
Application of Deep Neural Networks as a Prescreening Tool to Assign Individualized Absorption Models in Pharmacokinetic Analysis


Abstract
:1. Introduction
2. Methods
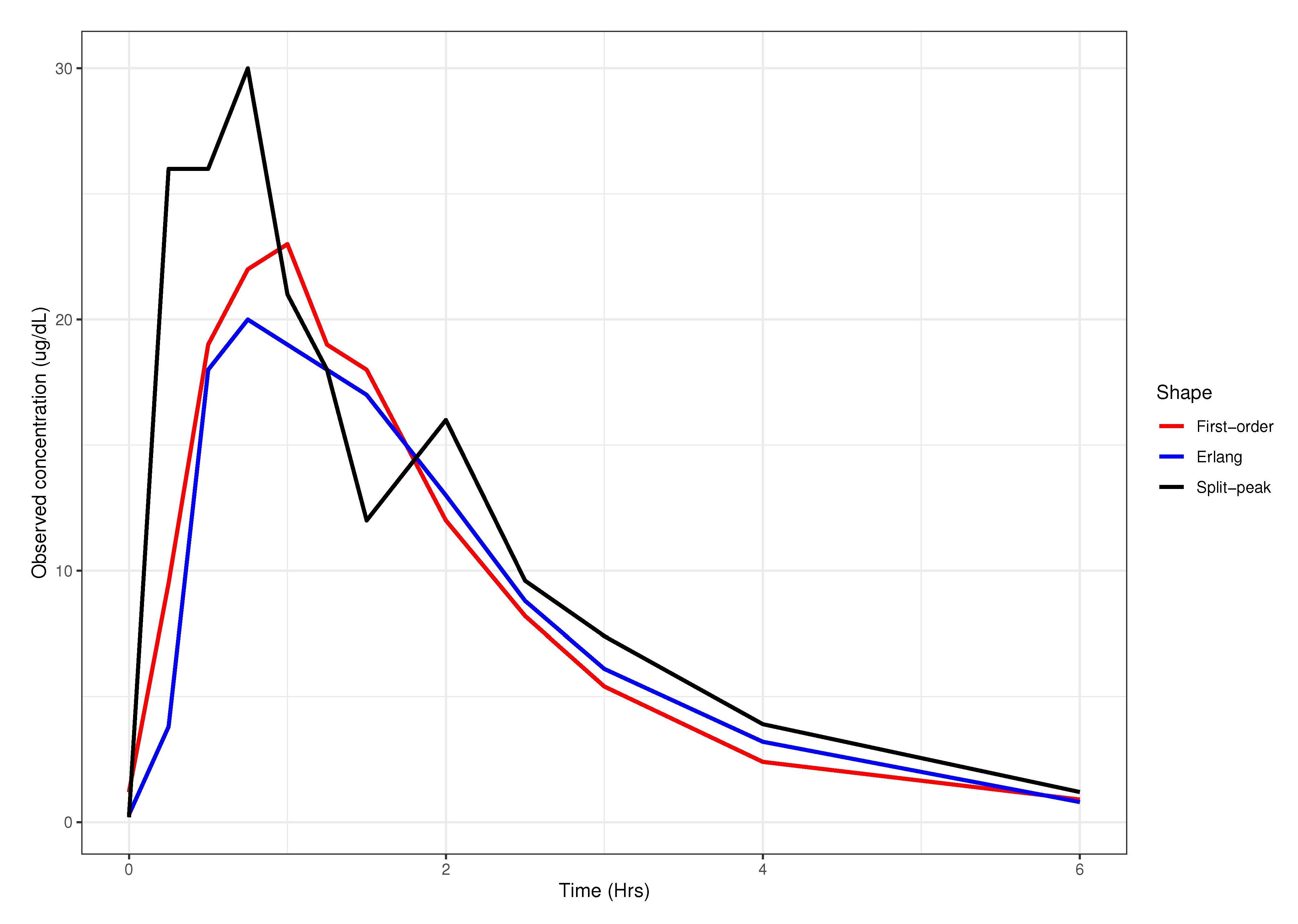
2.1. Observed Data and Visual Assignment
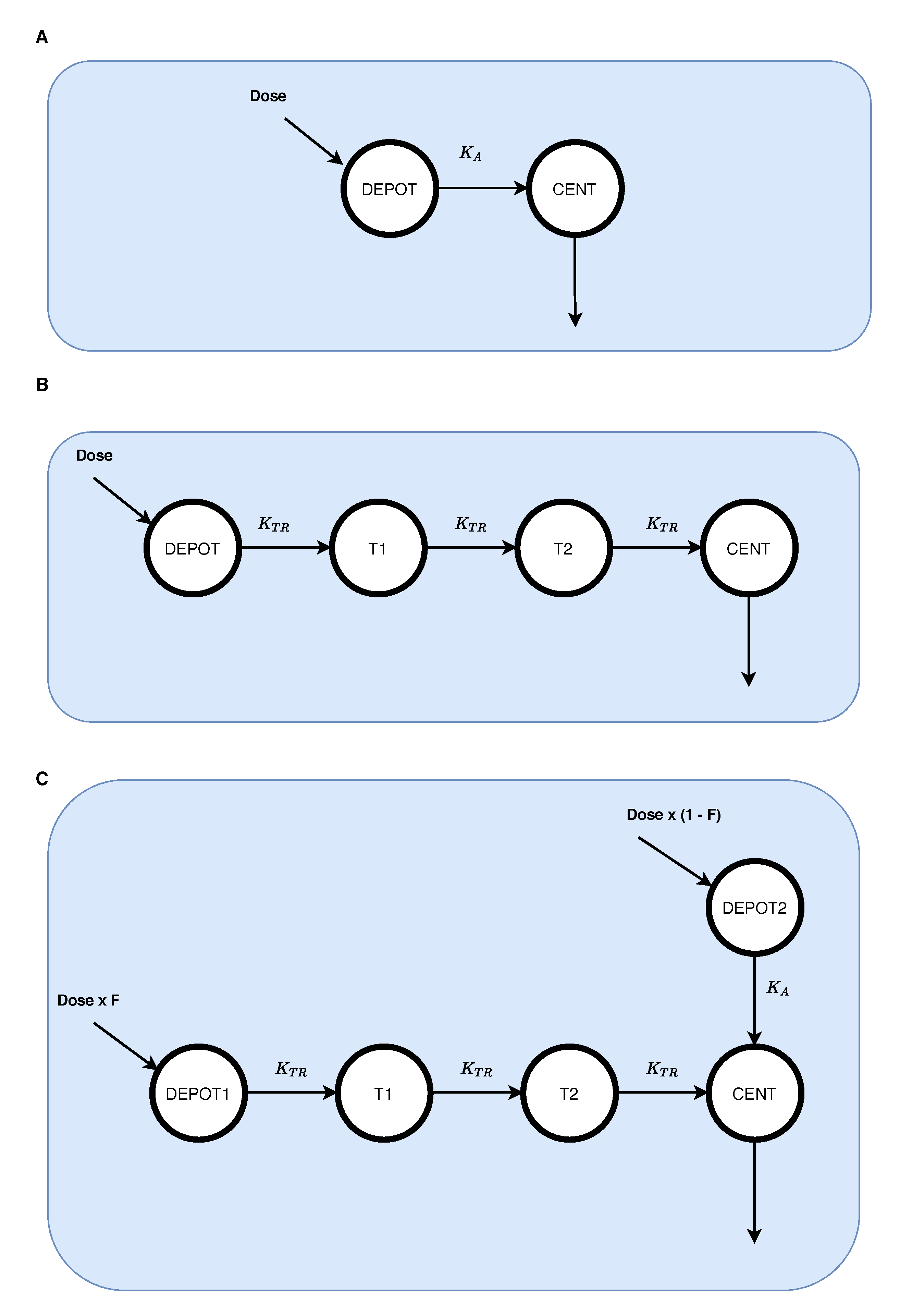
2.2. Estimation of Pharmacokinetic Model
2.3. Simulation of Training Profiles
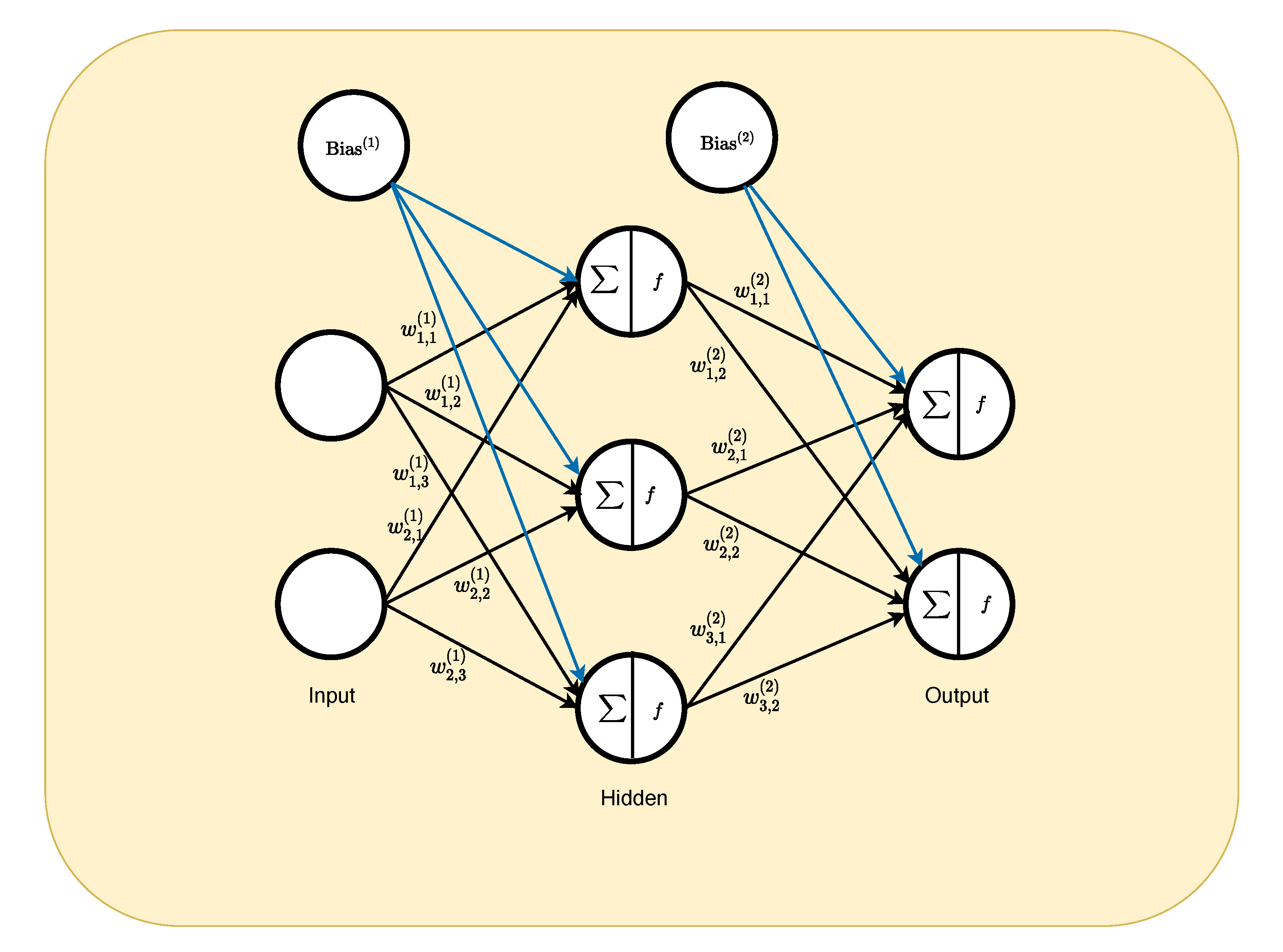
2.4. Deep Learning Algorithm
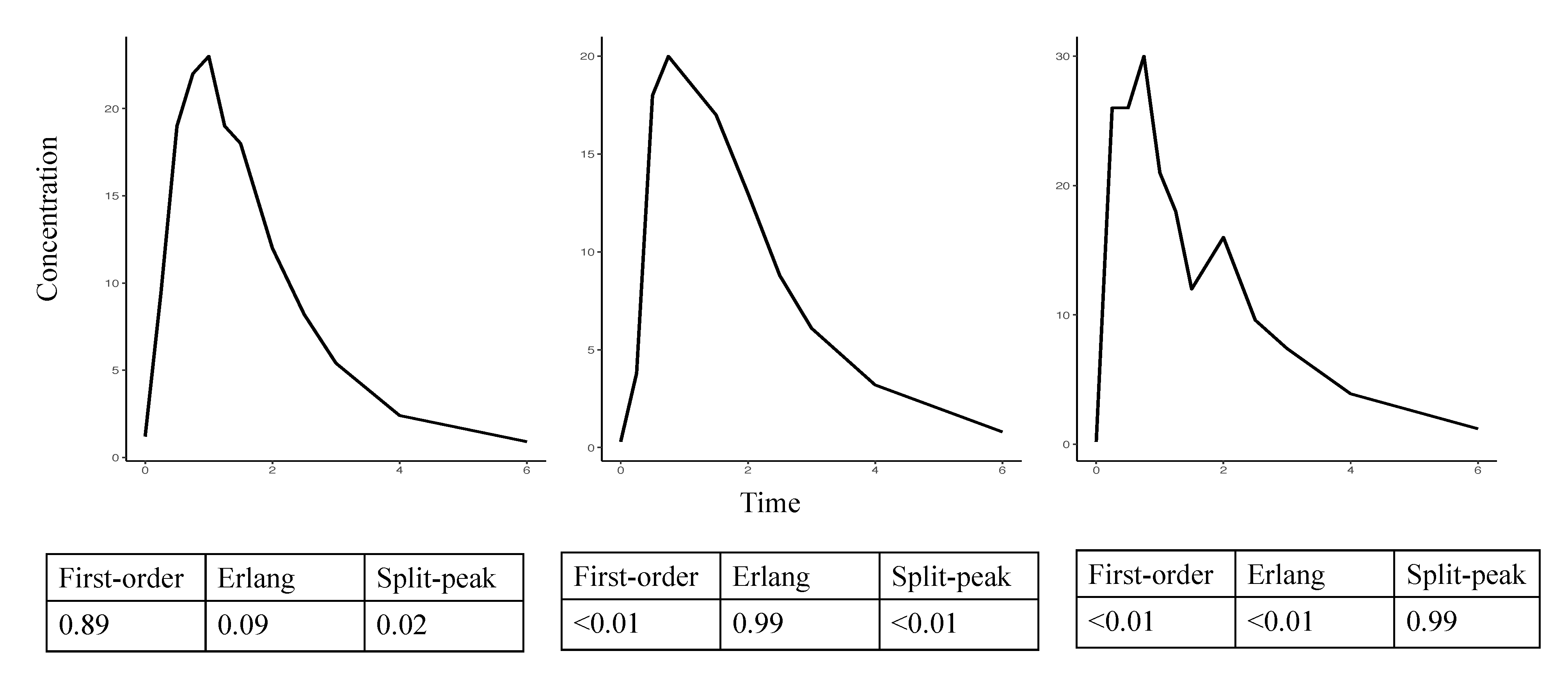
2.5. Evaluation
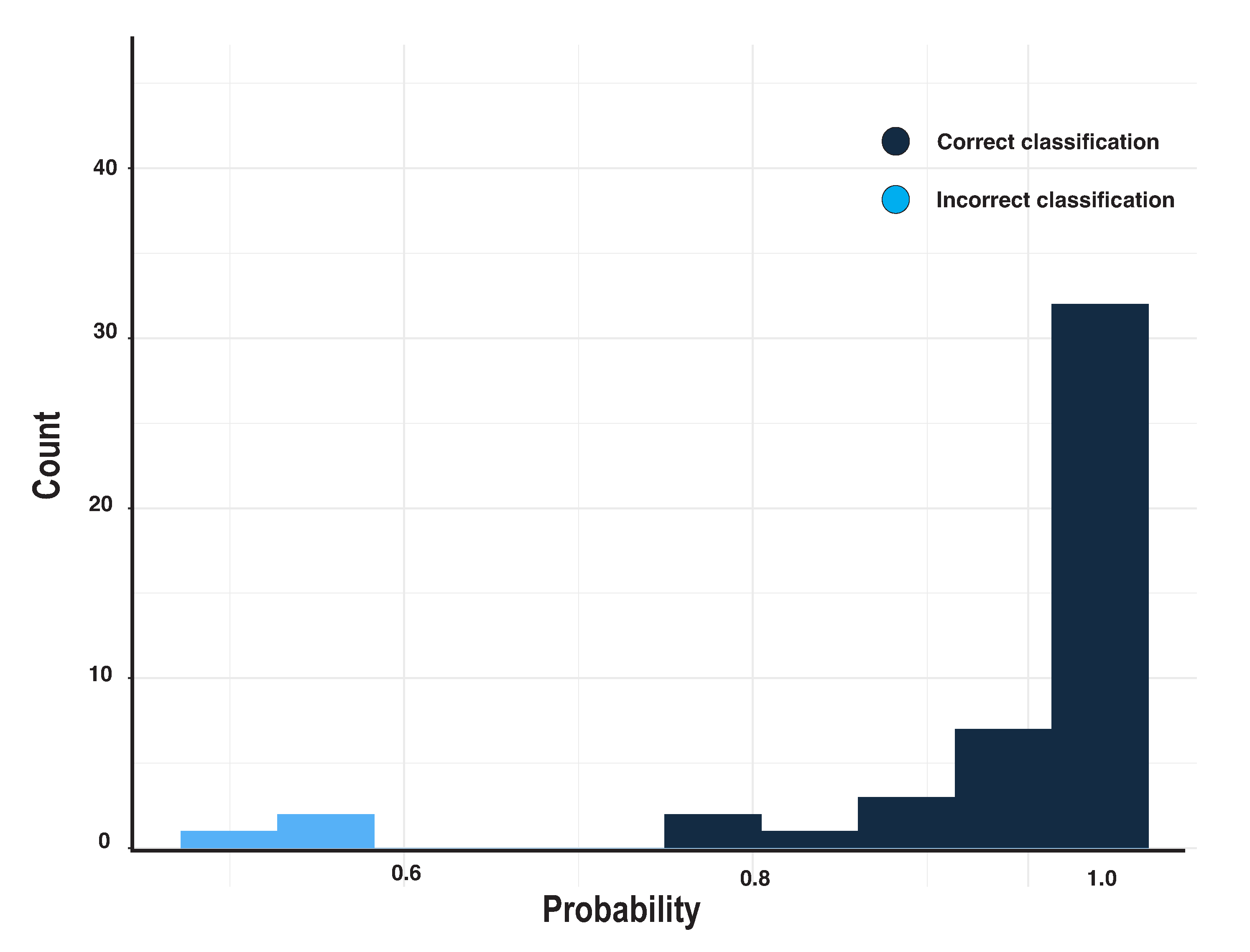
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wade, J.R.; Kelman, A.W.; Howie, C.A.; Whiting, B. Effect of misspecification of the absorption process on subsequent parameter estimation in population analysis. J. Pharmacokinet. Biopharm. 1993, 21, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Hénin, E.; Bergstrand, M.; Standing, J.F.; Karlsson, M.O. A mechanism-based approach for absorption modeling: The gastro-intestinal transit time (GITT) model. AAPS J. 2012, 14, 155–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaila, N.; Straka, R.J.; Brundage, R.C. Mixture