Seth, B.; Yadav, A.; Nair, S.; Bhatnagar, P.; Karmakar, M.; Kumari, M.; Chauhan, L.K.; Patel, D.K.; et al. Curcumin-loaded nanoparticles potently induce adult neurogenesis and reverse cognitive deficits in Alzheimer’s disease model via canonical Wnt/beta-catenin pathway. ACS Nano 2014, 8, 76–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wan, X.; Zheng, X.; Shao, X.; Liu, Q.; Zhang, Q.; Qian, Y. Dual-functional nanoparticles targeting amyloid plaques in the brains of Alzheimer’s disease mice. Biomaterials 2014, 35, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Hanafy, A.S.; Farid, R.M.; ElGamal, S.S. Complexation as an approach to entrap cationic drugs into cationic nanoparticles administered intranasally for Alzheimer’s disease management: Preparation and detection in rat brain. Drug Dev. Ind. Pharm. 2015, 41, 2055–2068. [Google Scholar] [CrossRef] [PubMed]
- Carradori, D.; Balducci, C.; Re, F.; Brambilla, D.; Le, D.B.; Flores, O.; Gaudin, A.; Mura, S.; Forloni, G.; Ordoñez-Gutierrez, L.; et al. Antibody-functionalized polymer nanoparticle leading to memory recovery in Alzheimer’s disease-like transgenic mouse model. Nanomedicine 2018, 14, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Muntimadugu, E.; Dhommati, R.; Jain, A.; Challa, V.G.; Shaheen, M.; Khan, W. Intranasal delivery of nanoparticle encapsulated tarenflurbil: A potential brain targeting strategy for Alzheimer’s disease. Eur. J. Pharm. Sci. 2016, 92, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Skaat, H.; Corem-Slakmon, E.; Grinberg, I.; Last, D.; Goez, D.; Mardor, Y.; Margel, S. Antibody-conjugated, dual-modal, near-infrared fluorescent iron oxide nanoparticles for antiamyloidgenic activity and specific detection of amyloid-beta fibrils. Int. J. Nanomed. 2013, 8, 4063–4076. [Google Scholar] [CrossRef]
- Agyare, E.K.; Jaruszewski, K.M.; Curran, G.L.; Rosenberg, J.T.; Grant, S.C.; Lowe, V.J.; Ramakrishnan, S.; Paravastu, A.K.; Poduslo, J.F.; Kandimalla, K.K. Engineering theranostic nanovehicles capable of targeting cerebrovascular amyloid deposits. J. Control. Release 2014, 185, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.K.; Chan, P.S.; Fan, S.; Kwan, S.M.; Yeung, K.L.; Wang, Y.X.; Chow, A.H.; Wu, E.X.; Baum, L. Curcumin-conjugated magnetic nanoparticles for detecting amyloid plaques in Alzheimer’s disease mice using magnetic resonance imaging (MRI). Biomaterials 2015, 44, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Viola, K.L.; Sbarboro, J.; Sureka, R.; De, M.; Bicca, M.A.; Wang, J.; Vasavada, S.; Satpathy, S.; Wu, S.; Joshi, H.; et al. Towards non-invasive diagnostic imaging of early-stage Alzheimer’s disease. Nat. Nanotechnol. 