Nicardipine Loaded Solid Phospholipid Extrudates for the Prevention of Cerebral Vasospasms: In Vitro Characterization



Abstract
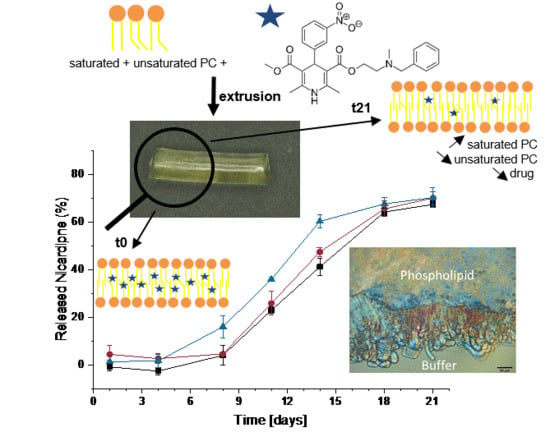
:
1. Introduction
2. Materials and Methods
2.1. Phospholipids
2.2. Drug and Further Materials
2.3. Manufacturing of Mixtures
2.4. Manufacturing of Implants by Extrusion
2.5. Microscopy
2.6. Differential Scanning Calorimetry
2.7. Texture Analysis/Mechanical Stiffness
2.8. Rheological Measurements
2.9. Quantification of Phosphatidylcholine Types and Nicardipine by ELSD-HPLC
2.10. In Vitro Erosion and Release
3. Results
3.1. Thermal Characterisation of Lipid and Drug-Lipid-Mixtures
3.2. Determination of Sample Composition by HPLC-ELSD
3.3. Mechanical Characterization of the Samples and Erosion Studies
3.4. Drug Release Studies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mäder, K.; Lehner, E.; Liebau, A.; Plontke, S.K. Controlled drug release to the inner ear: Concepts, materials, mechanisms, and performance. Hear. Res. 2018, 368, 49–66. [Google Scholar] [CrossRef]
- Omeis, I.; Neil, J.A.; Jayson, N.A.; Murali, R.; Abrahams, J.M. Treatment of cerebral vasospasm with biocompatible controlled-release systems for intracranial drug delivery. Neurosurgery 2008, 63, 1011–1019. [Google Scholar] [CrossRef]
- Mäder, K.; Gallez, B.; Liu, K.J.; Swartz, H.M. Non-invasive in vivo characterization of release processes in biodegradable polymers by low-frequency electron paramagnetic resonance spectroscopy. Biomaterials 1996, 17, 457–461. [Google Scholar] [CrossRef]
- Schädlich, A.; Kempe, S.; Mäder, K. Non-invasive in vivo characterization of microclimate pH inside in situ forming PLGA implants using multispectral fluorescence imaging. J. Control. Release 2014, 179, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems—A review. Int. J. Pharm. 2011, 415, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Zlomke, C.; Barth, M.; Mäder, K. Polymer degradation induced drug precipitation in PLGA implants—Why less is sometimes more. Eur. J. Pharm. Biopharm. 2019, 139, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Lucke, A.; Kiermaier, J.; Göpferich, A. Peptide Acylation by Poly(α-Hydroxy Esters). Pharm. Res. 2002, 19, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Wersig, T.; Hacker, M.C.; Kressler, J.; Mäder, K. Poly(glycerol adipate)—Indomethacin drug conjugates-synthesis and in vitro characterization. Int. J. Pharm. 2017, 531, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Steinman, N.Y.; Domb, A.J. Injectable pasty biodegradable polymers derived from castor oil and hydroxy-acid lactones. J. Pharmacol. Exp. Ther. 2019, 370, 736–741. [Google Scholar] [CrossRef]
- Vollrath, M.; Engert, J.; Winter, G. Long-term release and stability of pharmaceutical proteins delivered from solid lipid implants. Eur. J. Pharm. Biopharm. 2017, 117, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; McGoverin, C.M.; Gordon, K.C.; Winter, G.; Rades, T.; Myschik, J.; Strachan, C.J. Studies on the lipase-induced degradation of lipid-based drug delivery systems. Part II—Investigations on the mechanisms leading to collapse of the lipid structure. Eur. J. Pharm. Biopharm. 2013, 84, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Van Hoogevest, P.; Wendel, A. The use of natural and synthetic phospholipids as pharmaceutical excipients. Eur. J. Lipid Sci. Technol. 2014, 116, 1088–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angst, M.S.; Drover, D.R. Pharmacology of drugs formulated with DepoFoam™: A sustained release drug delivery system for parenteral administration using multivesicular liposome technology. Clin. Pharmacokinet. 2006, 45, 1153–1176. [Google Scholar] [CrossRef] [PubMed]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Breitsamer, M.; Winter, G. Vesicular phospholipid gels as drug delivery systems for small molecular weight drugs, peptides and proteins: State of the art review. Int. J. Pharm. 2019, 557, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rahnfeld, L.; Thamm, J.; Steiniger, F.; van Hoogevest, P.; Luciani, P. Study on the in situ aggregation of liposomes with negatively charged phospholipids for use as injectable depot formulation. Colloids Surf. B Biointerfaces 2018, 168, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Kolbina, M.; Schulte, A.; van Hoogevest, P.; Körber, M.; Bodmeier, R. Evaluation of Hydrogenated Soybean Phosphatidylcholine Matrices Prepared by Hot Melt Extrusion for Oral Controlled Delivery of Water-Soluble Drugs. AAPS PharmSciTech 2019, 20, 159. [Google Scholar] [CrossRef] [PubMed]
