Pulmonary Targeting of Inhalable Moxifloxacin Microspheres for Effective Management of Tuberculosis

 ,
,  ,
,  , , and
, , and
Abstract
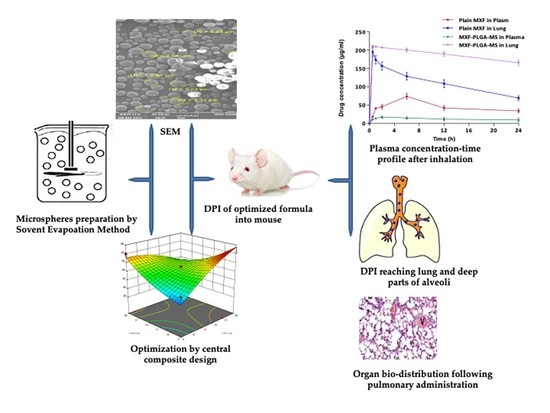
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Formulation and Optimization of Microspheres Loaded with MXF
2.2.1. Preparation of MXF-Loaded Microspores
2.2.2. Experimental Design
2.3. Structural Characterization of MXF-PLGA-MS
2.3.1. Drug Crystallinity Study
2.3.2. Thermal Analysis
2.3.3. Fourier Transform Infrared Spectroscopy (FTIR)
2.4. Evaluation of Prepared Microspheres
2.4.1. Particle Size
2.4.2. Particle Morphology
2.4.3. Drug Loading and Drug Entrapment Efficiency
HPLC Method for MXF Determination
2.4.4. Determination of Moisture Content
2.4.5. Flow Properties Tests
2.4.6. Determination of Aerodynamic Diameter
2.5. In Vitro Release Studies
2.6. Alamar Blue Dye for Testing Anti-TB Activity
2.7. In Vivo Studies
2.7.1. Animals
2.7.2. Plasma Pharmacokinetic Study of MXF-PLGA-MS
2.7.3. Lung Biodistribution Study
2.8. Stability Studies
2.9. Statistical Analysis
3. Results
3.1. Formulation of Moxifloxacin-Loaded PLGA Microspheres (MXF-PLGA-MS)
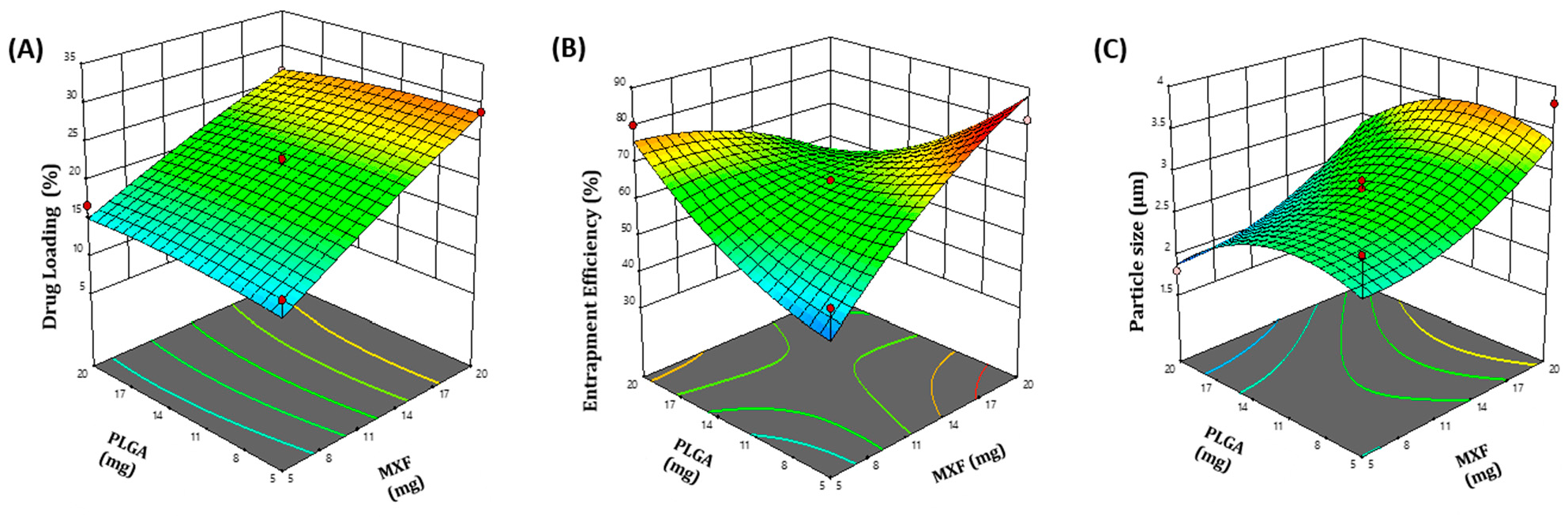
3.1.1. Central Composite Design Experiment and Response Surface Analysis
3.1.2. Optimization of MXC-PLGA-MS Formulation
3.2. Characterization of Optimized MXF-Loaded PLGA Microspheres as Dry Powder Inhalation
3.2.1. Particle Size
3.2.2. Particle Morphology
3.2.3. Moisture Content
3.2.4. Flow Properties
3.2.5. Aerodynamic Diameter
3.2.6. Drug–Polymer Interaction Studies
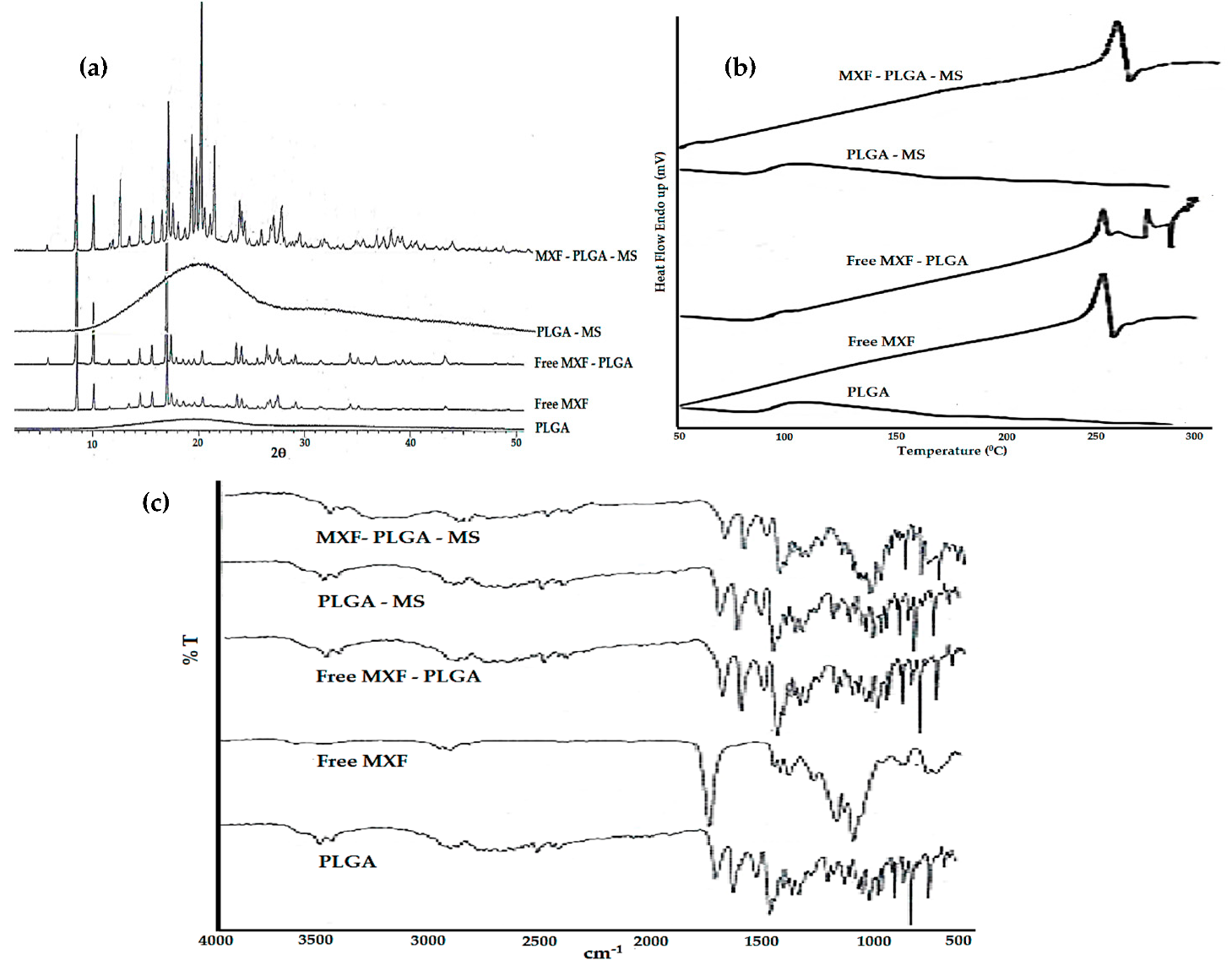
Crystallinity Study
Thermal Analysis
Fourier Transform Infrared Spectroscopy (FTIR)
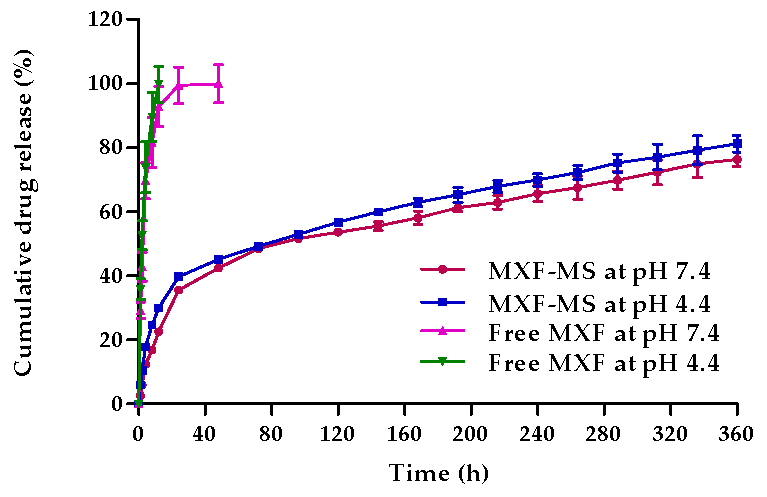
3.3. In Vitro Release Study
3.4. Anti-Tubercular Activity of Moxifloxacin
3.5. Stability Studies
3.6. In Vivo Studies
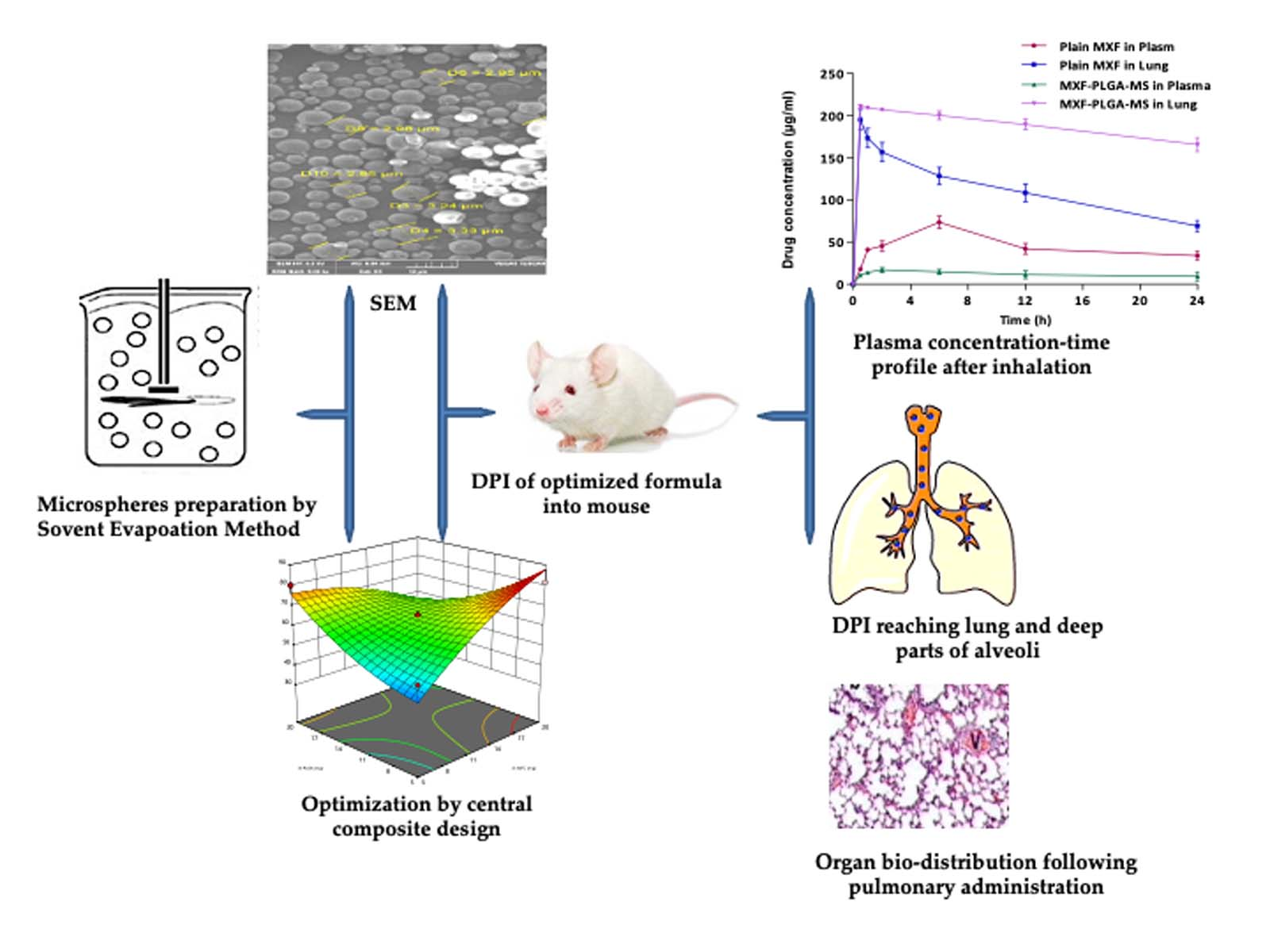
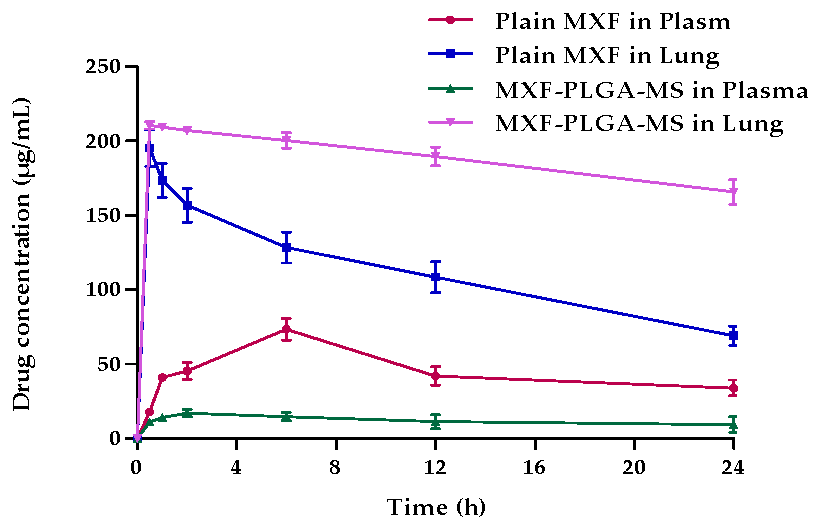
3.6.1. Pulmonary Pharmacokinetics
3.6.2. Bio-Distribution Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Mello, F.C.d.Q.; Silva, D.R.; Dalcolmo, M.P. Tuberculosis: Where are we? J. Bras. Pneumol. 2018, 44, 82. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2019. Available online: https://apps.who.int/iris/handle/10665/274453 (accessed on 13 July 2019).
