
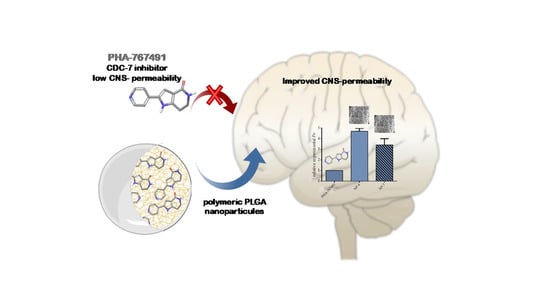
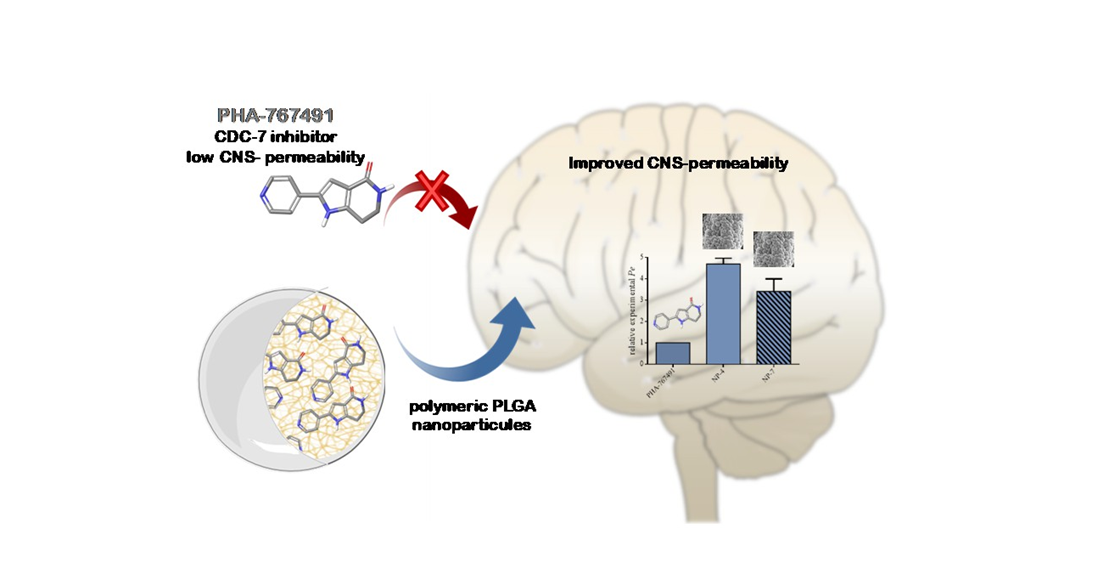
Increasing Brain Permeability of PHA-767491, a Cell Division Cycle 7 Kinase Inhibitor, with Biodegradable Polymeric Nanoparticles

 , , , ,
, , , , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
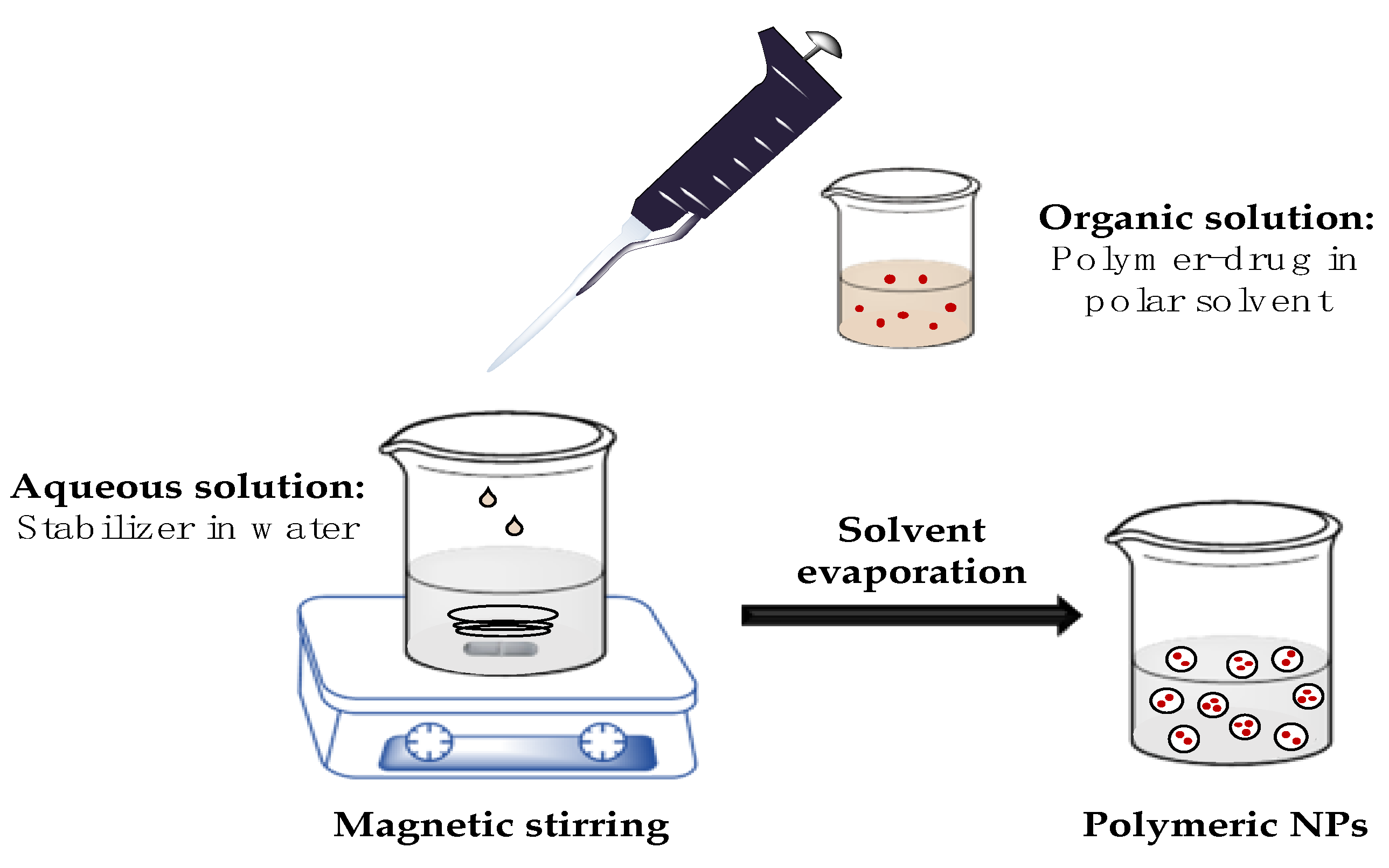
2.2.1. PLGA Nanoparticles Preparation
2.2.2. Characterization of PLGA Nanoparticles
Scanning Electron Microscopy (SEM)
Dynamic Light Scattering (DLS)
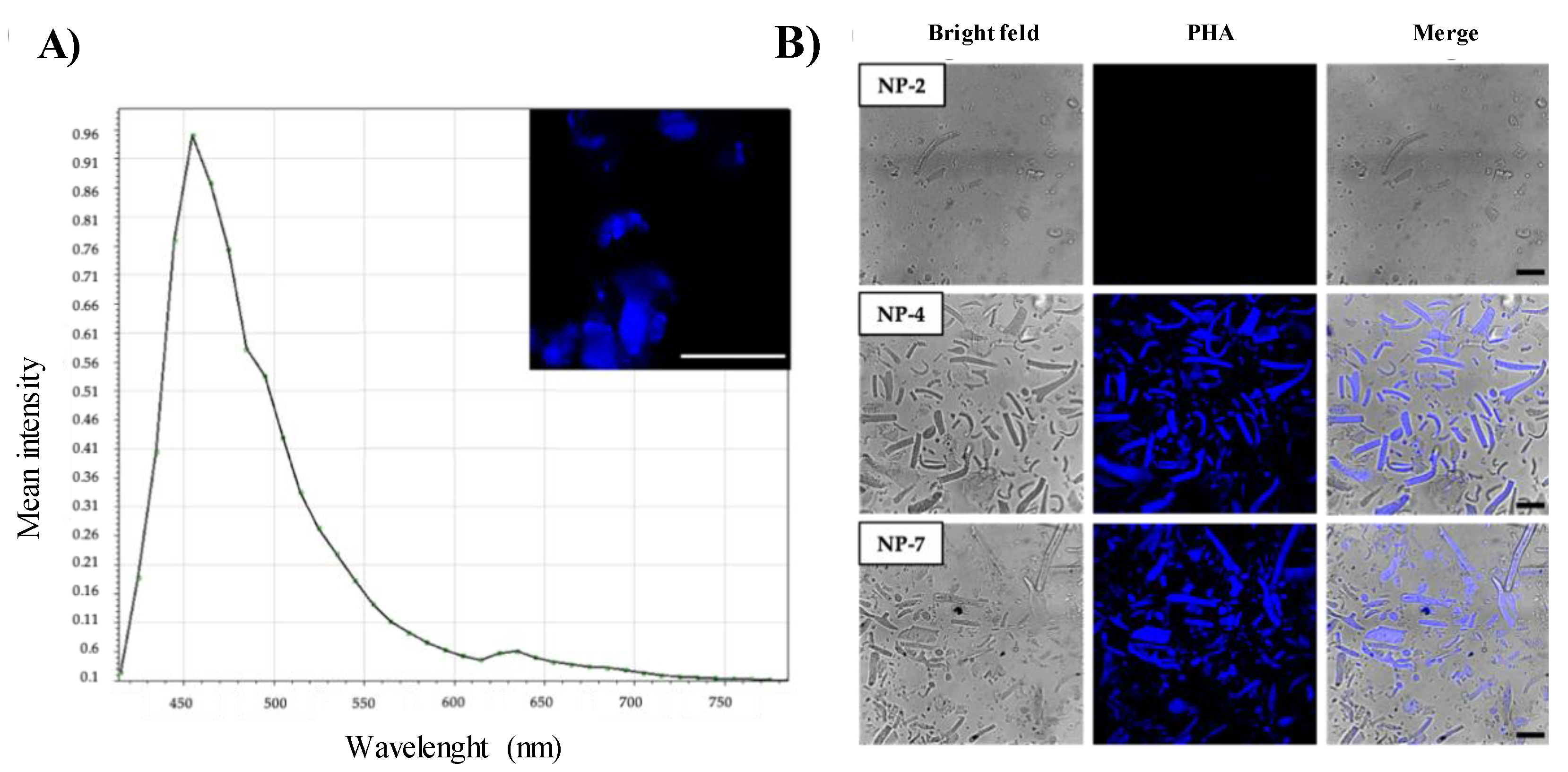
Confocal Laser Scanning Microscopy (CLSM)
Encapsulation Efficiency (EE%) and Loading Capacity (LC%)
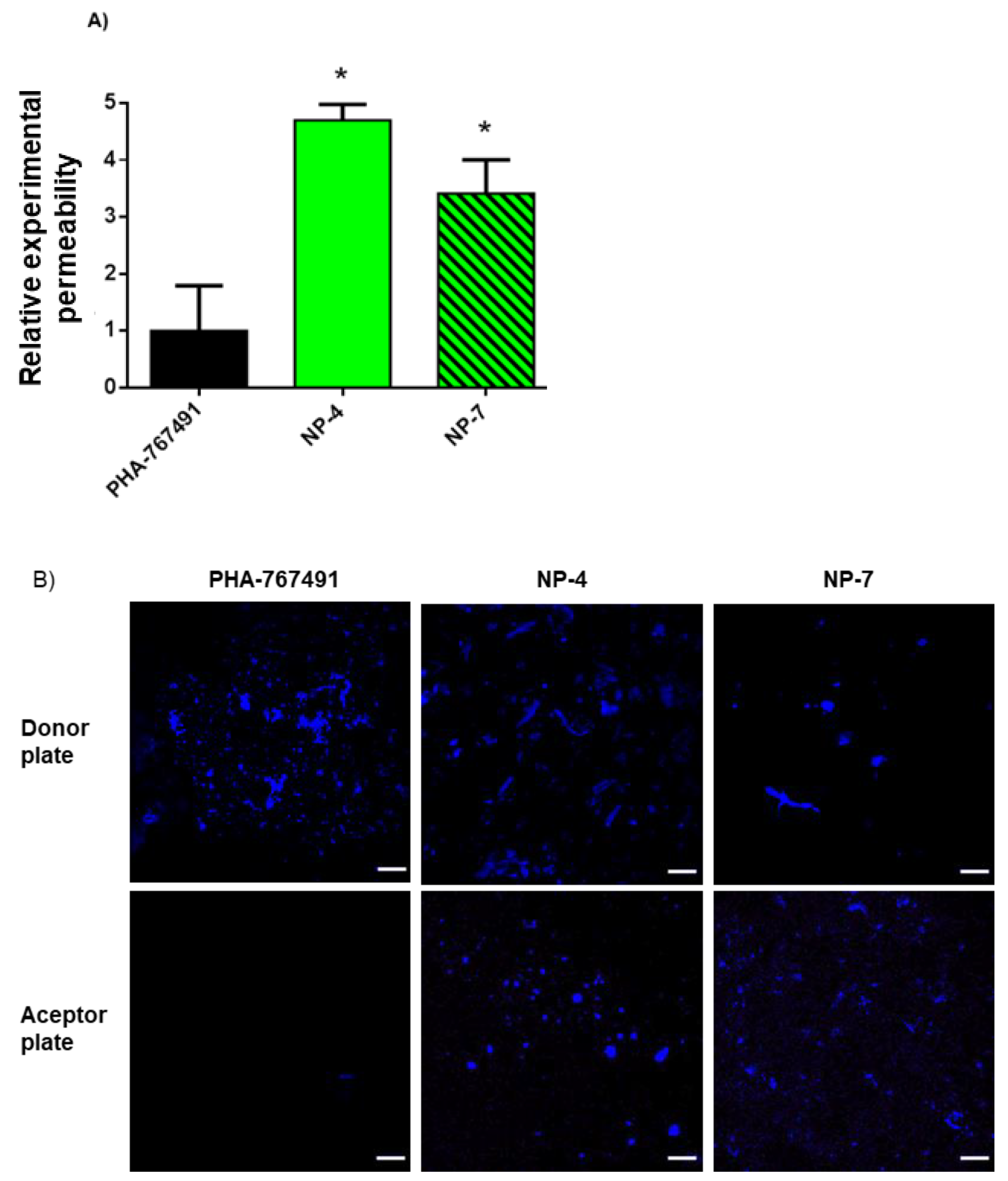
2.2.3. Permeability to the CNS
2.2.4. Neuronal Cell Culture
2.2.5. Immunofluorescence Analysis
3. Results and Discussion
3.1. Preparation of PLGA Nanoparticles
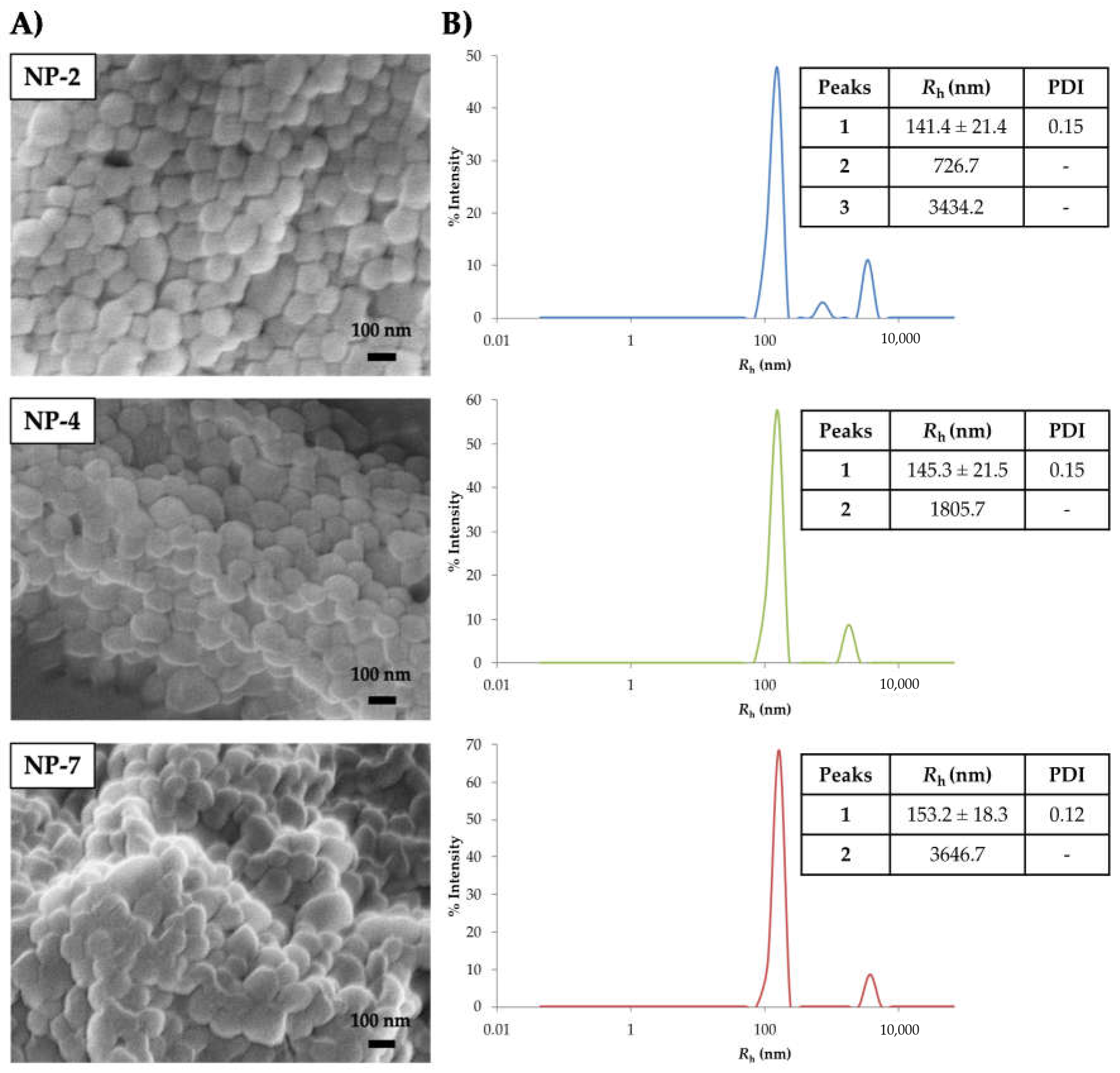
3.2. Characterization of PLGA Nanoparticles
3.2.1. Morphology and Size of Nanoparticles
3.2.2. Efficiency of Drug Encapsulation
3.3. Permeability to the CNS
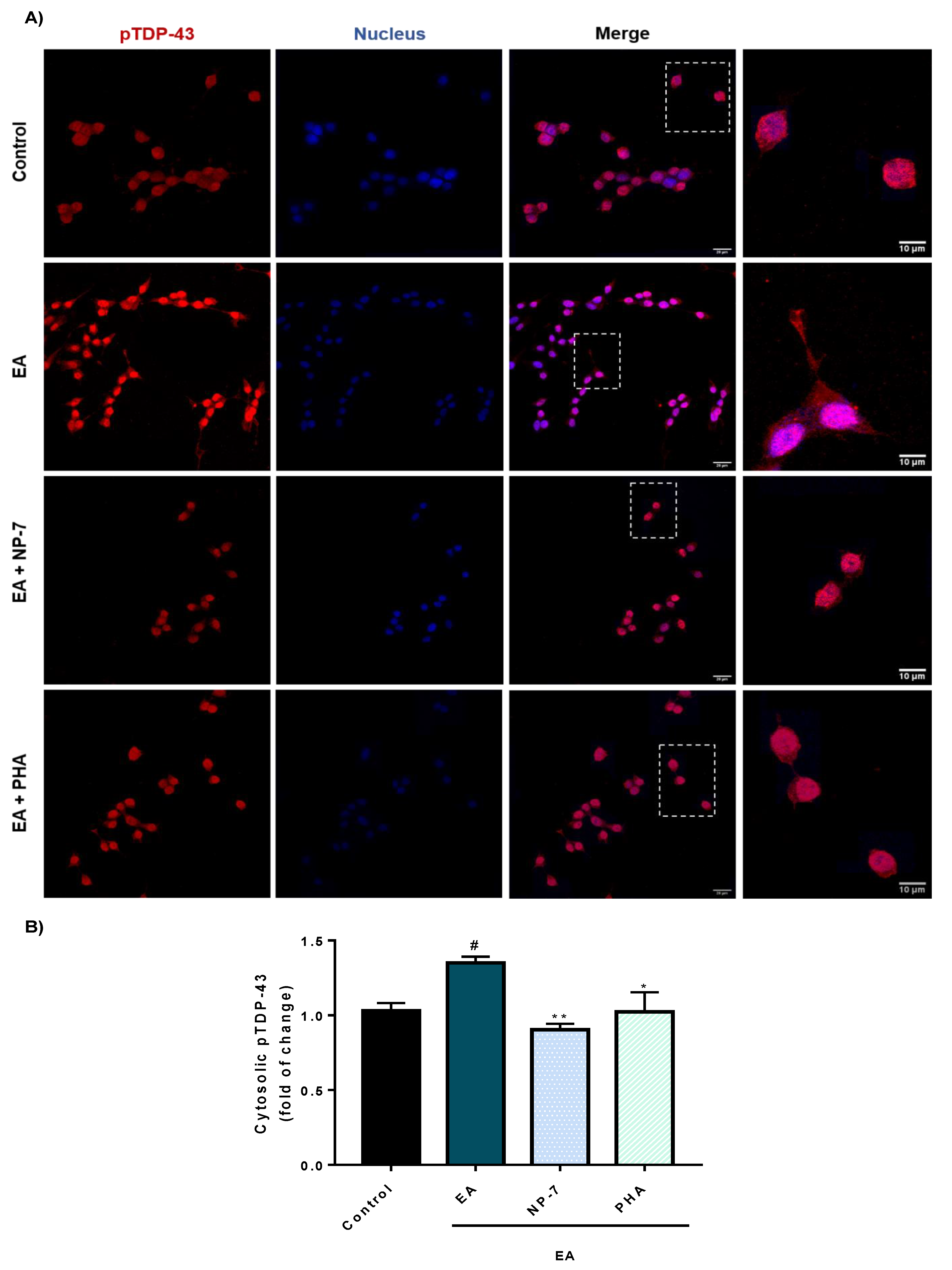
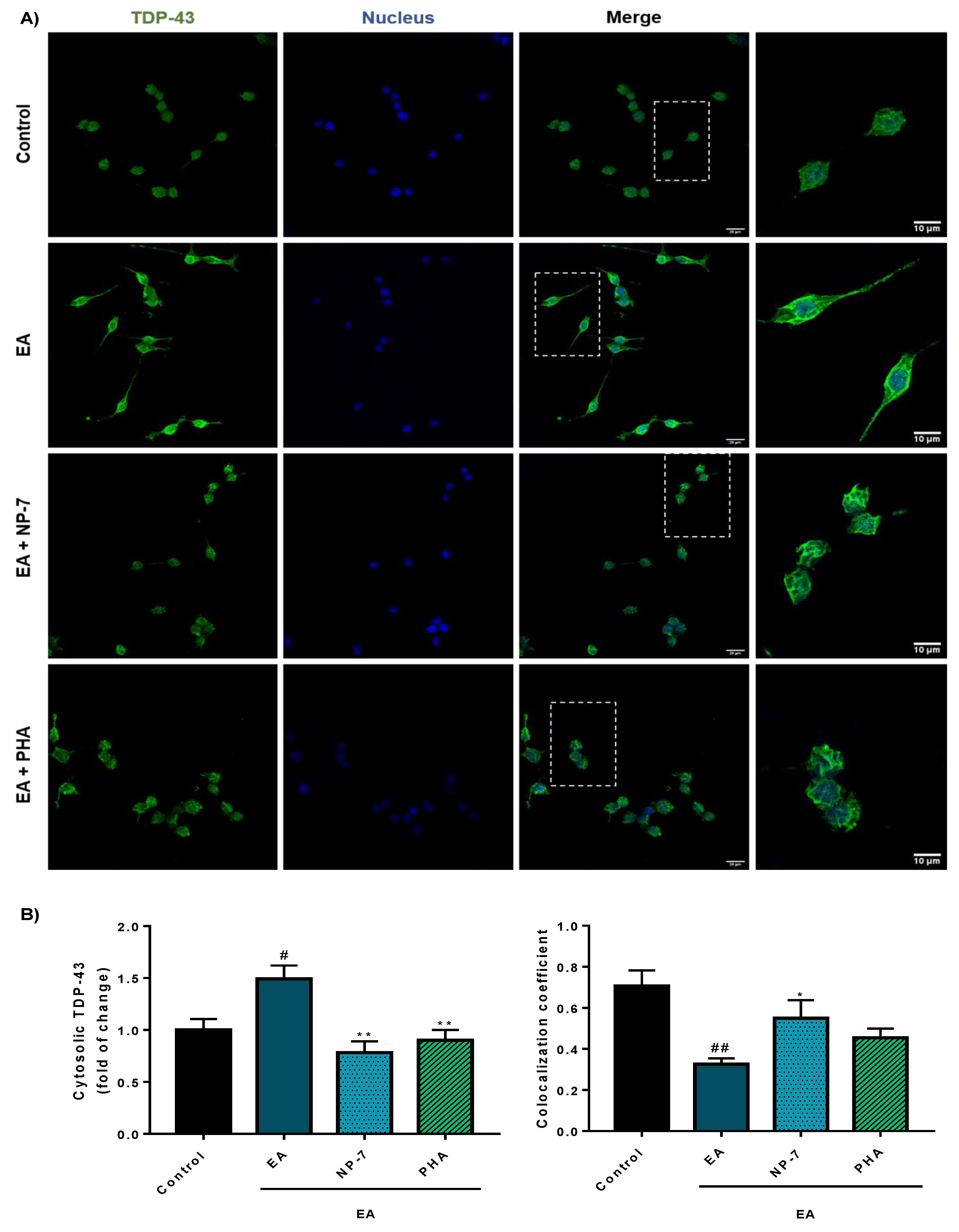
3.4. Neuroprotective Efficacy of PHA-Loaded PLGA Nanoparticles in a Cell Model of TDP-43 Phosphorylation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vanotti, E.; Amici, R.; Bargiotti, A.; Berthelsen, J.; Bosotti, R.; Ciavolella, A.; Cirla, A.; Cristiani, C.; D’Alessio, R.; Forte, B.; et al. Cdc7 kinase inhibitors: Pyrrolopyridinones as potential antitumor agents. 