Extracellular Vesicle-Based Therapeutics: Preclinical and Clinical Investigations

 , , and
, , and 
Abstract
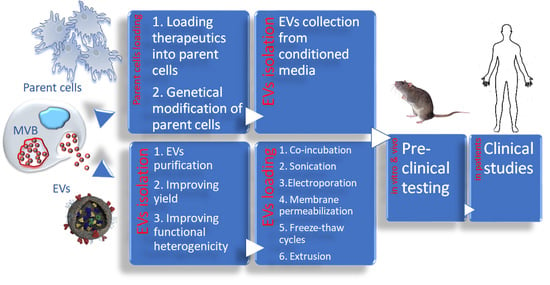
:
1. Introduction
2. Implications Related to Biological Activity Inherited from EV Origin
3. Improving Functional Heterogenicity and Yields of EVs Nanocarriers
4. Loading EVs with Therapeutic Cargo
4.1. Exogenous Loading of EVs
4.1.1. Co-Incubation
4.1.2. Sonication
4.1.3. Electroporation
4.1.4. Transient Permeabilization with Saponin
4.1.5. Freeze-Thaw and Extrusion Cycles
4.2. Exogenous Loading of Parent Cells
5. Challenges of Insufficient Targeting Efficiency of EVs Formulations to Disease Tissues
6. EV-Based Drug Formulations in Clinic
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chong, S.Y.; Lee, C.K.; Huang, C.; Ou, Y.H.; Charles, C.J.; Richards, A.M.; Neupane, Y.R.; Pavon, M.V.; Zharkova, O.; Pastorin, G.; et al. Extracellular Vesicles in Cardiovascular Diseases: Alternative Biomarker Sources, Therapeutic Agents, and Drug Delivery Carriers. Int. J. Mol. Sci. 2019, 20, 3272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulanger, C.M.; Loyer, X.; Rautou, P.E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Zamani, P.; Fereydouni, N.; Butler, A.E.; Navashenaq, J.G.; Sahebkar, A. The therapeutic and diagnostic role of exosomes in cardiovascular diseases. Trends Cardiovasc. Med. 2019, 29, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Sowers, J.R. Targeting endothelial exosomes for the prevention of cardiovascular disease. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165833. [Google Scholar] [CrossRef] [PubMed]
- Tikhomirov, R.; Donnell, B.R.; Catapano, F.; Faggian, G.; Gorelik, J.; Martelli, F.; Emanueli, C. Exosomes: From Potential Culprits to New Therapeutic Promise in the Setting of Cardiac Fibrosis. Cells 2020, 9, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Hollabaugh, C.; Miller, J.; Geiger, P.C.; Flynn, B.C. Molecular Cardioprotection and the Role of Exosomes: The Future Is Not Far Away. J. Cardiothorac. Vasc. Anesth. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ramasubramanian, L.; Kumar, P.; Wang, A. Engineering Extracellular Vesicles as Nanotherapeutics for Regenerative Medicine. Biomolecules 2019, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Akbari, A.; Jabbari, N.; Sharifi, R.; Ahmadi, M.; Vahhabi, A.; Seyedzadeh, S.J.; Nawaz, M.; Szafert, S.; Mahmoodi, M.; Jabbari, E.; et al. Free and hydrogel encapsulated exosome-based therapies in regenerative medicine. Life Sci. 2020, 249, 117447. [Google Scholar] [CrossRef]
- Kumar, S.; Zhi, K.; Mukherji, A.; Gerth, K. Repurposing Antiviral Protease Inhibitors Using Extracellular Vesicles for Potential Therapy of COVID-19. Viruses 2020, 12, 486. [Google Scholar] [CrossRef]
- Bell, B.M.; Kirk, I.D.; Hiltbrunner, S.; Gabrielsson, S.; Bultema, J.J. Designer exosomes as next-generation cancer immunotherapy. Nanomedicine 2016, 12, 163–169. [Google Scholar] [CrossRef]
- Moore, C.; Kosgodage, U.; Lange, S.; Inal, J.M. The emerging role of exosome and microvesicle- (EMV-) based cancer therapeutics and immunotherapy. Int. J. Cancer 2017, 141, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Masaoutis, C.; Mihailidou, C.; Tsourouflis, G.; Theocharis, S. Exosomes in lung cancer diagnosis and treatment. From the translating research into future clinical practice. Biochimie 2018, 151, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Su, C. Design strategies and application progress of therapeutic exosomes. Theranostics 2019, 9, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Wu, H.; Li, M.; Chen, X.; Xu, X.; Ni, W.; Lu, C.; Ni, R.; Bao, B.; Xiao, M. Progress in the application of exosomes as therapeutic vectors in tumor-targeted therapy. Cytotherapy 2019, 21, 509–524. [Google Scholar] [CrossRef] [PubMed]
- D’Agnano, I.; Berardi, A.C. Extracellular Vesicles, A Possible Theranostic Platform Strategy for Hepatocellular Carcinoma-An Overview. Cancers 2020, 12, 261. [Google Scholar] [CrossRef] [Green Version]
- Scavo, M.P.; Depalo, N.; Tutino, V.; de Nunzio, V.; Ingrosso, C.; Rizzi, F.; Notarnicola, M.; Curri, M.L.; Giannelli, G. Exosomes for Diagnosis and Therapy in Gastrointestinal Cancers. Int. J. Mol. Sci. 2020, 21, 367. [Google Scholar] [CrossRef] [Green Version]
- Chung, I.M.; Rajakumar, G.; Venkidasamy, B.; Subramanian, U.; Thiruvengadam, M. Exosomes: Current use and future applications. Clin. Chim. Acta 2020, 500, 226–232. [Google Scholar] [CrossRef]
- Jurj, A.; Zanoaga, O.; Braicu, C.; Lazar, V.; Tomuleasa, C.; Irimie, A.; Berindan-Neagoe, I. A Comprehensive Picture of Extracellular Vesicles and Their Contents. Molecular Transfer to Cancer Cells. Cancers 2020, 12, 298. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.H.; Xiang, D.; Nguyen, T.N.; Tran, T.T.; Chen, Q.; Yin, W.; Zhang, Y.; Kong, L.; Duan, A.; Chen, K.; et al. Aptamer-guided extracellular vesicle theranostics in oncology. Theranostics 2020, 10, 3849–3866. [Google Scholar] [CrossRef]
- Sousa, C.; Pereira, I.; Santos, A.C.; Carbone, C.; Kovacevic, A.B.; Silva, A.M.; Souto, E.B. Targeting dendritic cells for the treatment of autoimmune disorders. Colloids Surf. B Biointerfaces 2017, 158, 237–248. [Google Scholar] [CrossRef]
