
Varied Bulk Powder Properties of Micro-Sized API within Size Specifications as a Result of Particle Engineering Methods
 , , and
, , and
Abstract
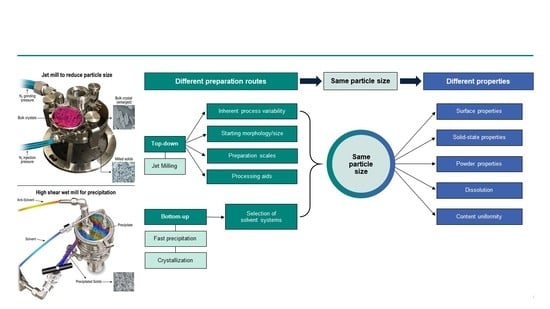
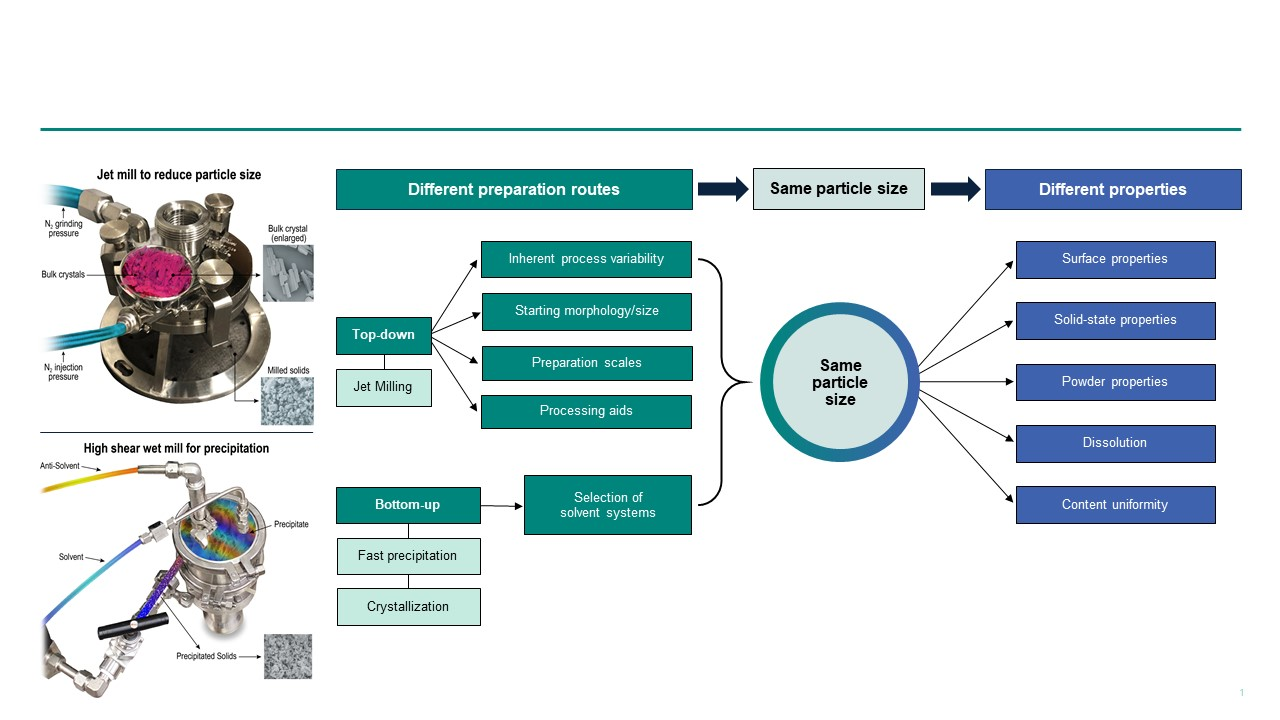
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
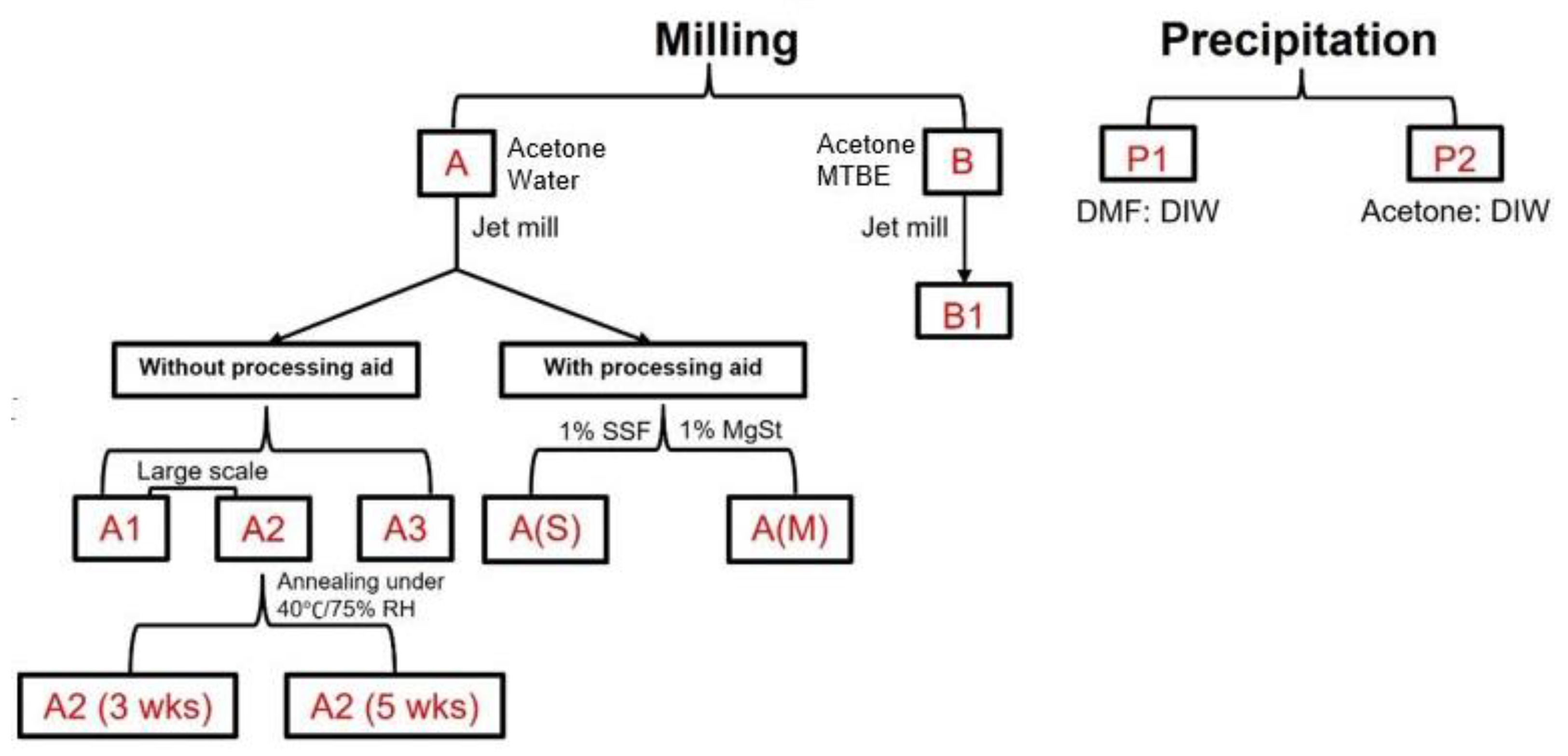