1. Synthesis and structure-activity relationships. J. Med. Chem. 2008, 51, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, A.; Valsasina, B.; Croci, V.; Menichincheri, M.; Rainoldi, S.; Marchesi, V.; Tibolla, M.; Tenca, P.; Brotherton, D.; Albanese, C.; et al. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat. Chem. Biol. 2008, 4, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Montagnoli, A.; Moll, J.; Colotta, F. Targeting cell division cycle 7 kinase: A new approach for cancer therapy. Clin. Cancer Res. 2010, 16, 4503–4508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liachko, N.F.; McMillan, P.J.; Guthrie, C.R.; Bird, T.D.; Leverenz, J.B.; Kraemer, B.C. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Ann. Neurol. 2013, 74, 39–52. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 2008, 13, 867–878. [Google Scholar]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Serlin, Y.; Shelef, I.; Knyazer, B.; Friedman, A. Anatomy and physiology of the blood-brain barrier. Semin. Cell Dev. Biol. 2015, 38, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar]
- Menichincheri, M.; Bargiotti, A.; Berthelsen, J.; Bertrand, J.A.; Bossi, R.; Ciavolella, A.; Cirla, A.; Cristiani, C.; Croci, V.; D’Alessio, R.; et al. First Cdc7 kinase inhibitors: Pyrrolopyridinones as potent and orally active antitumor agents. 2. Lead discovery. J. Med. Chem. 2009, 52, 293–307. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R.; Sriwastawa, B.; Bhati, L.; Pandey, S.; Pandey, P.; Bannerjee, S.K. Drug delivery systems: An updated review. Int. J. Pharm. Investig. 2012, 2, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamundeeswari, M.; Jeslin, J.; Verma, M.L. Nanocarriers for drug delivery applications. Environ. Chem. Lett. 2019, 17, 849–865. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. J. Formul. Manag. P T 2017, 42, 742–755. [Google Scholar]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, A.; Yadav, S.K.; Yadav, S.C. Biodegradable polymeric nanoparticles based drug delivery systems. Colloids Surf. B Biointerfaces 2010, 75, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Kreuter, J. Drug delivery to the central nervous system by polymeric nanoparticles: What do we know? Adv. Drug Deliv. Rev. 2014, 71, 2–14. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Préat, V. PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Fessi, H.; Puisieux, F.; Devissaguet, J.P.; Ammoury, N.; Benita, S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int. J. Pharm. 1989, 55, R1–R4. [Google Scholar] [CrossRef]