- Ha, D.; Yang, N.; Nadithe, V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: Current perspectives and future challenges. Acta Pharm. Sin. B 2016, 6, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vader, P.; Mol, E.A.; Pasterkamp, G.; Schiffelers, R.M. Extracellular vesicles for drug delivery. Adv. Drug Deliv. Rev. 2016, 106, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.; Quarto, R.; Tasso, R. Extracellular Vesicles as Natural, Safe and Efficient Drug Delivery Systems. Pharmaceutics 2019, 11, 557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batrakova, E.V.; Kim, M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control. Release 2015, 219, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Thery, C. Extracellular vesicles or exosomes? On primacy, precision, and popularity influencing a choice of nomenclature. J. Extracell. Vesicles 2019, 8, 1648167. [Google Scholar]
- Liu, D.; Kou, X.; Chen, C.; Liu, S.; Liu, Y.; Yu, W.; Yu, T.; Yang, R.; Wang, R.; Zhou, Y.; et al. Circulating apoptotic bodies maintain mesenchymal stem cell homeostasis and ameliorate osteopenia via transferring multiple cellular factors. Cell Res. 2018, 28, 918–933. [Google Scholar] [CrossRef] [Green Version]
- Van der Pol, E.; Coumans, F.A.; Grootemaat, A.E.; Gardiner, C.; Sargent, I.L.; Harrison, P.; Sturk, A.; van Leeuwen, T.G.; Nieuwland, R. Particle size distribution of exosomes and microvesicles determined by transmission electron microscopy, flow cytometry, nanoparticle tracking analysis, and resistive pulse sensing. J. Thromb. Haemost. 2014, 12, 1182–1192. [Google Scholar] [CrossRef]
- Vlassov, A.V.; Magdaleno, S.; Setterquist, R.; Conrad, R. Exosomes: Current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 2012, 1820, 940–948. [Google Scholar] [CrossRef]
- Turturici, G.; Tinnirello, R.; Sconzo, G.; Geraci, F. Extracellular membrane vesicles as a mechanism of cell-to-cell communication: Advantages and disadvantages. Am. J. Physiol. Cell Physiol. 2014, 306, C621–C633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryani, A.; Denecke, B. Exosomes as a Nanodelivery System: A Key to the Future of Neuromedicine? Mol. Neurobiol. 2016, 53, 818–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, S.; Samuel, M.; Kumar, S.; Mathivanan, S. Ticket to a bubble ride: Cargo sorting into exosomes and extracellular vesicles. Biochim. Biophys. Acta Proteins Proteom. 2019, 1867, 140203. [Google Scholar] [CrossRef] [PubMed]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tschuschke, M.; Kocherova, I.; Bryja, A.; Mozdziak, P.; Volponi, A.A.; Janowicz, K.; Sibiak, R.; Piotrowska-Kempisty, H.; Izycki, D.; Bukowska, D.; et al. Inclusion Biogenesis, Methods of Isolation and Clinical Application of Human Cellular Exosomes. J. Clin. Med. 2020, 9, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, H.I.; Vugmeyster, Y.; Mangal, N. Characterizing Exposure-Response Relationship for Therapeutic Monoclonal Antibodies in Immuno-Oncology and Beyond: Challenges, Perspectives, and Prospects. Clin. Pharmacol. Ther. 2020, 108, 1156–1170. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, J.; Stewart, T.; Sheng, L.; Li, N.; Bullock, K.; Song, N.; Shi, M.; Banks, W.A.; Zhang, J. Transmission of alpha-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: Another mechanism for initiation and progression of Parkinson’s disease? Acta Neuropathol. Commun. 2017, 5, 71. [Google Scholar] [CrossRef] [Green Version]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. Diagn. Assess. Dis. Monit. 2016, 3, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Liu, L.; Ma, F.; Wong, C.W.; Guo, X.E.; Chacko, J.V.; Farhoodi, H.P.; Zhang, S.X.; Zimak, J.; Segaliny, A.; et al. Elucidation of Exosome Migration across the Blood-Brain Barrier Model In Vitro. Cell. Mol. Bioeng. 2016, 9, 509–529. [Google Scholar] [CrossRef]
- Kuroda, H.; Tachikawa, M.; Yagi, Y.; Umetsu, M.; Nurdin, A.; Miyauchi, E.; Watanabe, M.; Uchida, Y.; Terasaki, T. Cluster of Differentiation 46 Is the Major Receptor in Human Blood-Brain Barrier Endothelial Cells for Uptake of Exosomes Derived from Brain-Metastatic Melanoma Cells (SK-Mel-28). Mol. Pharm. 2019, 16, 292–304. [Google Scholar] [CrossRef]
- Sun, D.; Zhuang, X.; Xiang, X.; Liu, Y.; Zhang, S.; Liu, C.; Barnes, S.; Grizzle, W.; Miller, D.; Zhang, H.G. A novel nanoparticle drug delivery system: The anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 2010, 18, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haney, M.J.; Klyachko, N.L.; Harrison, E.B.; Zhao, Y.; Kabanov, A.V.; Batrakova, E.V. TPP1 Delivery to Lysosomes with Extracellular Vesicles and their Enhanced Brain Distribution in the Animal Model of Batten Disease. Adv. Healthc. Mater. 2019, 8, e1801271. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef]
- Marcoux, G.; Duchez, A.C.; Cloutier, N.; Provost, P.; Nigrovic, P.A.; Boilard, E. Revealing the diversity of extracellular vesicles using high-dimensional flow cytometry analyses. Sci. Rep. 2016, 6, 35928. [Google Scholar] [CrossRef]
- Van der Pol, E.; Boing, A.N.; Harrison, P.; Sturk, A.; Nieuwland, R. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol. Rev. 2012, 64, 676–705. [Google Scholar] [CrossRef] [Green Version]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef]
- Andre, F.; Schartz, N.E.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef]
- Valadi, H.; Ekstrom, K.; Bossios, A.; Sjostrand, M.; Lee, J.J.; Lotvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, C.; Wang, S.; Wang, Z.; Jiang, J.; Wang, W.; Li, X.; Chen, J.; Liu, K.; Li, C.; et al. Exosomes Derived from Hypoxic Oral Squamous Cell Carcinoma Cells Deliver miR-21 to Normoxic Cells to Elicit a Prometastatic Phenotype. Cancer Res. 2016, 76, 1770–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogure, T.; Lin, W.L.; Yan, I.K.; Braconi, C.; Patel, T. Intercellular nanovesicle-mediated microRNA transfer: A mechanism of environmental modulation of hepatocellular cancer cell growth. Hepatology 2011, 54, 1237–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Qiao, B.; Gao, N.; Lin, N.; He, W. Oral squamous cell carcinoma-derived exosomes promote M2 subtype macrophage polarization mediated by exosome-enclosed miR-29a-3p. Am. J. Physiol. Cell Physiol. 2019, 316, C731–C740. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.C.; Kim, O.Y.; Yoon, C.M.; Choi, D.S.; Roh, T.Y.; Park, J.; Nilsson, J.; Lotvall, J.; Kim, Y.K.; Gho, Y.S. Bioinspired exosome-mimetic nanovesicles for targeted delivery of chemotherapeutics to malignant tumors. ACS Nano 2013, 7, 7698–7710. [Google Scholar] [CrossRef]