2015, 10, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Plissonneau, M.; Pansieri, J.; Heinrich-Balard, L.; Morfin, J.F.; Stransky-Heilkron, N.; Rivory, P.; Mowat, P.; Dumoulin, M.; Cohen, R.; Allemann, E.; et al. Gd-nanoparticles functionalization with specific peptides for ss-amyloid plaques targeting. J. Nanobiotechnol. 2016, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Sillerud, L.O.; Solberg, N.O.; Chamberlain, R.; Orlando, R.A.; Heidrich, J.E.; Brown, D.C.; Brady, C.I.; Vander Jagt, T.A.; Garwood, M.; Vander Jagt, D.L. SPION-enhanced magnetic resonance imaging of Alzheimer’s disease plaques in AbetaPP/PS-1 transgenic mouse brain. J. Alzheimers Dis. 2013, 34, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Dai, F.; Fan, Z.; Ma, G.; Tang, Q.; Zhang, X. Nanotheranostics: Congo Red/Rutin-MNPs with Enhanced Magnetic Resonance Imaging and H2O2-Responsive Therapy of Alzheimer’s Disease in APPswe/PS1dE9 Transgenic Mice. Adv. Mater. 2015, 27, 5499–5505. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.H.; Chang, Y.J.; Yoshiike, Y.; Chang, Y.C.; Chen, Y.R. Negatively charged gold nanoparticles inhibit Alzheimer’s amyloid-beta fibrillization, induce fibril dissociation, and mitigate neurotoxicity. Small 2012, 8, 3631–3639. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Y.; Zhang, L.; Sun, X.Y.; Li, C.L.; Qiu, Y.; Sun, H.P.; Tang, D.Q.; Liu, Y.W.; Yin, X.X. A sensitive colorimetric strategy for monitoring cerebral beta-amyloid peptides in AD based on dual-functionalized gold nanoplasmonic particles. Chem. Commun. 2015, 51, 8880–8883. [Google Scholar] [CrossRef] [PubMed]
- Adura, C.; Guerrero, S.; Salas, E.; Medel, L.; Riveros, A.; Mena, J.; Arbiol, J.; Albericio, F.; Giralt, E.; Kogan, M.J. Stable conjugates of peptides with gold nanorods for biomedical applications with reduced effects on cell viability. ACS Appl. Mater. Interfaces 2013, 5, 4076–4085. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, Y.; Yang, Y.; Sun, L.; Han, D.; Li, H.; Wang, C. Pharmacological and toxicological target organelles and safe use of single-walled carbon nanotubes as drug carriers in treating Alzheimer disease. Nanomedicine 2010, 6, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Luo, Y.; Derreumaux, P.; Wei, G. Carbon nanotube inhibits the formation of beta-sheet-rich oligomers of the Alzheimer’s amyloid-β(16-22) peptide. Biophys. J. 2011, 101, 2267–2276. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Yoo, G.; Chang, Y.W.; Kim, H.J.; Jose, J.; Kim, E.; Pyun, J.C.; Yoo, K.H. A carbon nanotube metal semiconductor field effect transistor-based biosensor for detection of amyloid-beta in human serum. Biosens. Bioelectron. 2013, 50, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Wang, L.R.; Sato, Y.; Jiang, Y.; Berg, M.; Yang, D.S.; Nixon, R.A.; Liang, X.J. Single-walled carbon nanotubes alleviate autophagic/lysosomal defects in primary glia from a mouse model of Alzheimer’s disease. Nano Lett. 2014, 14, 5110–5117. [Google Scholar] [CrossRef] [PubMed]