- Amini, Y.; Amel Jamehdar, S.; Sadri, K.; Zare, S.; Musavi, D.; Tafaghodi, M. Different methods to determine the encapsulation efficiency of protein in PLGA nanoparticles. Biomed. Mater. Eng. 2017, 28, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, G.T. High throughput artificial membrane permeability assay for blood-brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease. Int. J. Mol. Sci. 2019, 20, 204. [Google Scholar]
- Crucho, C.I.C.; Barros, M.T. Polymeric nanoparticles: A study on the preparation variables and characterization methods. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 80, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Dinarvand, R.; Sepehri, N.; Manoochehri, S.; Rouhani, H.; Atyabi, F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int. J. Nanomed. 2011, 6, 877–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Sela, E.; Teitlboim, S.; Chorny, M.; Koroukhov, N.; Danenberg, H.D.; Gao, J.; Golomb, G. Single and double emulsion manufacturing techniques of an amphiphilic drug in PLGA nanoparticles: Formulations of mithramycin and bioactivity. J. Pharm. Sci. 2009, 98, 1452–1462. [Google Scholar] [CrossRef]
- Ahmadi, F.; Derakhshandeh, K.; Jalalizadeh, A.; Mostafaie, A.; Hosseinzadeh, L. Encapsulation in PLGA-PEG enhances 9- nitro-camptothecin cytotoxicity to human ovarian carcinoma cell line through apoptosis pathway. Res. Pharm. Sci. 2015, 10, 161–168. [Google Scholar] [PubMed]
- Budhian, A.; Siegel, S.J.; Winey, K.I. Haloperidol-loaded PLGA nanoparticles: Systematic study of particle size and drug content. Int. J. Pharm. 2007, 336, 367–375. [Google Scholar] [CrossRef]
- Iqbal, M.; Zafar, N.; Fessi, H.; Elaissari, A. Double emulsion solvent evaporation techniques used for drug encapsulation. Int. J. Pharm. 2015, 496, 173–190. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, Z.L. Scanning Microscopy for Nanotechnology; Springer: New York, NY, USA, 2007. [Google Scholar]
- Pecora, R. Dynamic Light Scattering Measurement of Nanometer Particles in Liquids. J. Nanopart. Res. 2000, 2, 123–131. [Google Scholar] [CrossRef]
- Reddy, M.K.; Labhasetwar, V. Nanoparticle-mediated delivery of superoxide dismutase to the brain: An effective strategy to reduce ischemia-reperfusion injury. FASEB J. 2009, 23, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Pygall, S.R.; Whetstone, J.; Timmins, P.; Melia, C.D. Pharmaceutical applications of confocal laser scanning microscopy: The physical characterisation of pharmaceutical systems. Adv. Drug Deliv. Rev. 2007, 59, 1434–1452. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, H.; Kalashnikova, I.; White, M.A.; Sherman, M.; Rytting, E. Preparation, characterization, and transport of dexamethasone-loaded polymeric nanoparticles across a human placental in vitro model. Int. J. Pharm. 2013, 454, 149–157. [Google Scholar] [CrossRef] [Green Version]
- Cui, Y.N.; Xu, Q.X.; Davoodi, P.; Wang, D.P.; Wang, C.H. Enhanced intracellular delivery and controlled drug release of magnetic PLGA nanoparticles modified with transferrin. Acta. Pharmacol. Sin. 2017, 38, 943–953. [Google Scholar] [CrossRef] [Green Version]
- Pérez, D.I.; Pistolozzi, M.; Palomo, V.; Redondo, M.; Fortugno, C.; Gil, C.; Felix, G.; Martinez, A.; Bertucci, C. 5-Imino-1,2-4-thiadiazoles and quinazolines derivatives as glycogen synthase kinase 3β (GSK-3β) and phosphodiesterase 7 (PDE7) inhibitors: Determination of blood-brain barrier penetration and binding to human serum albumin. Eur. J. Pharm. Sci. 2012, 45, 677–684. [Google Scholar] [CrossRef]
- Nkabinde, L.A.; Shoba-Zikhali, L.N.; Semete-Makokotlela, B.; Kalombo, L.; Swai, H.S.; Hayeshi, R.; Naicker, B.; Hillie, T.K.; Hamman, J.H. Permeation of PLGA nanoparticles across different in vitro models. Curr. Drug Deliv. 2012, 9, 617–627. [Google Scholar] [CrossRef]