- Lienhardt, C.; Vernon, A.; Raviglione, M.C. New drugs and new regimens for the treatment of tuberculosis: Review of the drug development pipeline and implications for national programmes. Curr. Opin. Pulm. Med. 2010, 16, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Lienhardt, C.; McIlleron, H.; Nunn, A.J.; Wang, X. Global tuberculosis drug development pipeline: The need and the reality. Lancet 2010, 375, 2100–2109. [Google Scholar] [CrossRef]
- Ouchi, Y.; Mukai, T.; Koide, K.; Yamaguchi, T.; Park, J.-H.; Kim, H.; Yokoyama, K.; Tamaru, A.; Gordon, S.V.; Nakajima, C.; et al. WQ-3810: A new fluoroquinolone with a high potential against fluoroquinolone-resistant Mycobacterium tuberculosis. Tuberculosis 2020, 120, 101891. [Google Scholar] [CrossRef] [PubMed]
- Türe, A.; Kulabaş, N.; Dingiş, S.İ.; Birgül, K.; Bozdeveci, A.; Alpay Karaoğlu, Ş.; Krishna, V.S.; Sriram, D.; Küçükgüzel, İ. Design, synthesis and molecular modeling studies on novel moxifloxacin derivatives as potential antibacterial and antituberculosis agents. Bioorg. Chem. 2019, 88, 102965. [Google Scholar] [CrossRef]
- Manika, K.; Chatzika, K.; Zarogoulidis, K.; Kioumis, I. Moxifloxacin in multidrug-resistant tuberculosis: Is there any indication for therapeutic drug monitoring? Eur. Resp. J. 2012, 40, 1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, S.H. The role of moxifloxacin in tuberculosis therapy. Eur. Resp. Review 2016, 25, 19. [Google Scholar] [CrossRef] [Green Version]
- Naidoo, A.; Naidoo, K.; McIlleron, H.; Essack, S.; Padayatchi, N. A Review of Moxifloxacin for the Treatment of Drug-Susceptible Tuberculosis. J. Clin. Pharmacol. 2017, 57, 1369–1386. [Google Scholar] [CrossRef]
- Deshpande, D.; Srivastava, S.; Nuermberger, E.; Pasipanodya, J.G.; Swaminathan, S.; Gumbo, T. A Faropenem, Linezolid, and Moxifloxacin Regimen for Both Drug-Susceptible and Multidrug-Resistant Tuberculosis in Children: FLAME Path on the Milky Way. Clin. Infect. Dis. 2016, 63, S95–S101. [Google Scholar] [CrossRef]
- Thakur, A.K.; Chellappan, D.K.; Dua, K.; Mehta, M.; Satija, S.; Singh, I. Patented therapeutic drug delivery strategies for targeting pulmonary diseases. Expert Opin. Ther. Pat. 2020, 30, 375–387. [Google Scholar] [CrossRef]