- Lisi, S.; Scarano, S.; Fedeli, S.; Pascale, E.; Cicchi, S.; Ravelet, C.; Peyrin, E.; Minunni, M. Toward sensitive immuno-based detection of tau protein by surface plasmon resonance coupled to carbon nanostructures as signal amplifiers. Biosens. Bioelectron. 2017, 93, 289–292. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NP | Core/Type | Surface Ligands | Cargo | Applications in Ad | Ref. |
|---|---|---|---|---|---|
 | LP (PC + CH) | PEG | Galantamine | Allowed intranasal administration of galantamine, which improved its pharmacodynamic and pharmacokinetic properties. | [54] |
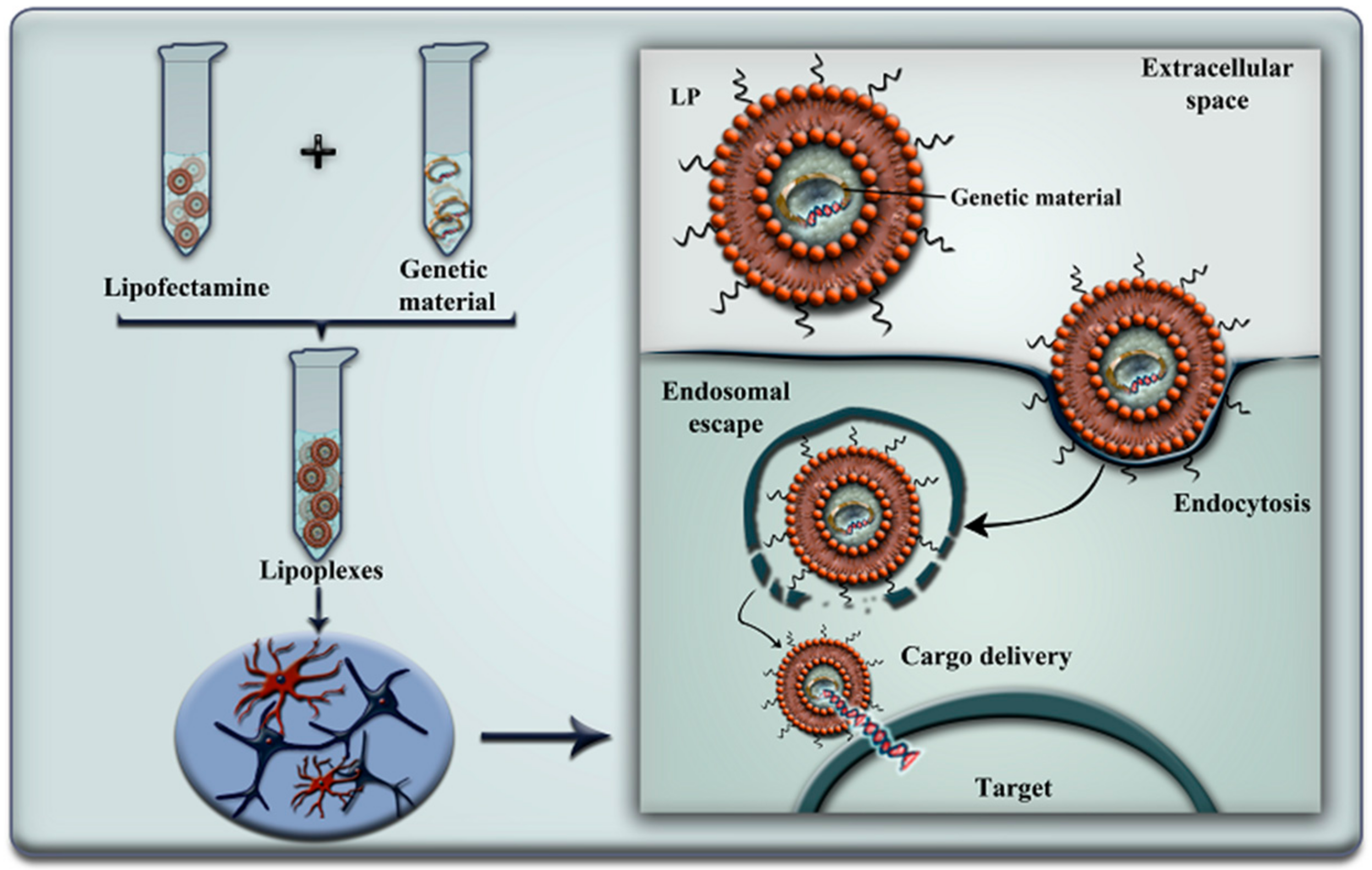
| Lipofectamine 2000® | Unknown | Cofilin siRNA | Reversed mitochondrial superoxide production and Ca2+ deregulation mediated by cofilin in response to Aβ stimulation. | [37,38] | |
| LP (PC + CH) | CPP + PEG | Rivastigmine | Improved rivastigmine distribution in hippocampus and cortex by intranasal administration compared to free drug and intravenous administration. Also diminished adverse effects. | [55] | |
| Nano-LP (DSPC + CH) | TrF-mAb + PEG | Curcumin | Retardation of Aβ aggregation. Could be used for Aβ plaques labelling due to the affinity between curcumin and Aβ peptide. | [56] | |
| LP (SPG + CH) | Phosphatidic acid/Cardiolipin | None | Reduced Aβ peptide amount in the plasma in a rodent model which may modify Aβ levels in the brain. | [57] | |
| LP (SPG + CH) | Phosphatidic acid + ApoE | None | Increased Aβ clearance from the brain. | [58] | |
| Nano-LP (SPG + CH) | RI-OR2-TAT + Maleimide-PEG | None | Inhibited the formation of Aβ oligomers and fibrils in vitro, reduced activated microglial cells, and increased the number of neurons. | [59] | |
| LP (DSPC + CH) | PEGOX26 mAb, 19B8MAb | None | LPs coupled with OX26 mAb, through the streptavidin-biotin complex, were able to reach the rat brain after tail vein injection. | [60] | |
 | Piperine SLN | Polysorbate 80 | Donepezil | Improved cognitive function and diminished Aβ plaques and tangles. | [61] |
| NLC (pεC, CTG) | Polysorbate 80 | Indomethacin | The encapsulation of indomethacin allowed a higher drug concentration in brain, which results in improved behavior in rats after Aβ injection. This seems to be due to a reduction of microglial activation. | [62] | |