- Crivori, P.; Cruciani, G.; Carrupt, P.A.; Testa, B. Predicting blood-brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 2000, 43, 2204–2216. [Google Scholar] [CrossRef]
- Rojas-Prats, E.; Martinez-Gonzalez, L.; Gonzalo-Consuegra, C.; Liachko, N.F.; Perez, C.; Ramírez, D.; Kraemer, B.C.; Martin-Requero, Á.; Perez, D.I.; Gil, C.; et al. Targeting nuclear protein TDP-43 by cell division cycle kinase 7 inhibitors: A new therapeutic approach for amyotrophic lateral sclerosis. Eur. J. Med. Chem. 2020, 112968. [Google Scholar]
- Alquezar, C.; Salado, I.G.; de la Encarnación, A.; Pérez, D.I.; Moreno, F.; Gil, C.; de Munain, A.L.; Martínez, A.; Martín-Requero, Á. Targeting TDP-43 phosphorylation by Casein Kinase-1δ inhibitors: A novel strategy for the treatment of frontotemporal dementia. Mol. Neurodegener 2016, 11, 36. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
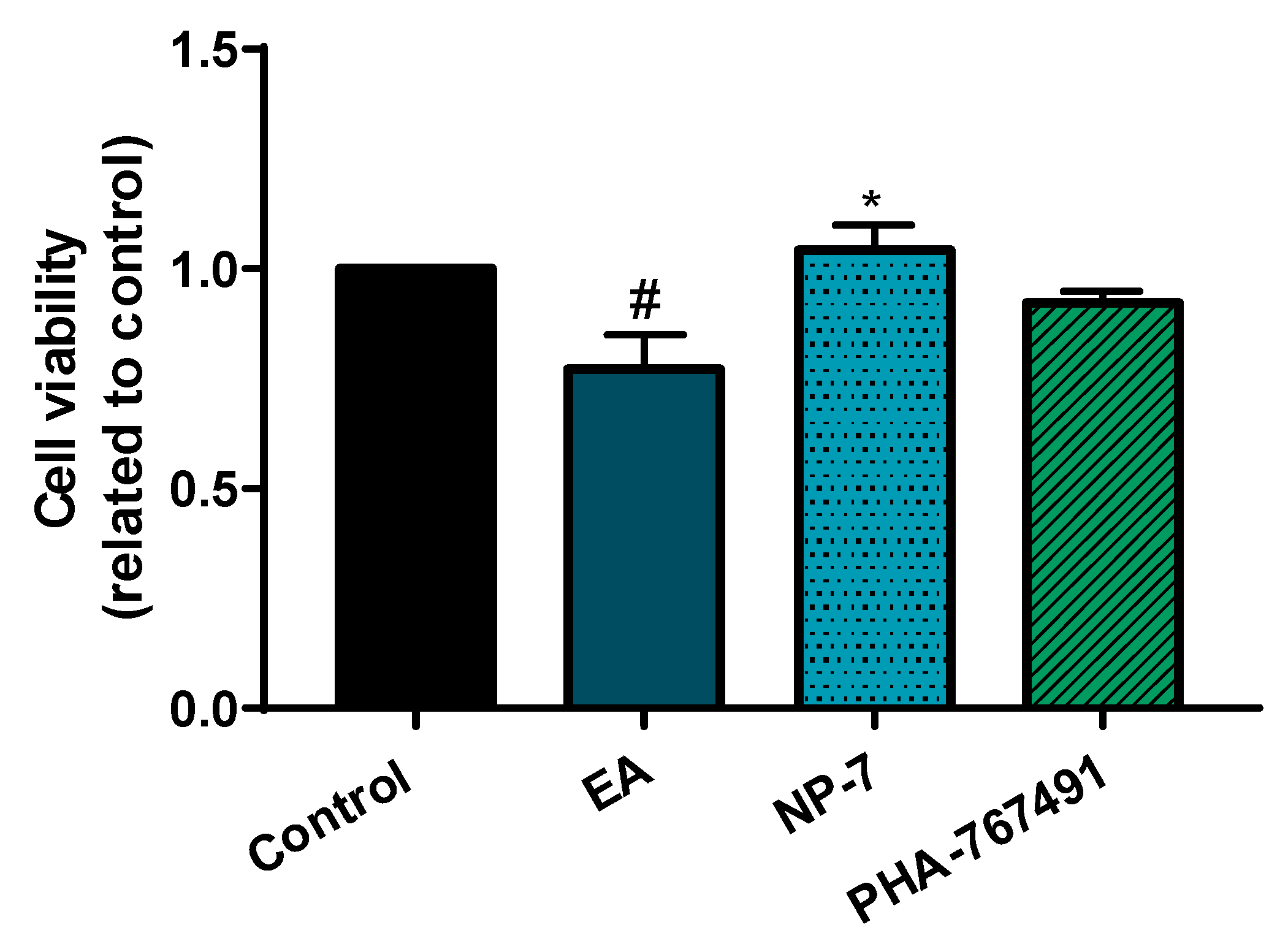{kind=link}
{kind=link}
{kind=link}
| Formulations | Raw Materials | Final NPs (mg) | ||
|---|---|---|---|---|
| PHA (mg) | PLGA (mg) | PVA (mg) | ||
| NP-1 | - | 51.4 | 400.0 | 31.0 |
| NP-2 | - | 52.4 | 400.5 | 32.6 |
| NP-3 | - | 52.5 | 401.5 | 24.4 |
| NP-4 | 5.0 | 50.2 | 400.1 | 32.8 |
| NP-5 | 5.6 | 51.7 | 403.6 | 42.4 |
| NP-6 | 10.4 | 51.5 | 401.6 | 51.8 |
| NP-7 | 10.2 | 50.5 | 402.4 | 41.1 |
| NP-8 | 10.1 | 51.0 | 401.5 | 46.8 |