- Kolbina, M.; Bodmeier, R.; Körber, M. Saturated phosphatidylcholine as matrix former for oral extended release dosage forms. Eur. J. Pharm. Sci. 2017, 108, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Zlomke, C.; Metz, H.; Mäder, K. Spectral-Spatial EPR Imaging of Phospholipid-mixtures. In Proceedings of the 4th Symposium on Phospholipids in Pharmaceutical Research, Heidelberg, Germany, 21 September 2015. [Google Scholar]
- Dorsch, N.W.C. Therapeutic approaches to vasospasm in subarachnoid hemorrhage. Curr. Opin. Crit. Care 2002, 8, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Kassell, N.F.; Sasaki, T.; Colohan, A.; Nazar, G. Cerebral vasospasm following aneurysmal subarachnoid hemorrhage. Stroke 1985, 16, 562–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouwers, J.F.; Gadella, B.M.; van Golde, L.M.; Tielens, A.G. Quantitative analysis of phosphatidylcholine molecular species using HPLC and light scattering detection. J. Lipid Res. 1998, 39, 344–353. [Google Scholar] [PubMed]
- Lee, W.-J.; Weng, S.-H.; Su, N.-W. Individual Phosphatidylcholine Species Analysis by RP-HPLC-ELSD for Determination of Polyenylphosphatidylcholine in Lecithins. J. Agric. Food Chem. 2015, 63, 3851–3858. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Calvo, E.; Muntó, M.; Wurst, K.; Ventosa, N.; Masciocchi, N.; Veciana, J. Polymorphs and solvates of nicardipine hydrochloride. Selective stabilization of different diastereomeric racemates. Mol. Pharm. 2011, 8, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Indulkar, A.S.; Box, K.J.; Taylor, R.; Ruiz, R.; Taylor, L.S. pH-Dependent Liquid-Liquid Phase Separation of Highly Supersaturated Solutions of Weakly Basic Drugs. Mol. Pharm. 2015, 12, 2365–2377. [Google Scholar] [CrossRef] [PubMed]
- Peterson, B.L.; Cummings, B.S. A review of chromatographic methods for the assessment of phospholipids in biological samples. Biomed. Chromatogr. 2006, 20, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Riehl, M.; Harms, M.; Hanefeld, A.; Mäder, K. Investigation of the stabilizer elimination during the washing step of charged PLGA microparticles utilizing a novel HPLC-UV-ELSD method. Eur. J. Pharm. Biopharm. 2015, 94, 468–472. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fatty Acid | Unsaturated Lecithin Lipoid S100 | Hydrogenated Lecithin Lipoid S-PC-3 |
|---|---|---|
| 16:0 (palmitic) | 15% | 13% |
| 18:0 (stearic) | 3% | 86% |
| 18:1 (oleic and isomers) | 12% | <1% |
| 18:2 (linoleic) | 62% | |
| 18:3 (linolenic) | 5% |
| Mixture Composition S100:S-PC-3 | Extrusion Speed in Rpm | Temperature Setting of the Three Heating Zones in °C |
|---|---|---|
| 70:30 | 100 | 26/26/28 |
| 60:40 | 100 | 28/28/30 |
| 50:50 | 100 | 32/32/36 |
| 40:60 | 100 | 34/34/36 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zlomke, C.; Albrecht, J.; Mäder, K. Nicardipine Loaded Solid Phospholipid Extrudates for the Prevention of Cerebral Vasospasms: In Vitro Characterization. Pharmaceutics 2020, 12, 817. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12090817
Zlomke C, Albrecht J, Mäder K. Nicardipine Loaded Solid Phospholipid Extrudates for the Prevention of Cerebral Vasospasms: In Vitro Characterization. Pharmaceutics. 2020; 12(9):817. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12090817
Chicago/Turabian StyleZlomke, Christin, Johannes Albrecht, and Karsten Mäder. 2020. "Nicardipine Loaded Solid Phospholipid Extrudates for the Prevention of Cerebral Vasospasms: In Vitro Characterization" Pharmaceutics 12, no. 9: 817. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12090817