- Coffman, L.G.; Choi, Y.J.; McLean, K.; Allen, B.L.; di Magliano, M.P.; Buckanovich, R.J. Human carcinoma-associated mesenchymal stem cells promote ovarian cancer chemotherapy resistance via a BMP4/HH signaling loop. Oncotarget 2016, 7, 6916–6932. [Google Scholar] [CrossRef] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. Concise Review: Crosstalk of Mesenchymal Stroma/Stem-Like Cells with Cancer Cells Provides Therapeutic Potential. Stem Cells 2018, 36, 951–968. [Google Scholar] [CrossRef] [Green Version]
- Vakhshiteh, F.; Atyabi, F.; Ostad, S.N. Mesenchymal stem cell exosomes: A two-edged sword in cancer therapy. Int. J. Nanomed. 2019, 14, 2847–2859. [Google Scholar] [CrossRef] [Green Version]
- Qi, J.; Zhou, Y.; Jiao, Z.; Wang, X.; Zhao, Y.; Li, Y.; Chen, H.; Yang, L.; Zhu, H.; Li, Y. Exosomes Derived from Human Bone Marrow Mesenchymal Stem Cells Promote Tumor Growth Through Hedgehog Signaling Pathway. Cell. Physiol. Biochem. 2017, 42, 2242–2254. [Google Scholar] [CrossRef]
- Wu, S.; Ju, G.Q.; Du, T.; Zhu, Y.J.; Liu, G.H. Microvesicles derived from human umbilical cord Wharton’s jelly mesenchymal stem cells attenuate bladder tumor cell growth in vitro and in vivo. PLoS ONE 2013, 8, e61366. [Google Scholar] [CrossRef]
- Bruno, S.; Camussi, G. Role of mesenchymal stem cell-derived microvesicles in tissue repair. Pediatr. Nephrol. 2013, 28, 2249–2254. [Google Scholar] [CrossRef]
- Bruno, S.; Collino, F.; Deregibus, M.C.; Grange, C.; Tetta, C.; Camussi, G. Microvesicles derived from human bone marrow mesenchymal stem cells inhibit tumor growth. Stem Cells Dev. 2013, 22, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Takahara, K.; Ii, M.; Inamoto, T.; Nakagawa, T.; Ibuki, N.; Yoshikawa, Y.; Tsujino, T.; Uchimoto, T.; Saito, K.; Takai, T.; et al. microRNA-145 Mediates the Inhibitory Effect of Adipose Tissue-Derived Stromal Cells on Prostate Cancer. Stem Cells Dev. 2016, 25, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Roccaro, A.M.; Sacco, A.; Maiso, P.; Azab, A.K.; Tai, Y.T.; Reagan, M.; Azab, F.; Flores, L.M.; Campigotto, F.; Weller, E.; et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J. Clin. Investig. 2013, 123, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Mitsialis, S.A.; Aslam, M.; Vitali, S.H.; Vergadi, E.; Konstantinou, G.; Sdrimas, K.; Fernandez-Gonzalez, A.; Kourembanas, S. Exosomes mediate the cytoprotective action of mesenchymal stromal cells on hypoxia-induced pulmonary hypertension. Circulation 2012, 126, 2601–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, R.C.; Yeo, R.W.; Tan, K.H.; Lim, S.K. Exosomes for drug delivery—A novel application for the mesenchymal stem cell. Biotechnol. Adv. 2013, 31, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G.; et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013, 10, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Katsuda, T.; Tsuchiya, R.; Kosaka, N.; Yoshioka, Y.; Takagaki, K.; Oki, K.; Takeshita, F.; Sakai, Y.; Kuroda, M.; Ochiya, T. Human adipose tissue-derived mesenchymal stem cells secrete functional neprilysin-bound exosomes. Sci. Rep. 2013, 3, 1197. [Google Scholar] [CrossRef] [Green Version]
- Xin, H.; Li, Y.; Chopp, M. Exosomes/miRNAs as mediating cell-based therapy of stroke. Front. Cell. Neurosci. 2014, 8, 377. [Google Scholar] [CrossRef] [Green Version]
- Ilmer, M.; Vykoukal, J.; Boiles, A.R.; Coleman, M.; Alt, E. Two sides of the same coin: Stem cells in cancer and regenerative medicine. FASEB J. 2014, 28, 2748–2761. [Google Scholar] [CrossRef]
- Emanueli, C.; Shearn, A.I.; Angelini, G.D.; Sahoo, S. Exosomes and exosomal miRNAs in cardiovascular protection and repair. Vascul. Pharmacol. 2015, 71, 24–30. [Google Scholar] [CrossRef] [Green Version]
- Kang, K.; Ma, R.; Cai, W.; Huang, W.; Paul, C.; Liang, J.; Wang, Y.; Zhao, T.; Kim, H.W.; Xu, M.; et al. Exosomes Secreted from CXCR4 Overexpressing Mesenchymal Stem Cells Promote Cardioprotection via Akt Signaling Pathway following Myocardial Infarction. Stem Cells Int. 2015, 2015, 659890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; el Andaloussi, S.; Wood, M.J. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibrahim, A.G.; Cheng, K.; Marban, E. Exosomes as critical agents of cardiac regeneration triggered by cell therapy. Stem Cell Rep. 2014, 2, 606–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeyaseelan, K.; Lim, K.Y.; Armugam, A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke 2008, 39, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.J.; Lim, K.Y.; Kaur, P.; Sepramaniam, S.; Armugam, A.; Wong, P.T.; Jeyaseelan, K. microRNAs Involved in Regulating Spontaneous Recovery in Embolic Stroke Model. PLoS ONE 2013, 8, e66393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusardi, T.A.; Murphy, S.J.; Phillips, J.I.; Chen, Y.; Davis, C.M.; Young, J.M.; Thompson, S.J.; Saugstad, J.A. MicroRNA responses to focal cerebral ischemia in male and female mouse brain. Front. Mol. Neurosci. 2014, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Zhu, Z.; Wang, H.; Yu, Y.; Chen, W.; Waqas, A.; Wang, Y.; Chen, L. Exosomes derived from human neural stem cells stimulated by interferon gamma improve therapeutic ability in ischemic stroke model. J. Adv. Res. 2020, 24, 435–445. [Google Scholar] [CrossRef]
