A Novel Bivalent Mannosylated Targeting Ligand Displayed on Nanoparticles Selectively Targets Anti-Inflammatory M2 Macrophages

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
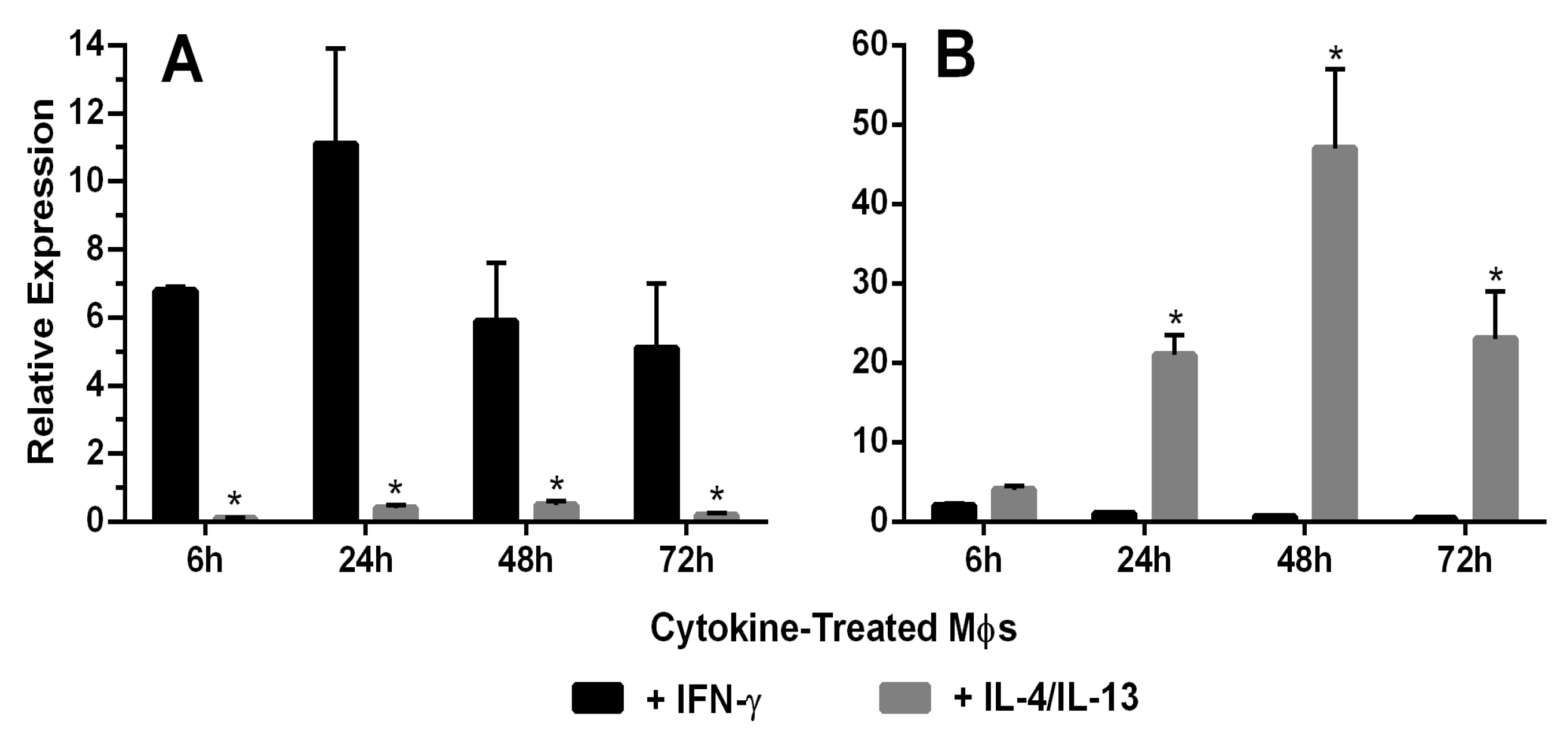
2.2. Quantitative Real-Time PCR
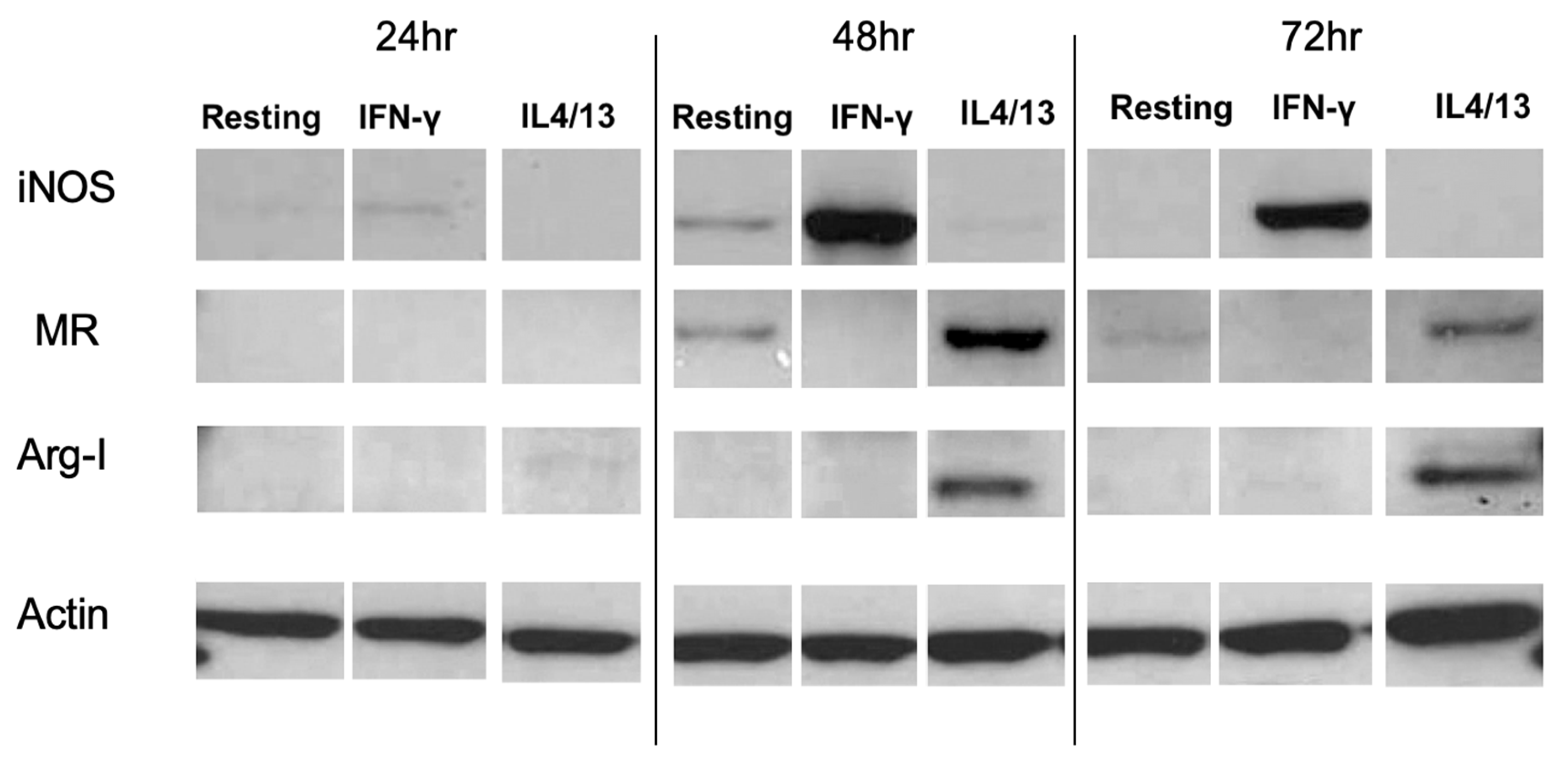
2.3. Western Blotting
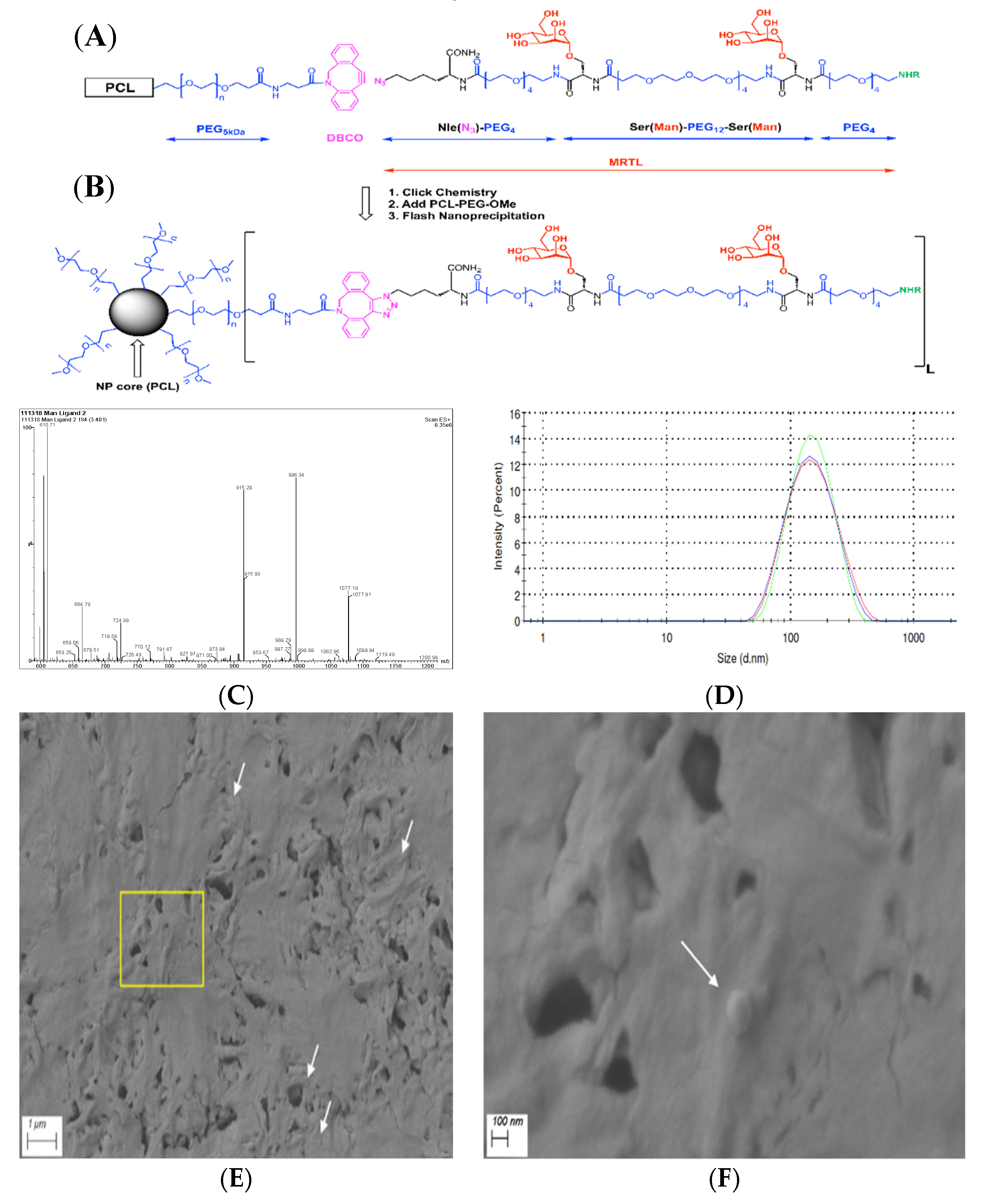
2.4. Preparation of Bivalent Mannose Receptor Targeting Ligand (MRTL)
2.5. Preparation and Characterization of Plain Nanoparticles (NPs) and NPs Displaying MRTL
2.6. Rat Peritoneal MP Isolation and Phenotypic Activation
2.7. Macrophage Uptake
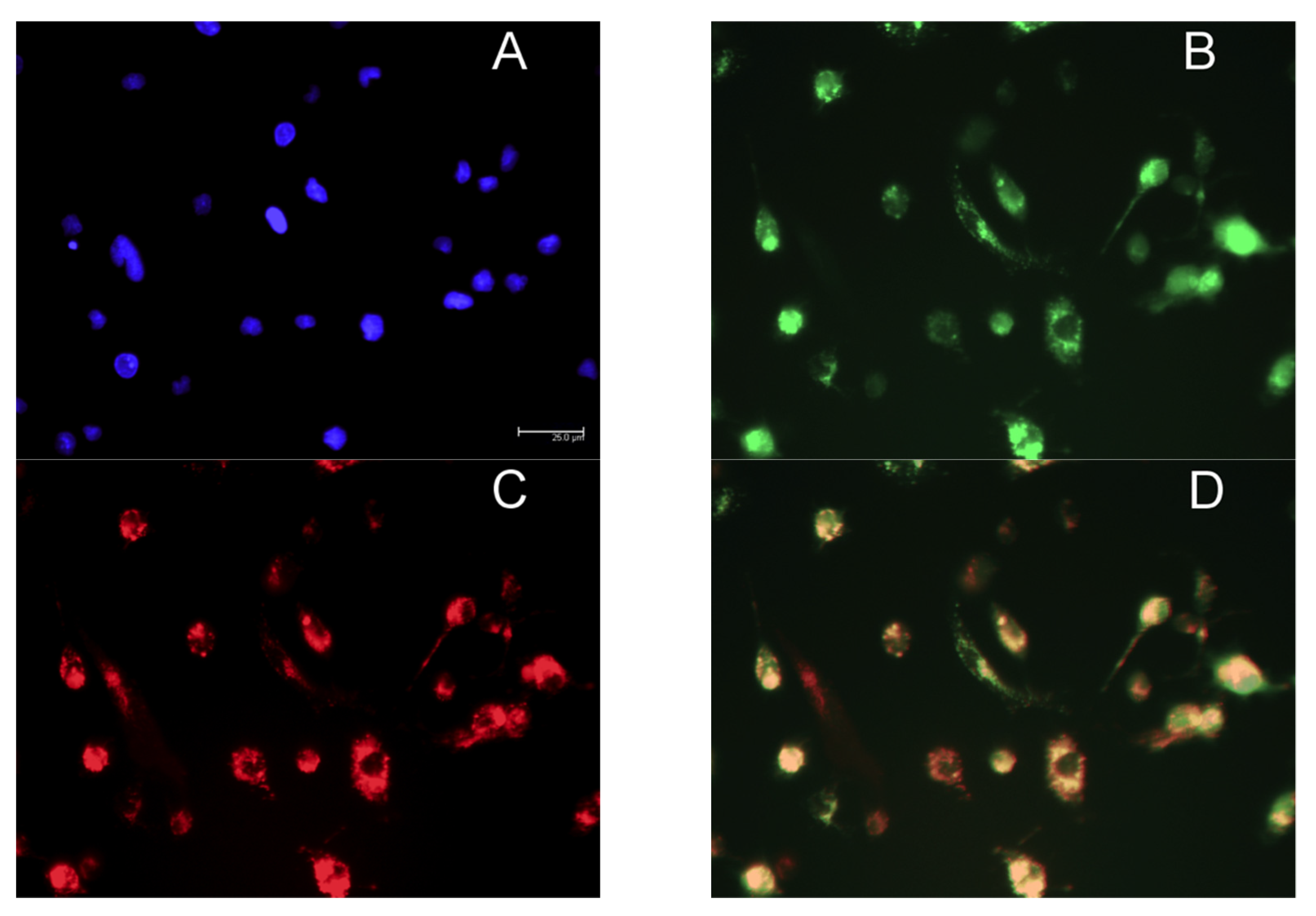
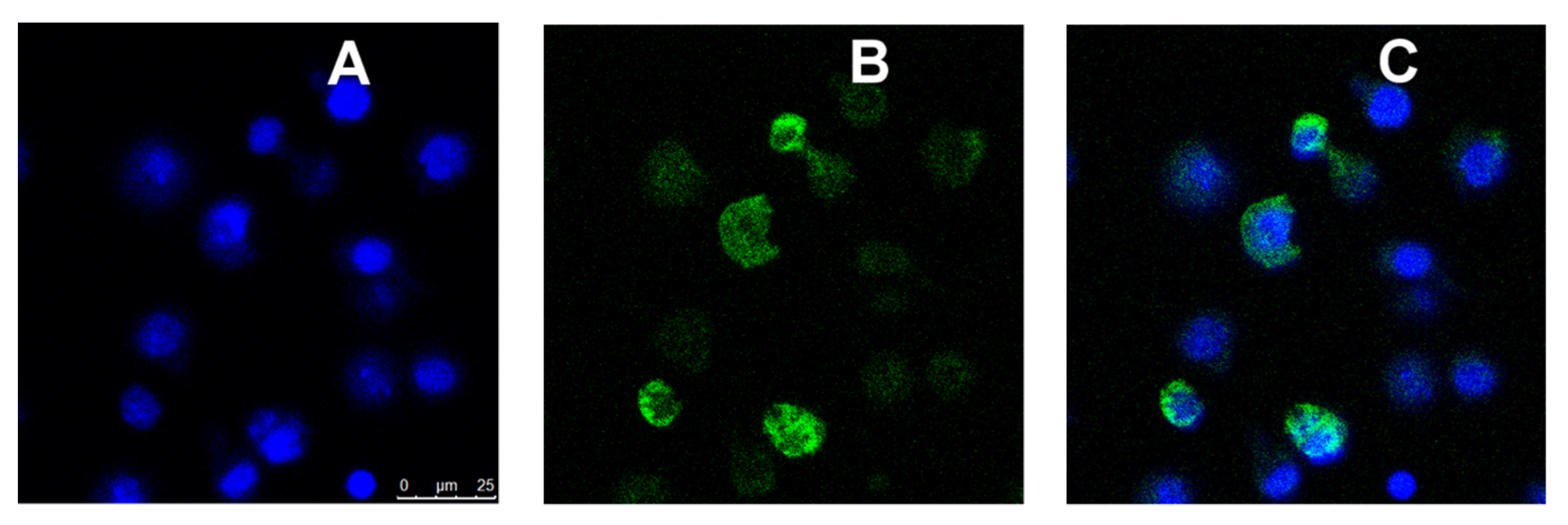
2.8. Confocal Microscopy
2.9. Flow Cytometry
3. Results
3.1. Synthesis of the Optimized Mannosylated Ligand (MRTL)
3.2. Fabrication and Characterization of MRTL-Nanoparticles
3.3. Characterization of Activated Rat Peritoneal MPs
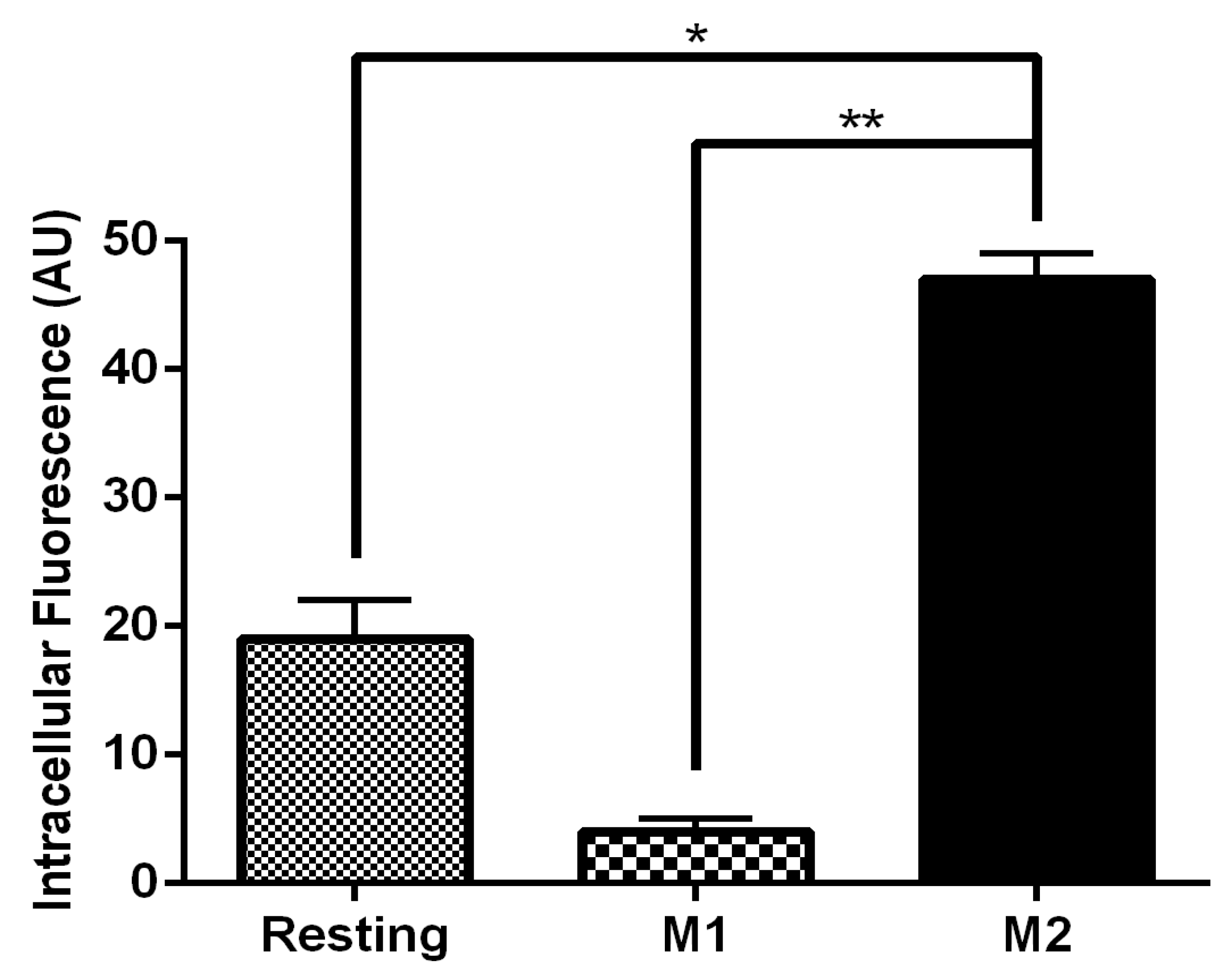
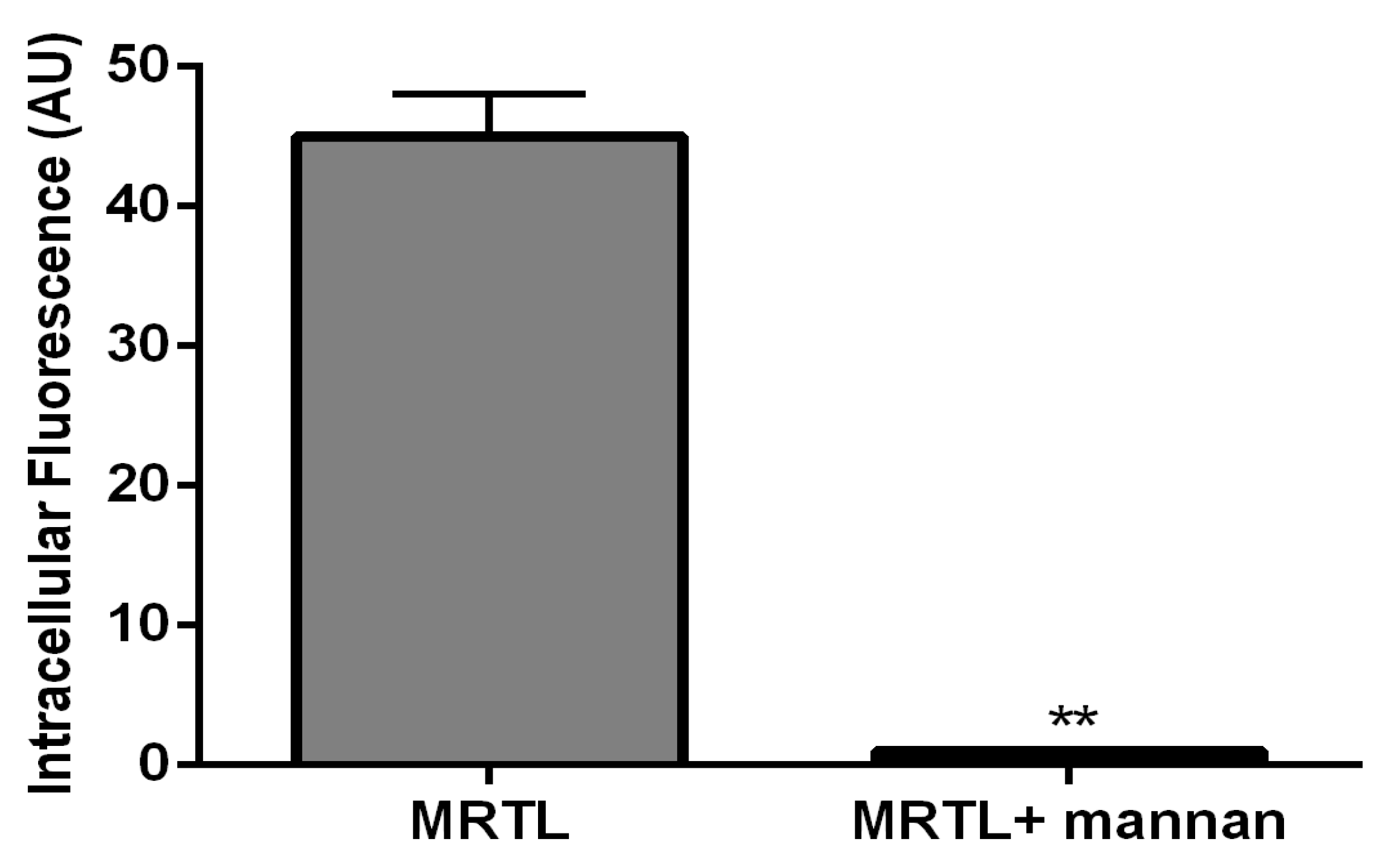
3.4. MR-Mediated MRTL Uptake into Activated MPs
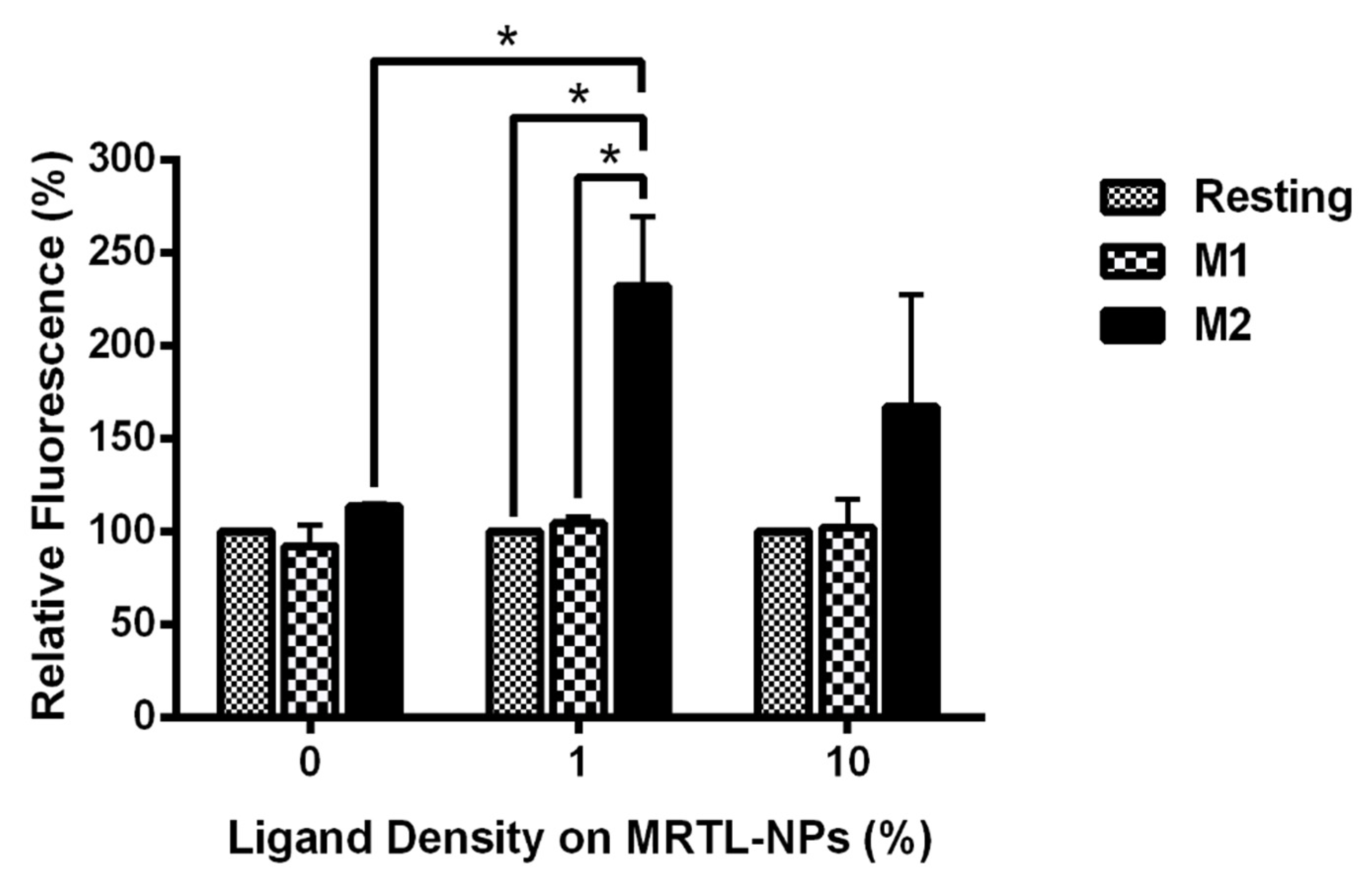
3.5. MR-Mediated Uptake of MRTL-NPs by Activated MPs
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murray, P.J. Macrophage polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Amiji, M.M. Targeted delivery systems for biological therapies of inflammatory diseases. Expert Opin. Drug Deliv. 2015, 12, 393–414. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K. Immune system and atherosclerotic disease: Heterogeneity of leukocyte subsets participating in the pathogenesis of atherosclerosis. Circ. J. 2009, 73, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- Heilbronn, L.K.; Campbell, L.V. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr. Pharm. Des. 2008, 14, 1225–1230. [Google Scholar] [CrossRef]