2.2.1. Preparation of Samples
2.2.2. Particle Size Distribution
2.2.3. X-ray Diffractometry
2.2.4. Scanning Electron Microscopy (SEM)
2.2.5. Surface Area Analysis
2.2.6. Tabletability
2.2.7. Shear Cell Testing
2.2.8. Intrinsic Dissolution Rate
2.2.9. Contact Angle
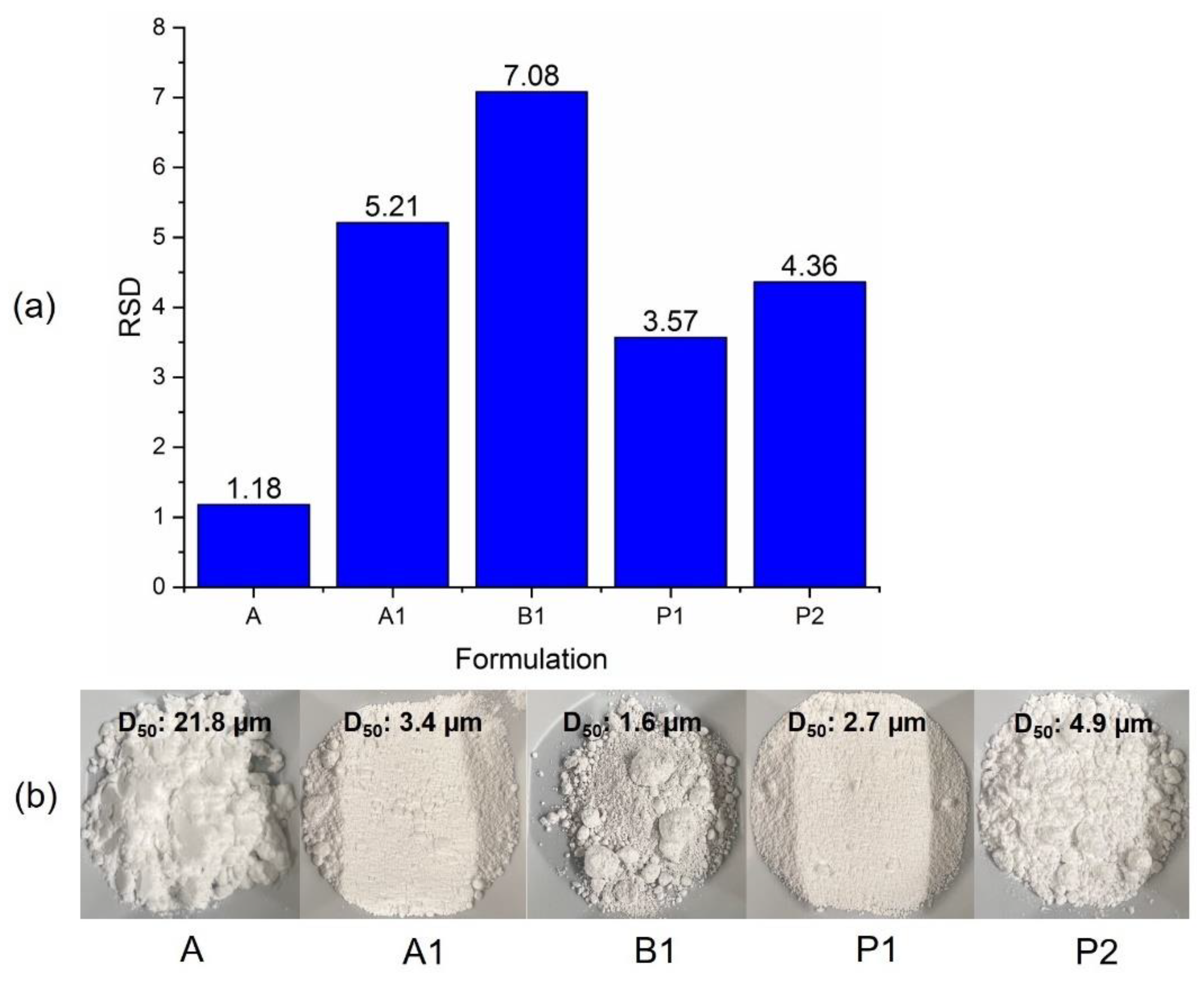
2.2.10. Content Uniformity (CU)