models and subpopulation classification: A pharmacokinetic simulation study and application to metoprolol CYP2D6 phenotype. J. Pharmacokinet. Pharmacodyn. 2007, 34, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.P. Machine Learning: A Probabilistic Perspective; MIT Press: Cambridge, MA, USA, 2012. [Google Scholar]
- Jones, W.; Alasoo, K.; Fishman, D.; Parts, L. Computational biology: Deep learning. Emerg. Top. Life Sci. 2017, 1, 257–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angermueller, C.; Pärnamaa, T.; Parts, L.; Stegle, O. Deep learning for computational biology. Mol. Syst. Biol. 2016, 12, 878. [Google Scholar] [CrossRef] [PubMed]
- McComb, M.; Bies, R.; Ramanathan, M. Machine learning in pharmacometrics: Opportunities and challenges. Br. J. Clin. Pharmacol. 2021, 1–18. [Google Scholar] [CrossRef]
- Gong, X.; Hu, M.; Zhao, L. Big Data Toolsets to Pharmacometrics: Application of Machine Learning for Time-to-Event Analysis. Clin. Transl. Sci. 2018, 11, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Liu, Q.; Feng, G.; Wang, Y.; Liu, C.; Gopalakrishnan, M.; Liu, X.; Gong, Y.; Zhu, H. A novel approach for personalized response model: Deep learning with individual dropout feature ranking. J. Pharmacokinet. Pharmacodyn. 