- Sica, A.; Larghi, P.; Mancino, A.; Rubino, L.; Porta, C.; Totaro, M.G.; Rimoldi, M.; Biswas, S.K.; Allavena, P.; Mantovani, A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008, 18, 349–355. [Google Scholar] [CrossRef]
- Herbein, G.; Varin, A. The macrophage in HIV-1 infection: From activation to deactivation? Retrovirology 2010, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Cassol, E.; Cassetta, L.; Alfano, M.; Poli, G. Macrophage polarization and HIV-1 infection. J. Leukoc. Biol. 2010, 87, 599–608. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Erratum: Exploring the full spectrum of macrophage activation (vol 8, pg 958, 2008). Nat. Rev. Immunol. 2010, 10, 460. [Google Scholar] [CrossRef] [Green Version]
- Laskin, D.L.; Sunil, V.R.; Gardner, C.R.; Laskin, J.D. Macrophages and tissue injury: Agents of defense or destruction? Annu. Rev. Pharm. Toxicol. 2011, 51, 267–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultze, J.L. Reprogramming of macrophages—New opportunities for therapeutic targeting. Curr. Opin. Pharm. 2016, 26, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage polarization in chronic inflammatory diseases: Killers or builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [PubMed]
- Porcheray, F.; Viaud, S.; Rimaniol, A.C.; Leone, C.; Samah, B.; Dereuddre-Bosquet, N.; Dormont, D.; Gras, G. Macrophage activation switching: An asset for the resolution of inflammation. Clin. Exp. Immunol. 2005, 142, 481–489. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Genard, G.; Lucas, S.; Michiels, C. Reprogramming of tumor-associated macrophages with anticancer therapies: Radiotherapy versus chemo- and immunotherapies. Front. Immunol. 2017, 8, 828. [Google Scholar] [CrossRef] [Green Version]
- Franz, S.; Allenstein, F.; Kajahn, J.; Forstreuter, I.; Hintze, V.; Moller, S.; Simon, J.C. Artificial extracellular matrices composed of collagen I and high-sulfated hyaluronan promote phenotypic and functional modulation of human pro-inflammatory M1 macrophages. Acta Biomater. 2013, 9, 5621–5629. [Google Scholar] [CrossRef]
- Hoeksema, M.A.; Stoger, J.L.; de Winther, M.P. Molecular pathways regulating macrophage polarization: Implications for atherosclerosis. Curr. Atheroscler. Rep. 2012, 14, 254–263. [Google Scholar] [CrossRef] [Green Version]
- Hristodorov, D.; Mladenov, R.; Huhn, M.; Barth, S.; Thepen, T. Macrophage-targeted therapy: CD64-based immunotoxins for treatment of chronic inflammatory diseases. Toxins 2012, 4, 676–694. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Luo, Y.C.; Ji, W.J.; Zhang, L.; Dong, Y.; Ge, L.; Lu, R.Y.; Sun, H.Y.; Guo, Z.Z.; Yang, G.H.; et al. Modulation of mononuclear phagocyte inflammatory response by liposome-encapsulated voltage gated sodium channel inhibitor ameliorates myocardial ischemia/reperfusion injury in rats. PLoS ONE 2013, 8, e74390. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization-new prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Barron, L.; Wynn, T.A. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am. J. Physiol. Gastrointest Liver Physiol. 