- Bian, B.; Zhao, C.; He, X.; Gong, Y.; Ren, C.; Ge, L.; Zeng, Y.; Li, Q.; Chen, M.; Weng, C.; et al. Exosomes derived from neural progenitor cells preserve photoreceptors during retinal degeneration by inactivating microglia. J. Extracell. Vesicles 2020, 9, 1748931. [Google Scholar] [CrossRef] [Green Version]
- Batrakova, E.V.; Kim, M.S. Development and regulation of exosome-based therapy products. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 744–757. [Google Scholar] [CrossRef]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-‘t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 20360. [Google Scholar] [CrossRef]
- Tauro, B.J.; Greening, D.W.; Mathias, R.A.; Ji, H.; Mathivanan, S.; Scott, A.M.; Simpson, R.J. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods 2012, 56, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Shah, S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods 2015, 87, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Zacharias, W.; Gercel-Taylor, C. Exosome isolation for proteomic analyses and RNA profiling. Methods Mol. Biol. 2011, 728, 235–246. [Google Scholar] [PubMed]
- Patel, G.K.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.; Singh, S.; Singh, A.P. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci. Rep. 2019, 9, 5335. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Greening, D.W.; Zhu, H.J.; Takahashi, N.; Simpson, R.J. Extracellular vesicle isolation and characterization: Toward clinical application. J. Clin. Investig. 2016, 126, 1152–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lener, T.; Gimona, M.; Aigner, L.; Borger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef] [PubMed]
- Chevillet, J.R.; Kang, Q.; Ruf, I.K.; Briggs, H.A.; Vojtech, L.N.; Hughes, S.M.; Cheng, H.H.; Arroyo, J.D.; Meredith, E.K.; Gallichotte, E.N.; et al. Quantitative and stoichiometric analysis of the microRNA content of exosomes. Proc. Natl. Acad. Sci. USA 2014, 111, 14888–14893. [Google Scholar] [CrossRef] [Green Version]
- Moleirinho, M.G.; Silva, R.J.S.; Carrondo, M.J.T.; Alves, P.M.; Peixoto, C. Exosome-based therapeutics: Purification using semi-continuous multicolumn chromatography. Sep. Purif. Technol. 2019, 224, 515–523. [Google Scholar] [CrossRef]
- Navajas, R.; Corrales, F.J.; Paradela, A. Serum Exosome Isolation by Size-Exclusion Chromatography for the Discovery and Validation of Preeclampsia-Associated Biomarkers. Methods Mol. Biol. 2019, 1959, 39–50. [Google Scholar]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mager, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.; Hallbrink, M.; Seow, Y.; Bultema, J.J.; et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine 2015, 11, 879–883. [Google Scholar] [CrossRef] [Green Version]
- Witwer, K.W.; van Balkom, B.W.M.; Bruno, S.; Choo, A.; Dominici, M.; Gimona, M.; Hill, A.F.; de Kleijn, D.; Koh, M.; Lai, R.C.; et al. Defining mesenchymal stromal cell (MSC)-derived small extracellular vesicles for therapeutic applications. J. Extracell. Vesicles 2019, 8, 1609206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamparski, H.G.; Metha-Damani, A.; Yao, J.Y.; Patel, S.; Hsu, D.H.; Ruegg, C.; Le Pecq, J.B. Production and characterization of clinical grade exosomes derived from dendritic cells. J. Immunol. Methods 2002, 270, 211–226. [Google Scholar] [CrossRef]
- Ban, J.J.; Lee, M.; Im, W.; Kim, M. Low pH increases the yield of exosome isolation. Biochem. Biophys. Res. Commun. 2015, 461, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Emam, S.E.; Ando, H.; Lila, A.S.A.; Shimizu, T.; Ukawa, M.; Okuhira, K.; Ishima, Y.; Mahdy, M.A.; Ghazy, F.S.; Ishida, T. A Novel Strategy to Increase the Yield of Exosomes (Extracellular Vesicles) for an Expansion of Basic Research. Biol. Pharm. Bull. 2018, 41, 733–742. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, S.; Wang, Z. High yield, scalable and remotely drug-loaded neutrophil-derived extracellular vesicles (EVs) for anti-inflammation therapy. Biomaterials 2017, 135, 62–73. [Google Scholar] [CrossRef]
- Jhan, Y.Y.; Prasca-Chamorro, D.; Zuniga, G.P.; Moore, D.M.; Kumar, S.A.; Gaharwar, A.K.; Bishop, C.J. Engineered extracellular vesicles with synthetic lipids via membrane fusion to establish efficient gene delivery. Int. J. Pharm. 2020, 573, 118802. [Google Scholar] [CrossRef]
- Rayamajhi, S.; Nguyen, T.D.T.; Marasini, R.; Aryal, S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019, 94, 482–494. [Google Scholar] [CrossRef]
- De la Pena, H.; Madrigal, J.A.; Rusakiewicz, S.; Bencsik, M.; Cave, G.W.; Selman, A.; Rees, R.C.; Travers, P.J.; Dodi, I.A. Artificial exosomes as tools for basic and clinical immunology. J. Immunol. Methods 2009, 344, 121–132. [Google Scholar] [CrossRef]
- Thone, M.N.; Kwon, Y.J. Extracellular blebs: Artificially-induced extracellular vesicles for facile production and clinical translation. Methods 2020, 177, 135–145. [Google Scholar] [CrossRef]
- Li, D.; Yao, S.; Zhou, Z.; Shi, J.; Huang, Z.; Wu, Z. Hyaluronan decoration of milk exosomes directs tumor-specific delivery of doxorubicin. Carbohydr. Res. 2020, 493, 108032. [Google Scholar] [CrossRef]
- Ju, S.; Mu, J.; Dokland, T.; Zhuang, X.; Wang, Q.; Jiang, H.; Xiang, X.; Deng, Z.B.; Wang, B.; Zhang, L.; et al. Grape exosome-like nanoparticles induce intestinal stem cells and protect mice from DSS-induced colitis. Mol. Ther. 2013, 21, 1345–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Kalani, A.; Kamat, P.K.; Chaturvedi, P.; Tyagi, S.C.; Tyagi, N. Curcumin-primed exosomes mitigate endothelial cell dysfunction during hyperhomocysteinemia. Life Sci. 2014, 107, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Rani, S.; Ryan, A.E.; Griffin, M.D.; Ritter, T. Mesenchymal Stem Cell-derived Extracellular Vesicles: Toward Cell-free Therapeutic Applications. Mol. Ther. 2015, 23, 812–823. [Google Scholar] [CrossRef] [Green Version]