2020, 48, 165–179. [Google Scholar] [CrossRef]
- Saleh, M.I.; Alzubiedi, S. Dosage individualization of warfarin using artificial neural networks. Mol. Diagn. Ther. 2014, 18, 371–379. [Google Scholar] [CrossRef]
- Derbalah, A.; Al-Sallami, H.S.; Duffull, S.B. Reduction of quantitative systems pharmacology models using artificial neural networks. J. Pharmacokinet. Pharmacodyn. 2021. [Google Scholar] [CrossRef] [PubMed]
- Al-Kofahi, M.; Ahmed, M.A.; Jaber, M.M.; Tran, T.N.; Willis, B.A.; Zimmerman, C.L.; Gonzalez-Bolanos, M.T.; Brundage, R.C.; Sarafoglou, K. An integrated PK-PD model for cortisol and the 17-hydroxyprogesterone and androstenedione biomarkers in children with congenital adrenal hyperplasia. Br. J. Clin. Pharmacol. 2021, 87, 1098–1110. [Google Scholar] [CrossRef] [PubMed]
- Jaber, M.M.; Al-Kofahi, M.; Sarafoglou, K.; Brundage, R.C. Individualized Absorption Models in Population Pharmacokinetic Analyses. CPT Pharmacomet. Syst. Pharmacol. 2020, 9, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Elmokadem, A.; Riggs, M.M.; Baron, K.T. Quantitative Systems Pharmacology and Physiologically-Based Pharmacokinetic Modeling with mrgsolve: A Hands-On Tutorial. CPT Pharmacomet. Syst. Pharmacol. 2019, 8, 883–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juszczak, P.; Tax, D.M.J.; Duin, R.P.W. Feature scaling in support vector data description. Proc. ASCI 2002, 95–102. [Google Scholar]
- Nwankpa, C.; Ijomah, W.; Gachagan, A.; Marshall, S. Activation Functions: Comparison of trends in Practice and Research for Deep Learning. arXiv 2018, arXiv:1811.03378. [Google Scholar]
- Zhang, Z.; Sabuncu, M.R. Generalized cross entropy loss for training deep neural networks with noisy labels. In Advances in Neural Information Processing Systems; Curran Associates, Inc.: Red Hook, NY, USA, 2018; pp. 8778–8788. [Google Scholar]
- Alaiz-Moreton, H.; Aveleira-Mata, J.; Ondicol-Garcia, J.; Muñoz-Castañeda, A.L.; García, I.; Benavides, C. Multiclass Classification Procedure for Detecting Attacks on MQTT-IoT Protocol. Complexity 2019, 2019, 6516253. [Google Scholar] [CrossRef] [Green Version]