2011, 300, G723–G728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barron, L.; Wynn, T.A. Macrophage activation governs schistosomiasis-induced inflammation and fibrosis. Eur. J. Immunol. 2011, 41, 2509–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achyut, B.R.; Arbab, A.S. Myeloid cell signatures in tumor microenvironment predicts therapeutic response in cancer. Onco Targets 2016, 9, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; He, F.; Feng, F.; Liu, X.W.; Dong, G.Y.; Qin, H.Y.; Hu, X.B.; Zheng, M.H.; Liang, L.; Feng, L.; et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010, 70, 4840–4849. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.P.; Su, Y.C.; Lee, P.H.; Lei, H.Y. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy 2013, 9, 619–621. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Tian, W.; Cai, X.; Wang, X.; Dang, W.; Tang, H.; Cao, H.; Wang, L.; Chen, T. Hydrazinocurcumin Encapsuled nanoparticles “re-educate” tumor-associated macrophages and exhibit anti-tumor effects on breast cancer following STAT3 suppression. PLoS ONE 2013, 8, e65896. [Google Scholar] [CrossRef]
- Yaddanapudi, K.; Putty, K.; Rendon, B.E.; Lamont, G.J.; Faughn, J.D.; Satoskar, A.; Lasnik, A.; Eaton, J.W.; Mitchell, R.A. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. J. Immunol. 2013, 190, 2984–2993. [Google Scholar] [CrossRef]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [Green Version]
- Baer, C.; Squadrito, M.L.; Laoui, D.; Thompson, D.; Hansen, S.K.; Kiialainen, A.; Hoves, S.; Ries, C.H.; Ooi, C.H.; De Palma, M. Suppression of microRNA activity amplifies IFN-gamma-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 2016, 18, 790–802. [Google Scholar] [CrossRef]