- Passi, M.; Shahid, S.; Chockalingam, S.; Sundar, I.K.; Packirisamy, G. Conventional and Nanotechnology Based Approaches to Combat Chronic Obstructive Pulmonary Disease: Implications for Chronic Airway Diseases. Int. J. Nanomed. 2020, 15, 3803–3826. [Google Scholar] [CrossRef] [PubMed]
- Emami, F.; Mostafavi Yazdi, S.J.; Na, D.H. Poly (lactic acid)/poly (lactic-co-glycolic acid) particulate carriers for pulmonary drug delivery. J. Pharm. Investig. 2019, 49, 427–442. [Google Scholar] [CrossRef] [Green Version]
- Lalan, M.; Tandel, H.; Lalani, R.; Patel, V.; Misra, A. Inhalation Drug Therapy: Emerging Trends in Nasal and Pulmonary Drug Delivery. In Novel Drug Delivery Technologies: Innovative Strategies for Drug Re-positioning; Misra, A., Shahiwala, A., Eds.; Springer: Singapore, 2019; pp. 291–333. [Google Scholar]
- Kaur, K.; Gupta, A.; Narang, R.K.; Murthy, R.S.R. Novel drug delivery systems: Desired feat for tuberculosis. J. Adv. Pharm. Technol. Res. 2010, 1, 145–163. [Google Scholar]
- Clemens, D.L.; Lee, B.-Y.; Xue, M.; Thomas, C.R.; Meng, H.; Ferris, D.; Nel, A.E.; Zink, J.I.; Horwitz, M.A. Targeted intracellular delivery of antituberculosis drugs to Mycobacterium tuberculosis-infected macrophages via functionalized mesoporous silica nanoparticles. Antimicrob. Agents Chemother. 2012, 56, 2535–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, A.; Baboota, S.; Sahni, J.K.; Ali, J. Exploring targeted pulmonary delivery for treatment of lung cancer. Int. J. Pharm. Investig. 2013, 3, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Praphakar, R.A.; Shakila, H.; Azger Dusthackeer, V.N.; Munusamy, M.A.; Kumar, S.; Rajan, M. A mannose-conjugated multi-layered polymeric nanocarrier system for controlled and targeted release on alveolar macrophages. Polym. Chem. 2018, 9, 656–667. [Google Scholar] [CrossRef]
- de Boer, A.H.; Hagedoorn, P.; Hoppentocht, M.; Buttini, F.; Grasmeijer, F.; Frijlink, H.W. Dry powder inhalation: Past, present and future. Expert Opin. Drug Deliv. 2017, 14, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Abdelaziz, H.M.; Gaber, M.; Abd-Elwakil, M.M.; Mabrouk, M.T.; Elgohary, M.M.; Kamel, N.M.; Kabary, D.M.; Freag, M.S.; Samaha, M.W.; Mortada, S.M.; et al. Inhalable particulate drug delivery systems for lung cancer therapy: Nanoparticles, microparticles, nanocomposites and nanoaggregates. J. Control. Release 2018, 269, 374–392. [Google Scholar] [CrossRef]