- Bergstrand, M.; Söderlind, E.; Eriksson, U.G.; Weitschies, W.; Karlsson, M.O. A semi-mechanistic modeling strategy for characterization of regional absorption properties and prospective prediction of plasma concentrations following administration of new modified release formulations. Pharm. Res. 2012, 29, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Garcia, A.; Tan, W.; Li, J.; Haughey, M.; Masters, J.; Hibma, J.; Lin, S. Pharmacokinetic Models to Characterize the Absorption Phase and the Influence of a Proton Pump Inhibitor on the Overall Exposure of Dacomitinib. Pharmaceutics 2020, 12, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csajka, C.; Drover, D.; Verotta, D. The use of a sum of inverse gaussian functions to describe the absorption profile of drugs exhibiting complex absorption. Pharm. Res. 2005, 22, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Dokoumetzidis, A.; MacHeras, P. Fractional kinetics in drug absorption and disposition processes. J. Pharmacokinet. Pharmacodyn. 2009, 36, 165–178. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Draft Guidance on Methylphenidate Hydrochloride. 2015. Available online: https://www.accessdata.fda.gov/drugsatfdadocs/psg/MethylphenidateHydrochloridedraftOraltabERRLD21121RC07-18.pdf (accessed on 10 April 2021).
- U.S. Food and Drug Administration. Draft Guidance on Hydromorphone Hydrochloride. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Hydromorphone_ER_Tablets_021217_RC07-14.pdf (accessed on 9 April 2021).
- U.S. Food and Drug Administration. Draft Guidance on Amantadine Hydrochloride. 2011. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/psg/Amantadine_HCl_tab_76186_RC11-10.pdf (accessed on 10 April 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | First-Order | Erlang | Split-Peak |
|---|---|---|---|
| CL (L/h/70 kg) | 22.6 (29%) | ||
| V (L/70 kg) | 38.9 (21% | ||
| () | - | 8.2 (23%) | 5.2 (23%) |
| () | 3.3 (48%) | - | 7.6 (48%) |
| Fraction (%) | - | - | 78 (80%) |
| 16.5% |
| Data | N | Overall Accuracy | Overall Loss Value |
|---|---|---|---|
| Training | 21,000 | 99.7% | <0.01 |
| Validation | 9000 | 99.4% | <0.01 |
| External | 48 | 93.7% | 0.17 |
| DNN Prediction | Visual Assignment | ||
|---|---|---|---|
| First-Order | Erlang | Split-Peak | |
| First-order | 18 | 1 | 1 |
| Erlang | 0 | 21 | 1 |
| Split-peak | 0 | 0 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaber, M.M.; Yaman, B.; Sarafoglou, K.; Brundage, R.C. Application of Deep Neural Networks as a Prescreening Tool to Assign Individualized Absorption Models in Pharmacokinetic Analysis. Pharmaceutics 2021, 13, 797. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13060797
Jaber MM, Yaman B, Sarafoglou K, Brundage RC. Application of Deep Neural Networks as a Prescreening Tool to Assign Individualized Absorption Models in Pharmacokinetic Analysis. Pharmaceutics. 2021; 13(6):797. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13060797
Chicago/Turabian StyleJaber, Mutaz M., Burhaneddin Yaman, Kyriakie Sarafoglou, and Richard C. Brundage. 2021. "Application of Deep Neural Networks as a Prescreening Tool to Assign Individualized Absorption Models in Pharmacokinetic Analysis" Pharmaceutics 13, no. 6: 797. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics13060797