- Olsson, A.; Nakhle, J.; Sundstedt, A.; Plas, P.; Bauchet, A.L.; Pierron, V.; Bruetschy, L.; Deronic, A.; Torngren, M.; Liberg, D.; et al. Tasquinimod triggers an early change in the polarization of tumor associated macrophages in the tumor microenvironment. J. Immunother Cancer 2015, 3, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfano, M.; Graziano, F.; Genovese, L.; Poli, G. Macrophage polarization at the crossroad between HIV-1 infection and cancer development. Arter. Thromb. Vasc. Biol. 2013, 33, 1145–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Largent, B.L.; Walton, K.M.; Hoppe, C.A.; Lee, Y.C.; Schnaar, R.L. Carbohydrate-specific adhesion of alveolar macrophages to mannose-derivatized surfaces. J. Biol. Chem. 1984, 259, 1764–1769. [Google Scholar] [PubMed]
- Taylor, M.E.; Bezouska, K.; Drickamer, K. Contribution to ligand binding by multiple carbohydrate-recognition domains in the macrophage mannose receptor. J. Biol. Chem. 1992, 267, 1719–1726. [Google Scholar] [PubMed]
- Taylor, M.E.; Drickamer, K. Structural requirements for high affinity binding of complex ligands by the macrophage mannose receptor. J. Biol. Chem. 1993, 268, 399–404. [Google Scholar]
- Trujillo, J.R.; Rogers, R.; Molina, R.M.; Dangond, F.; McLane, M.F.; Essex, M.; Brain, J.D. Noninfectious entry of HIV-1 into peripheral and brain macrophages mediated by the mannose receptor. Proc. Natl. Acad. Sci. USA 2007, 104, 5097–5102. [Google Scholar] [CrossRef] [Green Version]