- Moustafa, I.O.F.; Ali, M.R.A.; Al Hallag, M.; Rabea, H.; Fink, J.B.; Dailey, P.; Abdelrahim, M.E.A. Lung deposition and systemic bioavailability of different aerosol devices with and without humidification in mechanically ventilated patients. Heart Lung 2017, 46, 464–467. [Google Scholar] [CrossRef]
- Mangal, S.; Nie, H.; Xu, R.; Guo, R.; Cavallaro, A.; Zemlyanov, D.; Zhou, Q.T. Physico-Chemical Properties, Aerosolization and Dissolution of Co-Spray Dried Azithromycin Particles with L-Leucine for Inhalation. Pharm. Res. 2018, 35, 28. [Google Scholar] [CrossRef] [Green Version]
- Hadiwinoto, G.D.; Lip Kwok, P.C.; Lakerveld, R. A review on recent technologies for the manufacture of pulmonary drugs. Ther. Deliv. 2018, 9, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Zellnitz, S.; Renner, N.; Cui, Y.; Scherließ, R.; Sommerfeld, M.; Steckel, H.; Urbanetz, N. The Importance of Interactions between Carrier and Drug Particles for the Application in Dry Powder Inhalers. In Particles in Contact: Micro Mechanics, Micro Process Dynamics and Particle Collective; Antonyuk, S., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 457–516. [Google Scholar]
- Mehta, P.; Bothiraja, C.; Kadam, S.; Pawar, A. Potential of dry powder inhalers for tuberculosis therapy: Facts, fidelity and future. Artif. Cells Nanomed. Biotechnol. 2018, 46, S791–S806. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Rashid, J.; Ahsan, F. Aerosolizable modified-release particles of montelukast improve retention and availability of the drug in the lungs. Eur. J. Pharm. Sci. 2017, 96, 560–570. [Google Scholar] [CrossRef] [PubMed]
- Allam, A.N.; Hamdalla, S.I.; Abdallah, O.Y. Chitosan-coated diacerein nanosuspensions as a platform for enhancing bioavailability and lowering side effects: Preparation, characterization, and ex vivo/in vivo evaluation. Int. J. Nanomed. 2017, 12, 4733–4745. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Choi, J.S.; Kim, I.; Oh, K.T.; Lee, E.S.; Park, E.-S.; Lee, K.C.; Youn, Y.S. Long-acting inhalable chitosan-coated poly(lactic-co-glycolic acid) nanoparticles containing hydrophobically modified exendin-4 for treating type 2 diabetes. Int. J. Nanomed. 2013, 8, 2975–2983. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Chen, L.; Li, H.; Deng, C.-L.; Chen, X.-F. Comparing Microspheres with Different Internal Phase of Polyelectrolyte as Local Drug Delivery System for Bone Tuberculosis Therapy. Biomed Res. Int. 2014, 2014, 297808. [Google Scholar] [CrossRef]