- Saari, H.; Lisitsyna, E.; Rautaniemi, K.; Rojalin, T.; Niemi, L.; Nivaro, O.; Laaksonen, T.; Yliperttula, M.; Vuorimaa-Laukkanen, E. FLIM reveals alternative EV-mediated cellular up-take pathways of paclitaxel. J Control. Release 2018, 284, 133–143. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Qu, M.; Lin, Q.; Huang, L.; Fu, Y.; Wang, L.; He, S.; Fu, Y.; Yang, S.; Zhang, Z.; Zhang, L.; et al. Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J. Control. Release 2018, 287, 156–166. [Google Scholar] [CrossRef]
- Li, Y.J.; Wu, J.Y.; Wang, J.M.; Hu, X.B.; Cai, J.X.; Xiang, D.X. Gemcitabine loaded autologous exosomes for effective and safe chemotherapy of pancreatic cancer. Acta Biomater. 2020, 101, 519–530. [Google Scholar] [CrossRef]
- Iessi, E.; Logozzi, M.; Lugini, L.; Azzarito, T.; Federici, C.; Spugnini, E.P.; Mizzoni, D.; di Raimo, R.; Angelini, D.F.; Battistini, L.; et al. Acridine Orange/exosomes increase the delivery and the effectiveness of Acridine Orange in human melanoma cells: A new prototype for theranostics of tumors. J. Enzyme Inhib. Med. Chem. 2017, 32, 648–657. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine 2018, 14, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Zhao, Y.; Jin, Y.S.; Li, S.M.; Bago, J.R.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Macrophage-Derived Extracellular Vesicles as Drug Delivery Systems for Triple Negative Breast Cancer (TNBC) Therapy. J. Neuroimmune Pharmacol. 2019, 15, 487–500. [Google Scholar] [CrossRef]
- Park, J.; Lee, H.; Youn, Y.S.; Oh, K.T.; Lee, E.S. Tumor-Homing pH-Sensitive Extracellular Vesicles for Targeting Heterogeneous Tumors. Pharmaceutics 2020, 12, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Ohno, S.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenaka, T.; Nakai, S.; Katayama, M.; Hirano, M.; Ueno, N.; Noguchi, K.; Takatani-Nakase, T.; Fujii, I.; Kobayashi, S.S.; Nakase, I. Effects of gefitinib treatment on cellular uptake of extracellular vesicles in EGFR-mutant non-small cell lung cancer cells. Int. J. Pharm. 2019, 572, 118762. [Google Scholar] [CrossRef]
- Wahlgren, J.; De, L.K.T.; Brisslert, M.; Sani, F.V.; Telemo, E.; Sunnerhagen, P.; Valadi, H. Plasma exosomes can deliver exogenous short interfering RNA to monocytes and lymphocytes. Nucleic Acids Res. 2012, 40, e130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Wickline, S.A.; Hood, J.L. Magnetic resonance imaging of melanoma exosomes in lymph nodes. Magn. Reson. Med. 2015, 74, 266–271. [Google Scholar] [CrossRef]
- Alyane, M.; Barratt, G.; Lahouel, M. Remote loading of doxorubicin into liposomes by transmembrane pH gradient to reduce toxicity toward H9c2 cells. Saudi Pharm. J. 2016, 24, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Sawada, K.; Miyamoto, M.; Shimizu, A.; Yamamoto, M.; Kinose, Y.; Nakamura, K.; Kawano, M.; Kodama, M.; Hashimoto, K.; et al. Exploring the potential of engineered exosomes as delivery systems for tumor-suppressor microRNA replacement therapy in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 527, 153–161. [Google Scholar] [CrossRef]
- Zhang, D.; Lee, H.; Wang, X.; Rai, A.; Groot, M.; Jin, Y. Exosome-Mediated Small RNA Delivery: A Novel Therapeutic Approach for Inflammatory Lung Responses. Mol. Ther. 2018, 26, 2119–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterzenbach, U.; Putz, U.; Low, L.H.; Silke, J.; Tan, S.S.; Howitt, J. Engineered Exosomes as Vehicles for Biologically Active Proteins. Mol. Ther. 2017, 25, 1269–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamichhane, T.N.; Jeyaram, A.; Patel, D.B.; Parajuli, B.; Livingston, N.K.; Arumugasaamy, N.; Schardt, J.S.; Jay, S.M. Oncogene Knockdown via Active Loading of Small RNAs into Extracellular Vesicles by Sonication. Cell. Mol. Bioeng. 2016, 9, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Albanese, M.; Chen, Y.-F.A.; Huls, C.; Gartner, K.; Tagawa, T.; Keppler, O.T.; Gobel, C.; Zeidler, R.; Hammerschmidt, W. Micro RNAs are minor constituents of extracellular vesicles and are hardly delivered to target cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Jones, A.; Troakes, C.; King, A.; Al-Sarraj, S.; van den Berg, L.H. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 124, 339–352. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Lakhal, S.; Mager, I.; Wood, M.J. Exosomes for targeted siRNA delivery across biological barriers. Adv. Drug Deliv. Rev. 2013, 65, 391–397. [Google Scholar] [CrossRef]
- Kooijmans, S.A.A.; Stremersch, S.; Braeckmans, K.; de Smedt, S.C.; Hendrix, A.; Wood, M.J.A.; Schiffelers, R.M.; Raemdonck, K.; Vader, P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control. Release 2013, 172, 229–238. [Google Scholar] [CrossRef]
- le Saux, S.; Aarrass, H.; Lai-Kee-Him, J.; Bron, P.; Armengaud, J.; Miotello, G.; Bertrand-Michel, J.; Dubois, E.; George, S.; Faklaris, O.; et al. Post-production modifications of murine mesenchymal stem cell (mMSC) derived extracellular vesicles (EVs) and impact on their cellular interaction. Biomaterials 2020, 231, 119675. [Google Scholar] [CrossRef]
- Villata, S.; Canta, M.; Cauda, V. EVs and Bioengineering: From Cellular Products to Engineered Nanomachines. Int. J. Mol. Sci. 2020, 21, 6048. [Google Scholar] [CrossRef]
- Osterman, C.J.; Lynch, J.C.; Leaf, P.; Gonda, A.; Bennit, H.R.F.; Griffiths, D.; Wall, N.R. Curcumin Modulates Pancreatic Adenocarcinoma Cell-Derived Exosomal Function. PLoS ONE 2015, 10, e0132845. [Google Scholar] [CrossRef] [Green Version]