- Irache, J.M.; Salman, H.H.; Gamazo, C.; Espuelas, S. Mannose-targeted systems for the delivery of therapeutics. Expert Opin. Drug Deliv. 2008, 5, 703–724. [Google Scholar] [CrossRef]
- Tiwari, S. Mannosylated Constructs as a Platform for cell-specific delivery of bioactive agents. Crit Rev. Drug Carr. Syst. 2018, 35, 157–194. [Google Scholar] [CrossRef]
- Chen, P.; Zhang, X.; Jia, L.; Prud’homme, R.K.; Szekely, Z.; Sinko, P.J. Optimal structural design of mannosylated nanocarriers for macrophage targeting. J. Control. Release 2014, 194, 341–349. [Google Scholar] [CrossRef] [Green Version]
- Kragol, G.; Otvos, L. Orthogonal solid-phase synthesis of tetramannosylated peptide constructs carrying three independent branched epitopes. Tetrahedron 2001, 57, 957–966. [Google Scholar] [CrossRef]
- Heon Lee, I.; Palombo, M.S.; Zhang, X.; Szekely, Z.; Sinko, P.J. Design and evaluation of a CXCR4 targeting peptide 4DV3 as an HIV entry inhibitor and a ligand for targeted drug delivery. Eur. J. Pharm. Biopharm. 2018, 138, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.K.; Prud’homme, R.K. Flash nanoprecipitation of organic actives and block copolymers using a confined impinging jets mixer. Aust. J. Chem. 2003, 56, 1021–1024. [Google Scholar] [CrossRef]
- Yeeprae, W.; Kawakami, S.; Yamashita, F.; Hashida, M. Effect of mannose density on mannose receptor-mediated cellular uptake of mannosylated O/W emulsions by macrophages. J. Control. Release 2006, 114, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Asthana, S.; Gupta, P.K.; Jaiswal, A.K.; Dube, A.; Chourasia, M.K. Overexpressed macrophage mannose receptor targeted nanocapsules- mediated cargo delivery approach for eradication of resident parasite: In vitro and in vivo studies. Pharm. Res. 2015, 32, 2663–2677. [Google Scholar] [CrossRef] [PubMed]