- Dua, K.; Rapalli, V.K.; Shukla, S.D.; Singhvi, G.; Shastri, M.D.; Chellappan, D.K.; Satija, S.; Mehta, M.; Gulati, M.; Pinto, T.D.J.A.; et al. Multi-drug resistant Mycobacterium tuberculosis & oxidative stress complexity: Emerging need for novel drug delivery approaches. Biomed. Pharmacother. 2018, 107, 1218–1229. [Google Scholar] [CrossRef]
- El-Sherbiny, I.M.; El-Baz, N.M.; Yacoub, M.H. Inhaled nano- and microparticles for drug delivery. Glob. Cardiol. Sci. Pract. 2015, 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Krasucka, D.M.; Kos, K.; Cybulski, W.A.; Mitura, A.; Łysiak, E.; Pietroń, W.J. Karl Fisher determination of residual moisture in veterinary vaccines—Practical implementation in market monitoring. Acta. Pol. Pharm. 2012, 69, 1364–1367. [Google Scholar]
- Beakawi Al-Hashemi, H.M.; Baghabra Al-Amoudi, O.S. A review on the angle of repose of granular materials. Powder Technol. 2018, 330, 397–417. [Google Scholar] [CrossRef]
- Sharma, A.; Vaghasiya, K.; Verma, R.K. Inhalable microspheres with hierarchical pore size for tuning the release of biotherapeutics in lungs. Micropor. Mesopor. Mat. 2016, 235, 195–203. [Google Scholar] [CrossRef]
- Simon, A.; Amaro, M.I.; Cabral, L.M.; Healy, A.M.; de Sousa, V.P. Development of a novel dry powder inhalation formulation for the delivery of rivastigmine hydrogen tartrate. Int. J. Pharm. 2016, 501, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, A.; Normandie, M.; Yousefi, M.; Saidi, M.S.; Ahmadi, G. Transport and deposition of pharmaceutical particles in three commercial spacer–MDI combinations. Comput. Biol. Med. 2014, 54, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wu, X.; Luo, J.; Fu, Y.; Zhao, L.; Ma, Y.; Li, Y.; Liang, Q.; Shang, Y.; Huang, H. Detection of pyrazinamide resistance of Mycobacterium tuberculosis using nicotinamide as a surrogate. Clin. Microbiol. Infect. 2017, 23, 835–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; Saxena, D.; Dwivedi, A.K.; Misra, A. Inhalable microparticles containing drug combinations to target alveolar macrophages for treatment of pulmonary tuberculosis. Pharm. Res. 2001, 18, 1405–1410. [Google Scholar] [CrossRef]