- Pascucci, L.; Cocce, V.; Bonomi, A.; Ami, D.; Ceccarelli, P.; Ciusani, E.; Vigano, L.; Locatelli, A.; Sisto, F.; Doglia, S.M.; et al. Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. J Control. Release 2014, 192, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.H.; Wan, Y.L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.L.; Lin, H.M.; Shang, C.Z.; Chen, Y.J.; Min, J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haney, M.J.; Suresh, P.; Zhao, Y.; Kanmogne, G.D.; Kadiu, I.; Sokolsky-Papkov, M.; Klyachko, N.L.; Mosley, R.L.; Kabanov, A.V.; Gendelman, H.E.; et al. Blood-borne macrophage-neural cell interactions hitchhike on endosome networks for cell-based nanozyme brain delivery. Nanomedicine 2012, 7, 815–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Haney, M.J.; Gupta, R.; Bohnsack, J.P.; He, Z.; Kabanov, A.V.; Batrakova, E.V. GDNF-transfected macrophages produce potent neuroprotective effects in Parkinson’s disease mouse model. PLoS ONE 2014, 9, e106867. [Google Scholar] [CrossRef] [PubMed]
- Mizrak, A.; Bolukbasi, M.F.; Ozdener, G.B.; Brenner, G.J.; Madlener, S.; Erkan, E.P.; Strobel, T.; Breakefield, X.O.; Saydam, O. Genetically engineered microvesicles carrying suicide mRNA/protein inhibit schwannoma tumor growth. Mol. Ther. 2013, 21, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.Y.; Lee, J.K.; Jeon, Y.K.; Kim, C.W. Exosome derived from epigallocatechin gallate treated breast cancer cells suppresses tumor growth by inhibiting tumor-associated macrophage infiltration and M2 polarization. BMC Cancer 2013, 13, 421. [Google Scholar] [CrossRef] [Green Version]
- Zeelenberg, I.S.; Ostrowski, M.; Krumeich, S.; Bobrie, A.; Jancic, C.; Boissonnas, A.; Delcayre, A.; le Pecq, J.B.; Combadiere, B.; Amigorena, S.; et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008, 68, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Haney, M.J.; Zhao, Y.; Harrison, E.B.; Mahajan, V.; Ahmed, S.; He, Z.; Suresh, P.; Hingtgen, S.D.; Klyachko, N.L.; Mosley, R.L.; et al. Specific transfection of inflamed brain by macrophages: A new therapeutic strategy for neurodegenerative diseases. PLoS ONE 2013, 8, e61852. [Google Scholar] [CrossRef] [Green Version]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, M.; Nawaz, M.; Papadimitriou, A.; Angerfors, A.; Camponeschi, A.; Na, M.; Holtta, M.; Skantze, P.; Johansson, S.; Sundqvist, M.; et al. Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nat. Commun. 2019, 10, 4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateescu, B.; Kowal, E.J.; van Balkom, B.W.; Bartel, S.; Bhattacharyya, S.N.; Buzas, E.I.; Buck, A.H.; de Candia, P.; Chow, F.W.; Das, S.; et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA—An ISEV position paper. J. Extracell. Vesicles 2017, 6, 1286095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiklander, O.P.; Nordin, J.Z.; O’Loughlin, A.; Gustafsson, Y.; Corso, G.; Mager, I.; Vader, P.; Lee, Y.; Sork, H.; Seow, Y.; et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles 2015, 4, 26316. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Wang, L.; Zhu, C.; Zheng, Q.; Wang, G.; Tong, J.; Fang, Y.; Xia, Y.; Cheng, G.; He, X.; et al. Aptamer-Conjugated Extracellular Nanovesicles for Targeted Drug Delivery. Cancer Res. 2018, 78, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emam, S.E.; Lila, A.S.A.; Elsadek, N.E.; Ando, H.; Shimizu, T.; Okuhira, K.; Ishima, Y.; Mahdy, M.A.; Ghazy, F.S.; Ishida, T. Cancer cell-type tropism is one of crucial determinants for the efficient systemic delivery of cancer cell-derived exosomes to tumor tissues. Eur. J. Pharm. Biopharm. 2019, 145, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Zhang, H.X.; He, C.P.; Fan, S.; Zhu, Y.L.; Qi, C.; Huang, N.P.; Xiao, Z.D.; Lu, Z.H.; Tannous, B.A.; et al. Surface functionalized exosomes as targeted drug delivery vehicles for cerebral ischemia therapy. Biomaterials 2018, 150, 137–149. [Google Scholar] [CrossRef]
- Guell, K.; Bix, G.J. Brain endothelial cell specific integrins and ischemic stroke. Expert Rev. Neurother. 2014, 14, 1287–1292. [Google Scholar] [CrossRef]
- Ishikawa, R.; Yoshida, S.; Sawada, S.I.; Sasaki, Y.; Akiyoshi, K. Preparation of engineered extracellular vesicles with full-length functional PD-1 membrane proteins by baculovirus expression system. Biochem. Biophys. Res. Commun. 2020, 526, 967–972. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef]
- Pachler, K.; Lener, T.; Streif, D.; Dunai, Z.A.; Desgeorges, A.; Feichtner, M.; Oller, M.; Schallmoser, K.; Rohde, E.; Gimona, M. A Good Manufacturing Practice-grade standard protocol for exclusively human mesenchymal stromal cell-derived extracellular vesicles. Cytotherapy 2017, 19, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Gimona, M.; Pachler, K.; Laner-Plamberger, S.; Schallmoser, K.; Rohde, E. Manufacturing of Human Extracellular Vesicle-Based Therapeutics for Clinical Use. Int. J. Mol. Sci. 2017, 18, 1190. [Google Scholar] [CrossRef] [PubMed]
- Mentkowski, K.I.; Snitzer, J.D.; Rusnak, S.; Lang, J.K. Therapeutic Potential of Engineered Extracellular Vesicles. AAPS J. 2018, 20, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghaei, K.; Tokhanbigli, S.; Asadzadeh, H.; Nmaki, S.; Zali, M.R.; Hashemi, S.M. Exosomes as a novel cell-free therapeutic approach in gastrointestinal diseases. J. Cell. Physiol. 2019, 234, 9910–9926. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Dorval, T.; Chaput, N.; Andre, F.; Caby, M.P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived-exosomes: Results of thefirst phase I clinical trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [Green Version]