2.2.11. Statistical Analysis
3. Results and Discussion
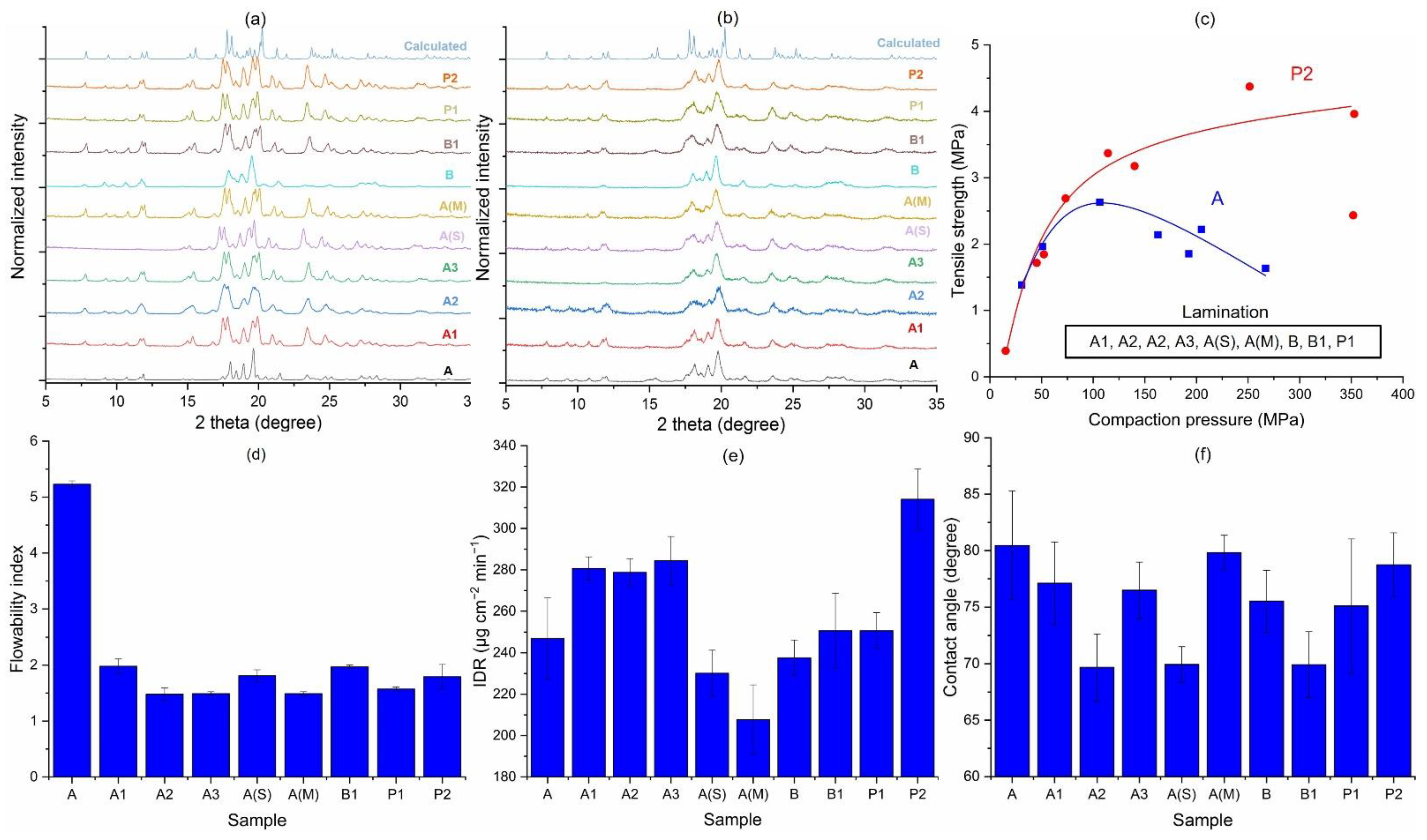
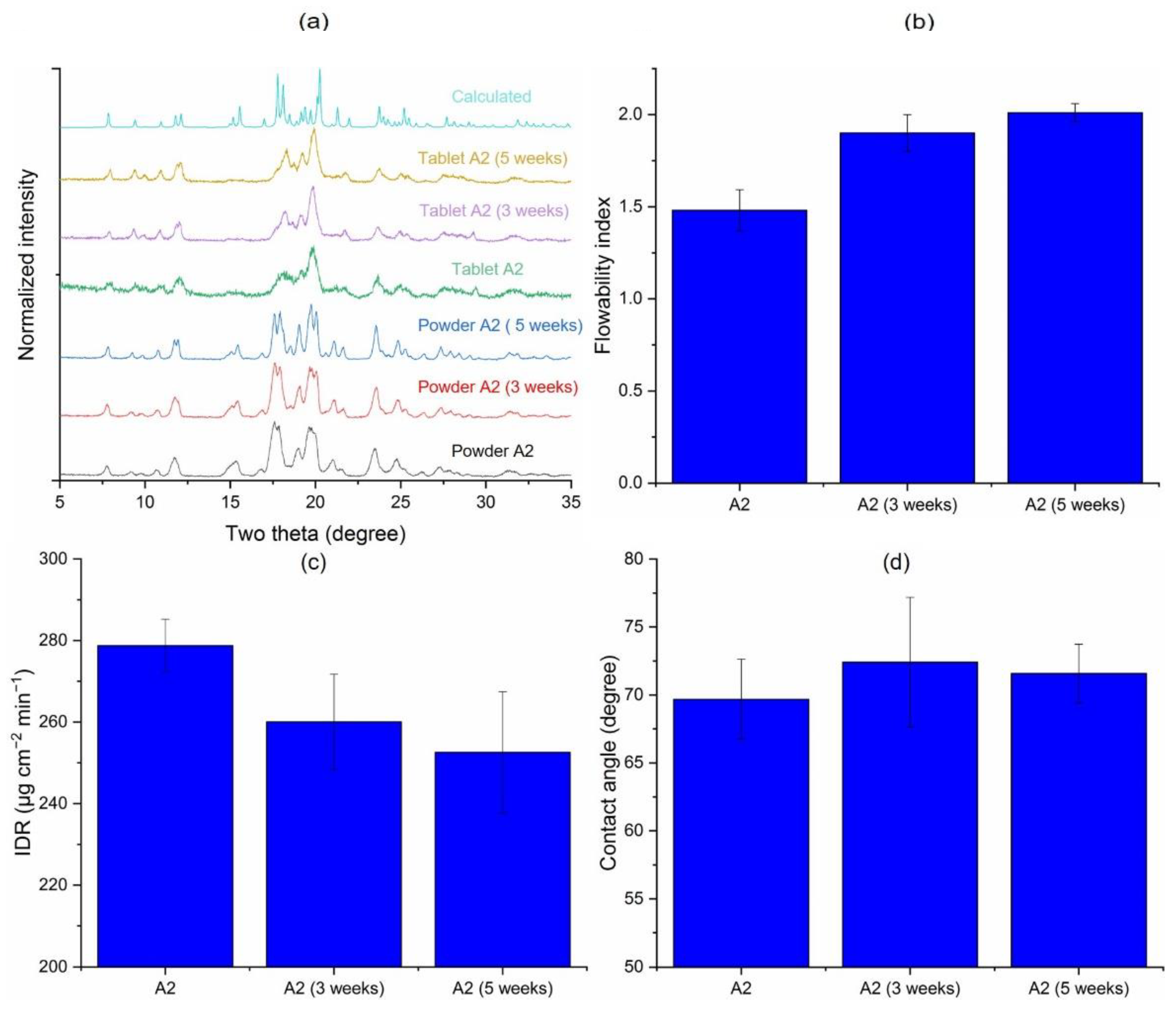
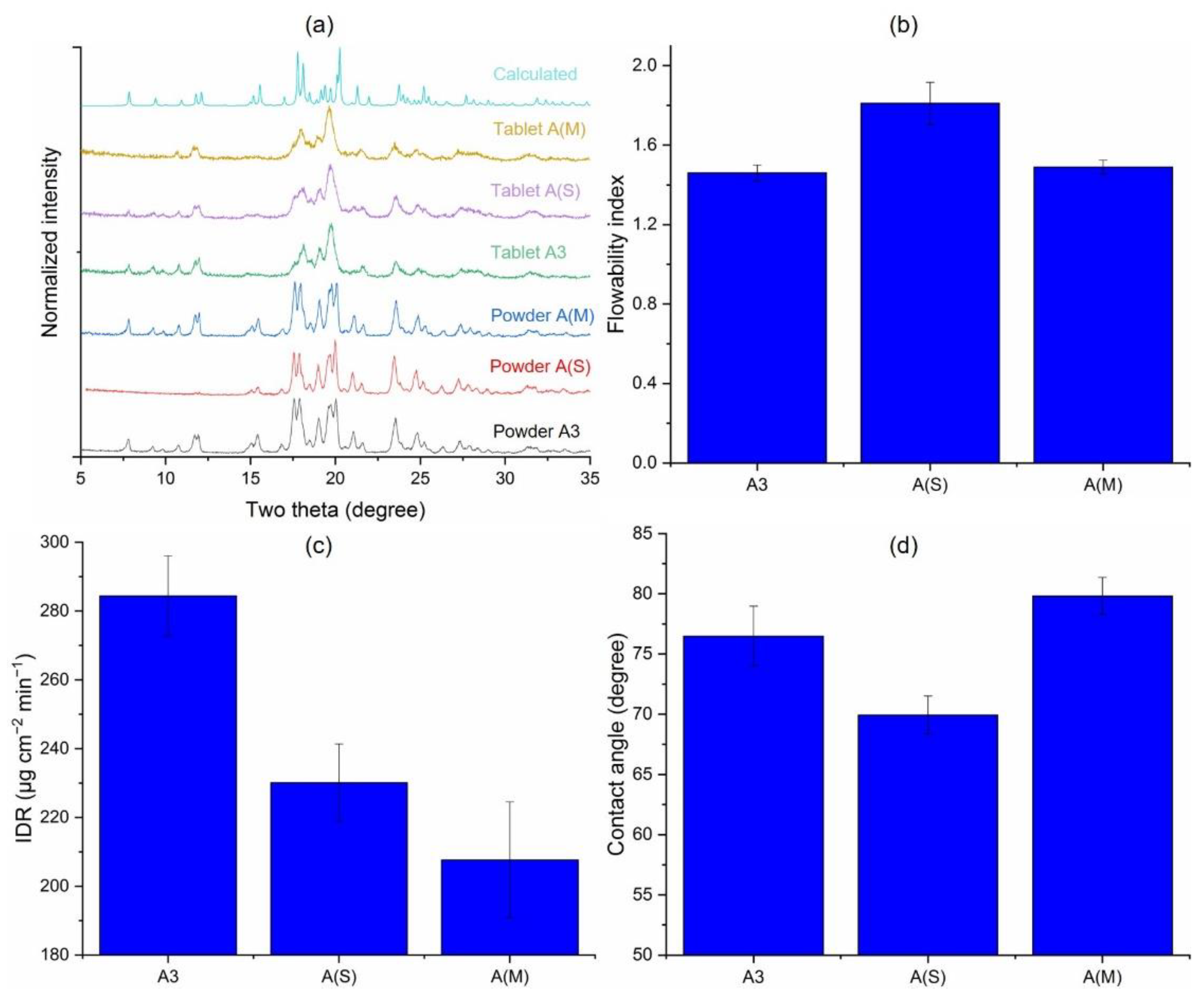