- Ghosh Dastidar, D.; Saha, S.; Chowdhury, M. Porous microspheres: Synthesis, characterisation and applications in pharmaceutical & medical fields. Int. J. Pharm. 2018, 548, 34–48. [Google Scholar] [CrossRef]
- Allam, A.N.; Komeil, I.A.; Abdallah, O.Y. Curcumin phytosomal softgel formulation: Development, optimization and physicochemical characterization. Acta Pharm. 2015, 65, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Makino, K.; Yamamoto, N.; Higuchi, K.; Harada, N.; Ohshima, H.; Terada, H. Phagocytic uptake of polystyrene microspheres by alveolar macrophages: Effects of the size and surface properties of the microspheres. Colloids Surf. B Biointerfaces 2003, 27, 33–39. [Google Scholar] [CrossRef]
- Champion, J.A.; Walker, A.; Mitragotri, S. Role of particle size in phagocytosis of polymeric microspheres. Pharm. Res. 2008, 25, 1815–1821. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.B.; Kang, J.H.; Han, C.S.; Kim, D.W.; Park, C.W. The Effect of Particle Size and Surface Roughness of Spray-Dried Bosentan Microparticles on Aerodynamic Performance for Dry Powder Inhalation. Pharmaceutics 2020, 12, 765. [Google Scholar] [CrossRef]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.-Y.; Chen, L.; Wu, C.-Y.; Chan, H.-K.; Freeman, T. The Effects of Relative Humidity on the Flowability and Dispersion Performance of Lactose Mixtures. Materials 2017, 10, 592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.J.; Lohade, A.A.; Parmar, J.J.; Hegde, D.D.; Soni, P.; Samad, A.; Menon, M.D. Development of Chitosan-based Dry Powder Inhalation System of Cisplatin for Lung Cancer. Indian J. Pharm. Sci. 2012, 74, 521–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdou, E.M.; Kandil, S.M.; Morsi, A.; Sleem, M.W. In-vitro and in-vivo respiratory deposition of a developed metered dose inhaler formulation of an anti-migraine drug. Drug Deliv. 2019, 26, 689–699. [Google Scholar] [CrossRef]
- Mahajan, H.S.; Gundare, S.A. Preparation, characterization and pulmonary pharmacokinetics of xyloglucan microspheres as dry powder inhalation. Carbohydr. Polym. 2014, 102, 529–536. [Google Scholar] [CrossRef]
- Yıldız-Peköz, A.; Ehrhardt, C. Advances in Pulmonary Drug Delivery. Pharmaceutics 2020, 12, 911. [Google Scholar] [CrossRef]