- Besse, B.; Charrier, M.; Lapierre, V.; Dansin, E.; Lantz, O.; Planchard, D.; le Chevalier, T.; Livartoski, A.; Barlesi, F.; Laplanche, A.; et al. Dendritic cell-derived exosomes as maintenance immunotherapy after first line chemotherapy in NSCLC. Oncoimmunology 2016, 5, e1071008. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Varela, M.; de Menezes-Neto, A.; Perez-Zsolt, D.; Gamez-Valero, A.; Segui-Barber, J.; Izquierdo-Useros, N.; Martinez-Picado, J.; Fernandez-Becerra, C.; Del Portillo, H.A. Proteomics study of human cord blood reticulocyte-derived exosomes. Sci. Rep. 2018, 8, 14046. [Google Scholar] [CrossRef] [Green Version]
- Hafiane, A.; Daskalopoulou, S.S. Extracellular vesicles characteristics and emerging roles in atherosclerotic cardiovascular disease. Metabolism 2018, 85, 213–222. [Google Scholar] [CrossRef]
- Singhto, N.; Kanlaya, R.; Nilnumkhum, A.; Thongboonkerd, V. Roles of Macrophage Exosomes in Immune Response to Calcium Oxalate Monohydrate Crystals. Front. Immunol. 2018, 9, 316. [Google Scholar] [CrossRef] [Green Version]
- TRice, F.; Donaldson, B.; Bouqueau, M.; Kampmann, B.; Holder, B. Macrophage-but not monocyte-derived extracellular vesicles induce placental pro-inflammatory responses. Placenta 2018, 69, 92–95. [Google Scholar]
- Ferreira, A.D.F.; Gomes, D.A. Stem Cell Extracellular Vesicles in Skin Repair. Bioengineering 2018, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Godoy, M.A.; Saraiva, L.M.; de Carvalho, L.R.P.; Vasconcelos-Dos-Santos, A.; Beiral, H.J.V.; Ramos, A.B.; Silva, L.R.P.; Leal, R.B.; Monteiro, V.H.S.; Braga, C.V.; et al. Mesenchymal stem cells and cell-derived extracellular vesicles protect hippocampal neurons from oxidative stress and synapse damage induced by amyloid-beta oligomers. J. Biol. Chem. 2018, 293, 1957–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobis-Wozowicz, S.; Kmiotek, K.; Kania, K.; Karnas, E.; Labedz-Maslowska, A.; Sekula, M.; Kedracka-Krok, S.; Kolcz, J.; Boruczkowski, D.; Madeja, Z.; et al. Diverse impact of xeno-free conditions on biological and regenerative properties of hUC-MSCs and their extracellular vesicles. J. Mol. Med. 2017, 95, 205–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taheri, B.; Soleimani, M.; Aval, S.F.; Esmaeili, E.; Bazi, Z.; Zarghami, N. Induced pluripotent stem cell-derived extracellular vesicles: A novel approach for cell-free regenerative medicine. J. Cell. Physiol. 2019, 234, 8455–8464. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Cheng, G.; Bobis-Wozowicz, S.; Zhao, L.; Kedracka-Krok, S.; Samanta, A.; Karnas, E.; Xuan, Y.T.; Skupien-Rabian, B.; Chen, X.; et al. Induced Pluripotent Stem Cell (iPSC)-Derived Extracellular Vesicles Are Safer and More Effective for Cardiac Repair Than iPSCs. Circ. Res. 2018, 122, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Najar, M.; Bouhtit, F.; Melki, R.; Afif, H.; Hamal, A.; Fahmi, H.; Merimi, M.; Lagneaux, L. Mesenchymal Stromal Cell-Based Therapy: New Perspectives and Challenges. J. Clin. Med. 2019, 8, 626. [Google Scholar] [CrossRef] [Green Version]
- Bagno, L.; Hatzistergos, K.E.; Balkan, W.; Hare, J.M. Mesenchymal Stem Cell-Based Therapy for Cardiovascular Disease: Progress and Challenges. Mol. Ther. 2018, 26, 1610–1623. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.L.; Crawford, J.R.; Dib, N.; Verkh, L.; Tankovich, N.; Cramer, S.C. Phase I/II Study of Safety and Preliminary Efficacy of Intravenous Allogeneic Mesenchymal Stem Cells in Chronic Stroke. Stroke 2019, 50, 2835–2841. [Google Scholar] [CrossRef]
- Mignot, G.; Roux, S.; Thery, C.; Segura, E.; Zitvogel, L. Prospects for exosomes in immunotherapy of cancer. J. Cell. Mol. Med. 2006, 10, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef]
- Mohammed, E.; Khalil, E.; Sabry, D. Effect of Adipose-Derived Stem Cells and Their Exo as Adjunctive Therapy to Nonsurgical Periodontal Treatment: A Histologic and Histomorphometric Study in Rats. Biomolecules 2018, 8, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, P.; Chen, X.; Guo, L.; Wang, Y.; Liu, X.; Liu, Y.; Zhou, T.; Huang, T.; Geng, S.; Luo, C.; et al. A potent immunomodulatory role of exosomes derived from mesenchymal stromal cells in preventing cGVHD. J. Hematol. Oncol. 2018, 11, 135. [Google Scholar] [CrossRef] [PubMed] [Green Version]

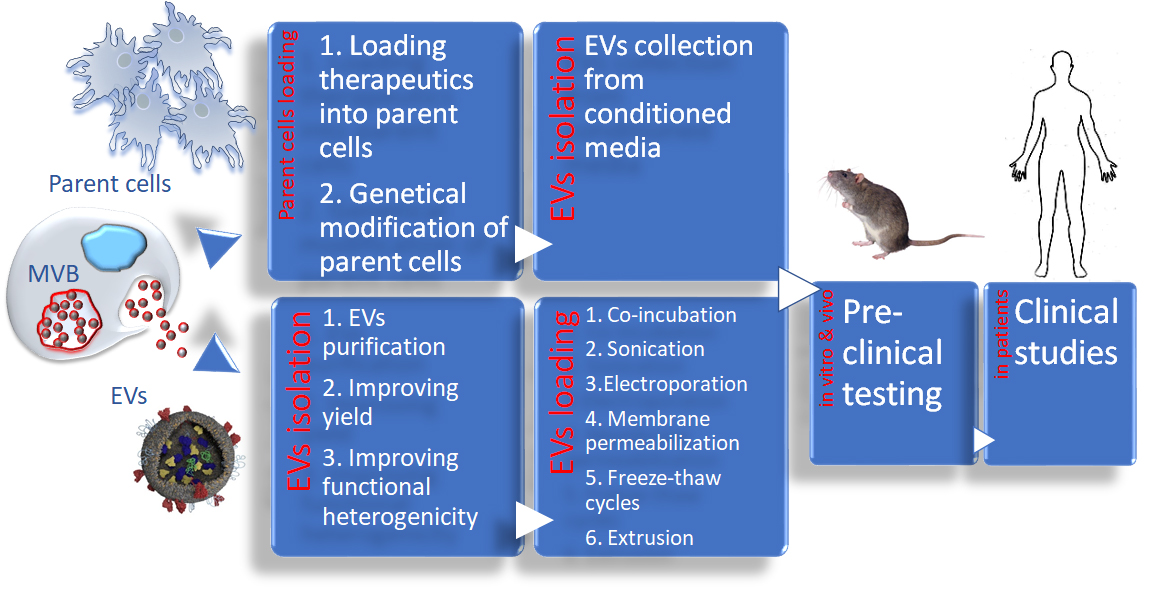{kind=link}
| Disease | Source of EVs | Therapeutic Cargo/Drug | Loading Method | Ref. |
|---|---|---|---|---|
| Inflammatory-related diseases | Neutrophil-derived EVs | Piceatannol | Co-incubation | [95] |
| Cancer | Bioinspired EV-mimetic nanovesicles | Doxorubicin | Co-incubation | [54] |
| Inflammatory-related diseases | EL-4 Cells | Curcumin | Co-incubation | [41] |
| Brain inflammatory-related diseases | Brain inflammation disease cells | Curcumin | Co-incubation | [102] |
| Cerebral diseases | Endothelial cells | Curcumin | Co-incubation | [103] |
| Cancer | Immature dendritic cells | Doxorubicin | Co-incubation | [104] |
| Prostate Cancer | PC-3 cells | Paclitaxel | Co-incubation | [106] |
| Parkinson’s Disease | Blood | Dopamine | Co-incubation | [108] |
| Melanoma | Me 30966 | Acridine Orange | Co-incubation | [110] |
| Cardiotoxicity | Liposomes | Doxorubicin | Co-incubation | [119] |
| Brain inflammatory-related diseases | Macrophages | Brain derived neurotrophic factor (BDNF) | Co-incubation | [45] |
| Pancreatic Cancer | Autologous EVs | Gemcitabine | Co-incubation and probe sonification | [109] |
| Triple Negative Breast Cancer | Macrophages | Paclitaxel and Doxorubicin | Probe sonification | [112] |