| NLC(LDL-mimic) | PEG + Lactoferrin | Curcumin | Targeted brain tissue and reduced malondialdehyde levels (indicator of oxidative stress) compared to curcumin solution. | [63] | |
| SLN | Pluronic | Galantamine | Improved memory process compared to free drug. | [64] | |
| SLN | Not specified | Chrysin | Restores lipid peroxidation and acetylcholine esterase activity that were increased after Aβ administration. | [65] | |
| SLN | OX26 mAb | Resveratrol | Targeted the BBB and prevented Aβ peptide fibrillation. | [66] | |
| SLN | CPP (RVG-9R) + Chitosan | BACE1 siRNA | Diminished Aβ peptide burden by silencing of β-secretase protein. | [67] | |
 | G3/4-CPD | Not specified | None | Disrupted Aβ and MAP-TAU aggregation at high concentrations and accelerated fibrils formation at low concentrations. | [46] |
| G3-GATG | Morpholine groups | None | Accelerated Aβ aggregation preventing the toxic effects of immature amyloid fibrils, which are more harmful than mature fibrils. | [68] | |
| G0-PAMAM | Tetra-maleimidopropionyl + Helical β-peptide foldamers | None | Protective effect against Aβ-induced LTP impairment. | [47] | |
| G3/4-PPI | Maltose/maltotriose | None | Maltose DDs reduced Aβ burden in APP/PS1 mice, while cationic maltose DDs provoked memory loss in wild-type mice. | [42] | |
 | Chitosan | Polysorbate 80 | Tacrine | Provided a diffusion-controlled release of the drug. | [69] |
| Chitosan | Not specified | Rivastigmine | Improved rivastigmine bioavailability and uptake in brain through intranasal administration. | [70] | |
| PLGA | Not specified | Curcumin | Reduced learning and memory impairments Aβ-induced through activation of Wnt/β-catenin pathway, which increases neurogenesis. | [71] | |
| PLA | PEG + TGN + OSH | None | NP was capable of target Aβ peptide and had low toxicity which suggested this NP as a possible vehicle to be used in AD treatment. | [72] | |
| Chitosan | Polysorbate 80 | Galantamine | Allowed intranasal administration of galantamine improving its brain uptake. | [73] | |
| P(HDCA-co-RCA-co-MePEGCA)/P(MePEGCA-co-Bio-PEGCA-co-HDCA) | PEGRhodamine/BiotinAβmAb | None | Tg2576 mice were intravenously injected with the NPs, resulting in improved results in the Novel Object Recognition test, which were similar to wild type mice. Although a low diffusion into the brain was found. | [74] | |
| PLGA | Not specified | Tarenflurbil | Improved pharmacokinetics and oral bioavailability compared to free tarenflurbil and could allow intranasal administration. | [75] | |
 | NIF-maghemite (Fe2O3) | AβmAb clone BAM10 | None | Detection (MRI and FI ex vivo) and disruption of Aβ fibrillation. | [76] |