| Formulations | PHAtotal (mg) | Total NPs (mg) | PHAentrapped (mg) | EE% | LC% |
|---|---|---|---|---|---|
| NP-4 | 5.0 | 32.8 | 0.7 | 14 | 2 |
| NP-5 | 5.6 | 42.4 | 0.8 | 14 | 2 |
| NP-6 | 10.4 | 51.8 | 1.6 | 15 | 3 |
| NP-7 | 10.2 | 41.1 | 1.2 | 12 | 3 |
| NP-8 | 10.1 | 46.8 | 1.9 | 18 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas-Prats, E.; Tosat-Bitrián, C.; Martínez-González, L.; Nozal, V.; Pérez, D.I.; Martínez, A. Increasing Brain Permeability of PHA-767491, a Cell Division Cycle 7 Kinase Inhibitor, with Biodegradable Polymeric Nanoparticles. Pharmaceutics 2021, 13, 180. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13020180
Rojas-Prats E, Tosat-Bitrián C, Martínez-González L, Nozal V, Pérez DI, Martínez A. Increasing Brain Permeability of PHA-767491, a Cell Division Cycle 7 Kinase Inhibitor, with Biodegradable Polymeric Nanoparticles. Pharmaceutics. 2021; 13(2):180. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13020180
Chicago/Turabian StyleRojas-Prats, Elisa, Carlota Tosat-Bitrián, Loreto Martínez-González, Vanesa Nozal, Daniel I. Pérez, and Ana Martínez. 2021. "Increasing Brain Permeability of PHA-767491, a Cell Division Cycle 7 Kinase Inhibitor, with Biodegradable Polymeric Nanoparticles" Pharmaceutics 13, no. 2: 180. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13020180