3.1. Solid-State Characterization
3.1.1. Particle Size Distribution and Specific Surface Area
3.1.2. Crystal Morphology
3.2. Crystallinity and Bulk Powder Properties
3.3. Comparison of Various Process Variables
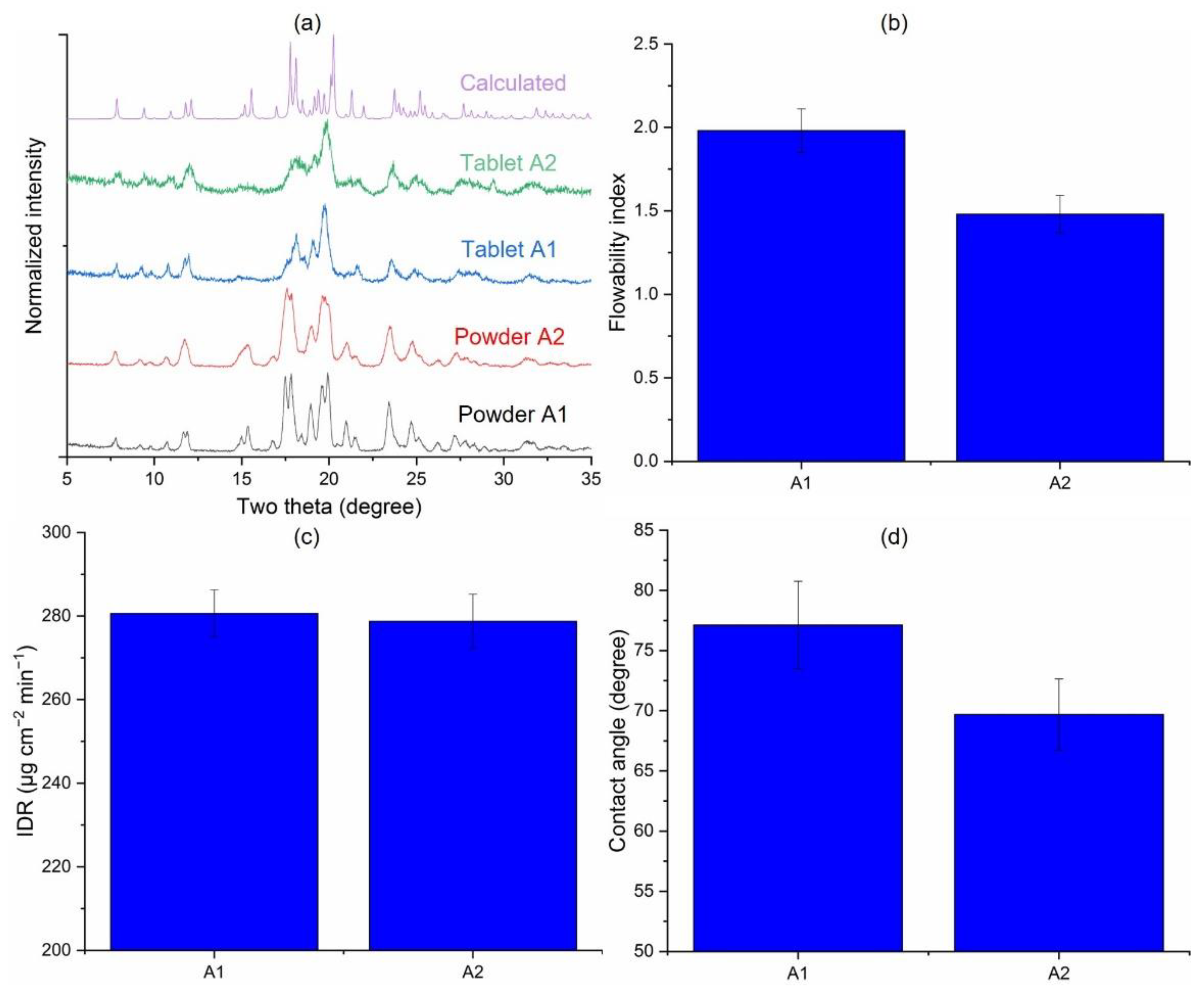
3.3.1. Inherent Process Variability of Jet Milling (A1 vs. A2)
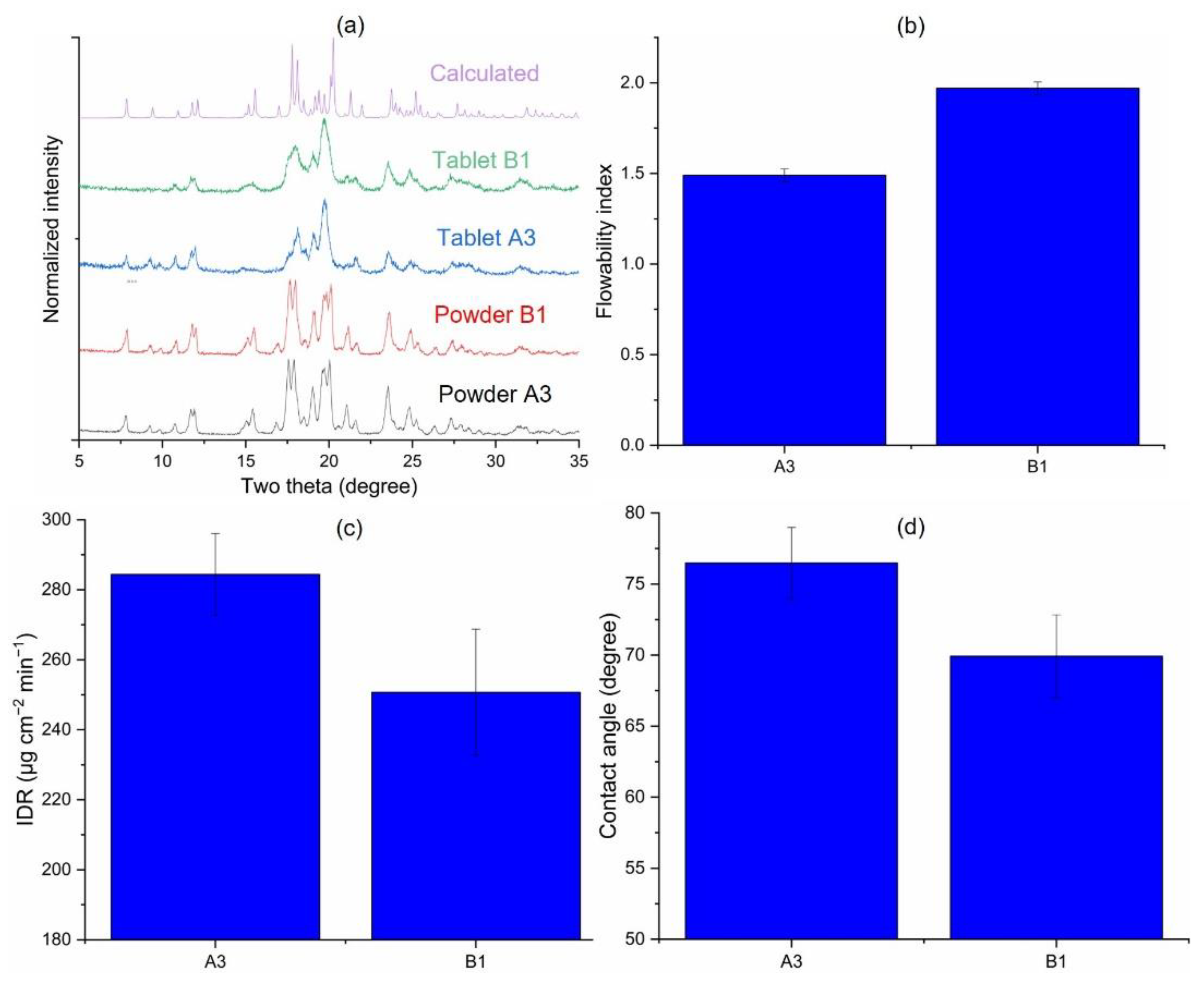