| Magnevist® (Gd-DTPA) | IgG-antiamyloid antibody + Chitosan + 125I | CTX | Contrast imaging of cerebrovascular amyloid (MRI, SPECT). Diminished pro-inflammatory cytokine compared with free cyclophosphamide. | [77] | |
| Magnetite (Fe3O4) | PEG/PVP + Curcumin | None | Detection of amyloid plaques by MRI. | [78] | |
| Magnetite (Fe3O4) | AβOmAb + Nitro-DOPA + PEG | None | Detection of Aβ oligomers as an early AD biomarker (MRI). | [79] | |
| AGuIX® (Gd3+) | KLVFF/LPFFD + PEG + Cyanine 5.5 | None | Selectively target Aβ1-42 fibrils and detects senile plaques (MRI). | [80] | |
| Magnetite (Fe3O4) | AβpAb/APPpAb | None | Imaging of Aβ plaques (MRI). | [81] | |
| Iron oxide (not specified) | DSPE-PEG-NHS + Congo Red | Rutin | Congo Red: detected senile plaques by specifically bind to Aβ; Rutin: Interfered with Aβ aggregation and neurotoxicity, is anti-inflammatory and antioxidant. | [82] | |
 | Au | Carboxyl-conjugated AuNPs (negative charged) | None | Disrupted Aβ fibrillation and fragmented the fibrils already formed. | [83] |
| Au | Cu2+:PEI/Hemin:PEI | None | Colorimetric detection of monomeric Aβ peptide (dual recognition: AuNP:PEI:Cu2+-Aβ-Hemin:PEI:AuNP). | [84] | |
| Au | Nanorods associated to CLPFFD or CTAB | None | Aβ detection and reduction of amyloidogenic process by NIR irradiation. | [85] | |
 | SWCNT | Not specified | Ach | Allows Ach delivery in the brain tissue. | [86] |
| SWCNT | Not specified | None | Prevents β-sheet formation by destabilization of prefibrillar β-sheet (shown by computational study). | [87] | |
| SWCNT | Cr/Au + linker + Aβ antibody | None | Construct of CNT-MESFET devices for Aβ peptide detection. | [88] | |
| SWCNT | Not specified | None | Restores normal autophagy by depressing mTOR activity and reversing lysosomal proteolytic dysfunction. | [89] | |
| MWCNT | Secondary mAbTAU | None | Use as mass enhancers in a classic sandwich assay for TAU immuno-detection. | [90] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De la Torre, C.; Ceña, V. The Delivery Challenge in Neurodegenerative Disorders: The Nanoparticles Role in Alzheimer’s Disease Therapeutics and Diagnostics. Pharmaceutics 2018, 10, 190. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10040190
De la Torre C, Ceña V. The Delivery Challenge in Neurodegenerative Disorders: The Nanoparticles Role in Alzheimer’s Disease Therapeutics and Diagnostics. Pharmaceutics. 2018; 10(4):190. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10040190
Chicago/Turabian StyleDe la Torre, Cristina, and Valentín Ceña. 2018. "The Delivery Challenge in Neurodegenerative Disorders: The Nanoparticles Role in Alzheimer’s Disease Therapeutics and Diagnostics" Pharmaceutics 10, no. 4: 190. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics10040190