- Liang, W.; Pan, H.W.; Vllasaliu, D.; Lam, J.K.W. Pulmonary Delivery of Biological Drugs. Pharmaceutics 2020, 12, 1025. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formula | Coded Values of Independent Values | Actual Values of Independent Values | Response Variables | ||||
|---|---|---|---|---|---|---|---|
| X1 | X2 | Drug Amount (mg) | Polymer Content (mg) | R1 (%) | R2 (%) | R3 (μm) | |
| F1 | −1 | 1 | 5 | 20 | 16.80 ± 0.92 | 80.1 ± 1.57 | 1.8 ± 1.48 |
| F2 | 0 | 0 | 12.5 | 12.5 | 21.67 ± 2.08 | 65.4 ± 1.22 | 2.8 ± 1.82 |
| F3 | 0 | 0 | 12.5 | 12.5 | 22.89 ± 1.46 | 65.4 ± 1.87 | 2.7 ± 1.93 |
| F4 | −1.414 | 0 | 1.8934 | 12.5 | 9.50 ± 1.97 | 39.5 ± 1.49 | 2.4 ± 0.78 |
| F5 | 0 | 1.414 | 12.5 | 23.1066 | 19.98 ± 1.85 | 79.2 ± 1.59 | 1.9 ± 1.38 |
| F6 | 1 | −1 | 20 | 5 | 28.90 ± 1.76 | 81.5 ± 2.01 | 3.8 ± 1.46 |
| F7 | 0 | 0 | 12.5 | 12.5 | 21.98 ± 1.45 | 65.4 ± 0.86 | 2.9 ± 1.67 |
| F8 | 1 | 1 | 20 | 20 | 26.50 ± 0.99 | 45.8 ± 1.06 | 2.8 ± 1.85 |
| F9 | 0 | −1.414 | 12.5 | 1.8934 | 19.87 ± 1.73 | 74.8 ± 1.38 | 1.8 ± 1.67 |
| F10 | 0 | 0 | 12.5 | 12.5 | 21.50 ± 2.05 | 65.4 ± 1.58 | 2.9 ± 1.86 |
| F11 | −1 | −1 | 5 | 5 | 15.90 ± 1.09 | 53.2 ± 0.79 | 2.9 ± 0.88 |
| F12 | 0 | 0 | 12.5 | 12.5 | 22.90 ± 1.81 | 65.4 ± 1.76 | 2.7 ± 0.79 |
| F13 | 1.414 | 0 | 23.1066 | 12.5 | 31.09 ± 0.94 | 75.8 ± 2.01 | 3.8 ± 1.93 |
| Parameters | Pure MXF | MXF-PLGA-MS |
|---|---|---|
| Bulk Density | 0.23 ± 0.03 g/mL | 1.42 ± 0.11 g/mL |
| Tapped Density | 0.34 ± 0.03 g/mL | 1.72 ± 0.16 g/mL |
| Carr’s Index | 32.35 | 17.44 |
| Hausner’s ratio | 1.47 | 1.21 |
| Angle of Repose (θ) | 42 ± 3° | 29 ± 2° |
| Parameters | Pure MXF | MXF-PLGA-MS |
|---|---|---|
| Recovered dose (RD) | 99.14 ± 1.02 μg | 90.79 ± 3.41 μg |
| Emitted dose (ED) | 81.86 ± 2.11 μg | 82.81 ± 1.67 μg |
| Fine particles dose (FPD) | 50.9 ± 1.05 μg | 66.07 ± 1.48 μg |
| Fine particle friction (FPF (%)) | 51.34 ± 146 | 72.77 ± 1.73 |
| Mass median aerodynamic diameter (MMAD) | 4.85 ± 1.57 µm | 2.85 ± 1.04 µm |
| Geometric standard deviation (GSD) | 1.37 ± 1.98 | 3.10 ± 1.23 |
| Pharmacokinetic Parameters | Plasma | Lung | ||
|---|---|---|---|---|
| Plain MXF | MXF-PLGA-MS | Plain MXF | MXF-PLGA-MS | |
| Cmax (μg/mL) | 65.92 ± 4.32 | 16.28 ± 2.41 | 181.39 ± 31.56 | 207.39 ± 23.48 |