| Cancer | BT-474 tumor cells/SK-N-MC tumor cells | Doxorubicin | Probe sonification | [113] |
| MDR Cancer | Macrophages | Paclitaxel | Sonification | [107] |
| Cancer | Hybrid EVs (EVs from macrophage hybridized with synthetic liposome) | Doxorubicin | Water bath sonification | [97] |
| Pulmonary metastases | Macrophages | Paclitaxel | Probe sonification and Electroporation | [111] |
| Brain-related diseases | Macrophages | TPP1 | Water bath sonification and permeabilization with saponin | [44] |
| Brain-related diseases | DC-derived EVs | siRNA | Electroporation | [114] |
| Breast Cancer | EVs targeted to EGFR-expressing cells | miRNA | Electroporation | [115] |
| Lung Cancer | EVs | Doxorubicin | Electroporation | [116] |
| Ovarian Cancer | Fibroblasts-derived EVs | Tumor suppressor miRNA | Electroporation | [120] |
| Lung diseases | Serum-derived EVs | siRNA | Permeabilization with saponin | [121] |
| Parkinson’s Disease | EVs secreted by monocytes and macrophages | Catalase | Co-incubation, probe sonification, permeabilization with saponin, freeze-thaw cycles, and extrusion | [43] |
| Disease | Source of EVs | Therapeutic Cargo/Drug | Loading Method | Ref. |
|---|---|---|---|---|
| Cancer | Bioinspired EV-mimetic nanovesicles | Dox | Loading of parent cells with therapeutic agent | [54] |
| Parkinson’s disease | EVs secreted by monocytes and macrophages | Catalase | Loading of parent cells with therapeutic agent | [43] |
| Pancreatic Cancer | PANC-1 cells | Curcumin | Loading of parent cells with therapeutic agent | [130] |
| Cancer | MSC-derived EVs | Paclitaxel | Loading of parent cells with therapeutic agent | [131] |
| Carcinoma | HepG2 cells | Paclitaxel, Etoposide, Carboplatin, Irinotecan, Epirubicin, Mitoxanthrone | Loading of parent cells with therapeutic agent | [132] |
| Neurodegene- rative diseases | Bone marrow-derived macrophages | Catalase | Loading of parent cells with therapeutic agent | [133] |
| Parkinson’s disease | GDNF-transfected macrophages | Catalase | Loading of parent cells with therapeutic agent and Transfection of parent cells | [134] |
| Glioblastomas | Isolated EVs from CD-UPRT-treated cells | Therapeutic CD-UPRT | Transfection of parent cells | [135] |
| Breast Cancer | Isolated EVs from EGCG-treated 4T1 cells | EGCG | Transfection of parent cells | [136] |
| Cancer | Isolated EVs from OVAC1C2-treated cells | OVAC1C2 | Transfection of parent cells | [137] |
| Neurodegene-rative diseases | Transfected macrophages | Catalase | Transfection of parent cells | [138] |
| Adeno-associated virus | VEVs | AAV vectors | Transfection of parent cells | [139] |
| EVs Source | Condition | Drug | Administration Route | Dose Reported | Phase | Study Identifier |
|---|---|---|---|---|---|---|
| MSCs | Cerebrovascular disorders/stroke | miR-124 | i.v. | 200 µg protein | 1/2 | NCT03384433 |
| MSCss | Alzheimer Disease | no | nasal drip | 5 μg–20 μg | 1/2 | NCT04388982 |
| MSCs | Periodontitis | no | local | not reported | early 1 | NCT04270006 |
| MSCs | Neuralgia | no | i.v. epineurally | 45 mg, 15 mg | n/a | NCT04202783 |
| MSCs | Depression | no | i.v. | 21 million cells | n/a | NCT04202770 |
| MSCs | Diabetes Mellitus Type 1 | no | i.v. | 1.2 × 1010 –1.51 × 1010 particles/kg | 2/3 | NCT02138331 |
| MSCs | Chronic Ulcer | no | topical | not reported | 1 | NCT04134676 |
| Plasma | Ulcer | no | topical | not reported | Early 1 | NCT02565264 |
| MSC | Dystrophic Epidermolysis Bullosa | no | topical | not reported | 1/2 | NCT04173650 |
| MSCs | Multiple Organ Failure | no | i.v. | 150 mg once a day for 14 times | n/a | NCT04356300 |
| MSCs | Healthy | no | inhalation | 2 × 108–20 × 108 particles/3 ml | 1 | NCT04313647 |
| Plant | Colon Cancer | curcumin | oral | not reported | 1 | NCT01294072 |
| Plant | Polycystic Ovary Syndrome | no | oral | not reported | n/a | NCT03493984 |
| Grape | Head and Neck Cancer Oral Mucositis | no | oral | not reported | 1 | NCT01668849 |
| DCs | Non Small Cell Lung Cancer | MHC class I- class II- cancer antigens | i.v. | 53–2422 μg protein/injection | 2 | NCT01159288 |
| MSCs | Pancreatic Adenocarcinoma | KRAS G12D siRNA | i.v. | days 1, 4, and 10 (dose not reported) | 1 | NCT03608631 |
| MSCs | SARS-CoV-2 pneumonia | no | inhalation | 2 × 108 particles/3 mL | 1 | NCT04276987 |
| MSCs | SARS-CoV-2 pneumonia | no | inhalation | 0.5 × 1010–2 × 1010 particles/3 ml | 1/2 | NCT04491240 |
| Bone marrow | SARS-CoV-2 pneumonia | no | i.v. | not reported | 2 | NCT04493242 |
| Human amniotic fluid | SARS-CoV-2 pneumonia | no | i.v. | 2 × 1010 –5 × 1010 particles | 1/2 | NCT04384445 |
| MSCs | Dry Eye | no | eye drop | 10 µg/drop | 1/2 | NCT04213248 |
| MSCs | Macular Holes | no | drop | 50 μg or 20 μg | early 1 | NCT03437759 |
| COVID-19 Specific T Cells | SARS-CoV-2 pneumonia | no | inhalation | 2 × 1018 particles/3 mL | 1 | NCT04389385 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klyachko, N.L.; Arzt, C.J.; Li, S.M.; Gololobova, O.A.; Batrakova, E.V. Extracellular Vesicle-Based Therapeutics: Preclinical and Clinical Investigations. Pharmaceutics 2020, 12, 1171. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12121171
Klyachko NL, Arzt CJ, Li SM, Gololobova OA, Batrakova EV. Extracellular Vesicle-Based Therapeutics: Preclinical and Clinical Investigations. Pharmaceutics. 2020; 12(12):1171. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12121171
Chicago/Turabian StyleKlyachko, Natalia L., Camryn J. Arzt, Samuel M. Li, Olesia A. Gololobova, and Elena V. Batrakova. 2020. "Extracellular Vesicle-Based Therapeutics: Preclinical and Clinical Investigations" Pharmaceutics 12, no. 12: 1171. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics12121171