- Garcon, N.; Gregoriadis, G.; Taylor, M.; Summerfield, J. Mannose-mediated targeted immunoadjuvant action of liposomes. Immunology 1988, 64, 743–745. [Google Scholar] [PubMed]
- Jain, S.K.; Gupta, Y.; Jain, A.; Saxena, A.R.; Khare, P.; Jain, A. Mannosylated gelatin nanoparticles bearing an anti-HIV drug didanosine for site-specific delivery. Nanomedicine 2008, 4, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Dutta, T.; Agashe, H.B.; Garg, M.; Balakrishnan, P.; Kabra, M.; Jain, N.K. Poly (propyleneimine) dendrimer based nanocontainers for targeting of efavirenz to human monocytes/macrophages in vitro. J. Drug Target 2007, 15, 89–98. [Google Scholar] [CrossRef]
- Scotton, C.; Locati, M.; Mantovani, A.; Sozzani, S. Arginase-1 and Ym1 are markers for murine, but not human, alternatively activated myeloid cells. J. Immunol. 2005, 174, 6561–6562. [Google Scholar]
- Tomioka, H.; Tatano, Y.; Maw, W.W.; Sano, C.; Kanehiro, Y.; Shimizu, T. Characteristics of suppressor macrophages induced by mycobacterial and protozoal infections in relation to alternatively activated M2 macrophages. Clin. Dev. Immunol. 2012, 2012, 635451. [Google Scholar] [CrossRef]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef]
- Novak, R.; Dabelic, S.; Dumic, J. Galectin-1 and galectin-3 expression profiles in classically and alternatively activated human macrophages. Biochim. et Biophys. Acta 2012, 1820, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Song, E.H.; Manganiello, M.J.; Chow, Y.H.; Ghosn, B.; Convertine, A.J.; Stayton, P.S.; Schnapp, L.M.; Ratner, D.M. In