| Tmax (h) | 4.88 ± 0.42 | 2.17 ± 0.18 | 0.13 ± 0.01 | 0.18 ± 0.01 |
| AUC0–24h (μg/mL*h) | 1142.07 ± 101.23 | 292.71 ± 22.78 | 2686.68 ± 231.45 | 4767.57 ± 230.85 |
| MRT (h) | 23.25 ± 0.89 | 33.77 ± 1.11 | 22.42 ± 0.76 | 277.39 ± 12.21 |
| Formulation | Organ | % Dose Detected | |||||
|---|---|---|---|---|---|---|---|
| 30 min | 1 h | 2 h | 6 h | 12 h | 24 h | ||
| Plain MXF | Plasma | 7.1 ± 1.21 | 16.36 ± 2.32 | 18.15 ± 1.84 | 29.37 ± 1.92 | 16.82 ± 1.29 | 13.58 ± 1.29 |
| Lung | 78.1 ± 2.42 | 69.35 ± 1.45 | 62.69 ± 1.32 | 51.34 ± 1.24 | 43.38 ± 1.43 | 27.63 ± 1.48 | |
| Spleen | 1.32 ± 1.29 | 2.63 ± 1.54 | 4.65 ± 1.33 | 4.18 ± 2.23 | 3.54 ± 1.29 | 4.21 ± 1.59 | |
| Liver | 1.87 ± 1.63 | 5.35 ± 0.54 | 9.78 ± 0.87 | 5.29 ± 2.51 | 15.24 ± 2.15 | 7.8 ± 1.53 | |
| Kidney | ND * | 0.45 ± 0.72 | 1.25 ± 1.29 | 1.99 ± 2.54 | 1.89 ± 1.48 | 1.23 ± 1.59 | |
| MXF-PLGA-MS | Plasma | 4.39 ± 1.58 | 5.62 ± 1.21 | 6.83 ± 1.29 | 5.87 ± 1.79 | 4.53 ± 1.68 | 3.69 ± 1.25 |
| Lung | 84.21 ± 2.59 | 83.68 ± 1.11 | 82.82 ± 2.24 | 80.12 ± 1.25 | 79.39 ± 2.17 | 78.26 ± 2.19 | |
| Spleen | 0.45 ± 1.23 | 0.74 ± 0.32 | 0.94 ± 0.36 | 1.55 ± 0.42 | 0.65 ± 1.59 | 1.15 ± 0.46 | |
| Liver | 1.62 ± 1.41 | 2.25 ± 1.81 | 2.78 ± 0.23 | 3.02 ± 1.11 | 2.57 ± 2.01 | 2.27 ± 1.48 | |
| Kidney | ND * | 0.92 ± 1.68 | 1.93 ± 1.11 | 2.51 ± 1.63 | 2.99 ± 0.57 | 0.44 ± 1.01 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vishwa, B.; Moin, A.; Gowda, D.V.; Rizvi, S.M.D.; Hegazy, W.A.H.; Abu Lila, A.S.; Khafagy, E.-S.; Allam, A.N. Pulmonary Targeting of Inhalable Moxifloxacin Microspheres for Effective Management of Tuberculosis. Pharmaceutics 2021, 13, 79. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010079
Vishwa B, Moin A, Gowda DV, Rizvi SMD, Hegazy WAH, Abu Lila AS, Khafagy E-S, Allam AN. Pulmonary Targeting of Inhalable Moxifloxacin Microspheres for Effective Management of Tuberculosis. Pharmaceutics. 2021; 13(1):79. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010079
Chicago/Turabian StyleVishwa, Bhavya, Afrasim Moin, D. V. Gowda, Syed M. D. Rizvi, Wael A. H. Hegazy, Amr S. Abu Lila, El-Sayed Khafagy, and Ahmed N. Allam. 2021. "Pulmonary Targeting of Inhalable Moxifloxacin Microspheres for Effective Management of Tuberculosis" Pharmaceutics 13, no. 1: 79. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13010079