vivo targeting of alveolar macrophages via RAFT-based glycopolymers. Biomaterials 2012, 33, 6889–6897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Addio, S.M.; Baldassano, S.; Shi, L.; Cheung, L.L.; Adamson, D.H.; Bruzek, M.; Anthony, J.E.; Laskin, D.L.; Sinko, P.J.; Prud’homme, R.K. Optimization of cell receptor-specific targeting through multivalent surface decoration of polymeric nanocarriers. J. Control. Release 2013, 168, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, P.; Zhang, X.; Venosa, A.; Lee, I.H.; Myers, D.; Holloway, J.A.; Prud’homme, R.K.; Gao, D.; Szekely, Z.; Laskin, J.D.; et al. A Novel Bivalent Mannosylated Targeting Ligand Displayed on Nanoparticles Selectively Targets Anti-Inflammatory M2 Macrophages. Pharmaceutics 2020, 12, 243. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12030243
Chen P, Zhang X, Venosa A, Lee IH, Myers D, Holloway JA, Prud’homme RK, Gao D, Szekely Z, Laskin JD, et al. A Novel Bivalent Mannosylated Targeting Ligand Displayed on Nanoparticles Selectively Targets Anti-Inflammatory M2 Macrophages. Pharmaceutics. 2020; 12(3):243. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12030243
Chicago/Turabian StyleChen, Peiming, Xiaoping Zhang, Alessandro Venosa, In Heon Lee, Daniel Myers, Jennifer A. Holloway, Robert K. Prud’homme, Dayuan Gao, Zoltan Szekely, Jeffery D. Laskin, and et al. 2020. "A Novel Bivalent Mannosylated Targeting Ligand Displayed on Nanoparticles Selectively Targets Anti-Inflammatory M2 Macrophages" Pharmaceutics 12, no. 3: 243. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12030243