3.3.2. Effects of Starting Material on Jet-Milled API (A3 vs. B1)
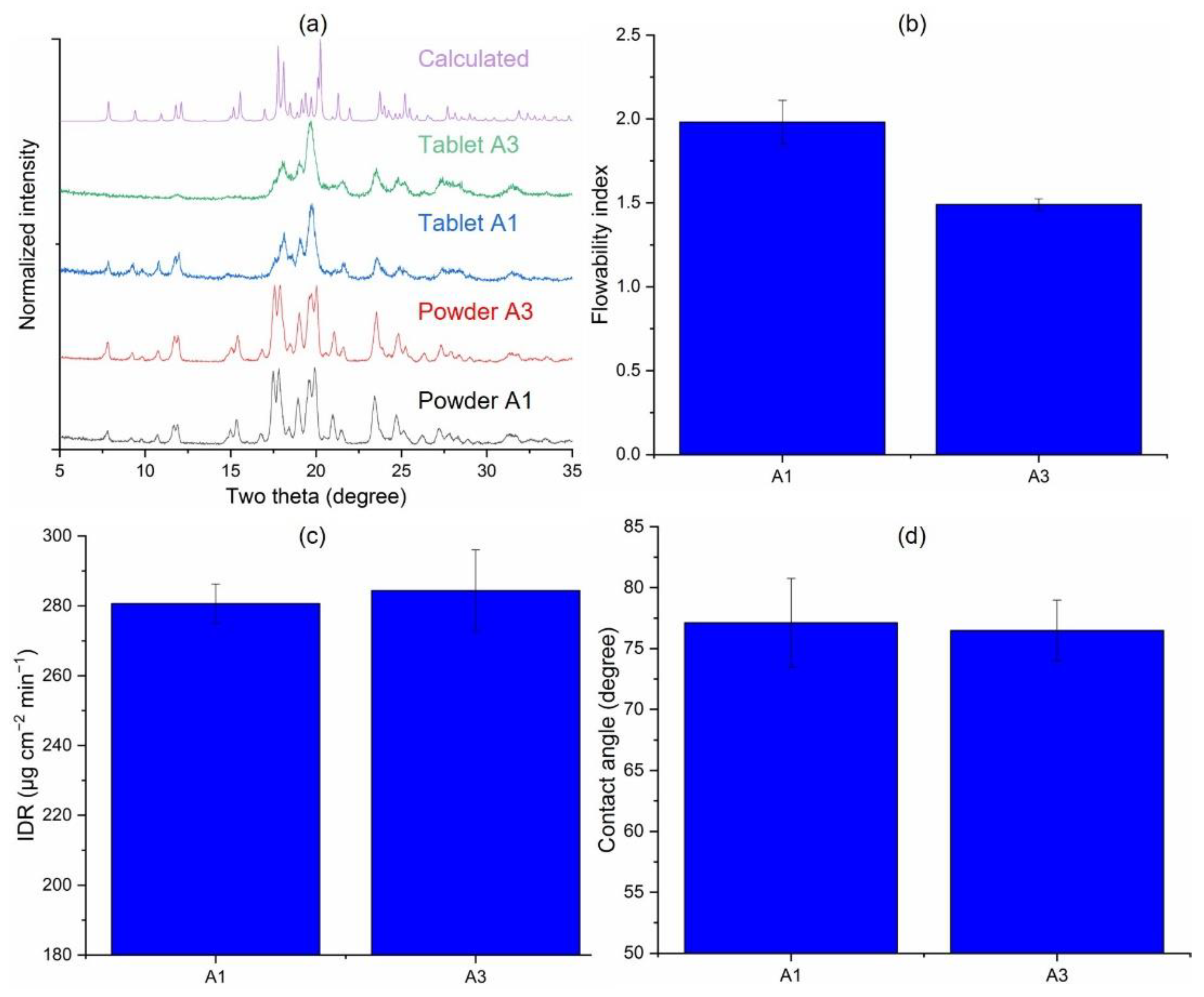
3.3.3. Effects of the Milling Scale (A1 vs. A3)
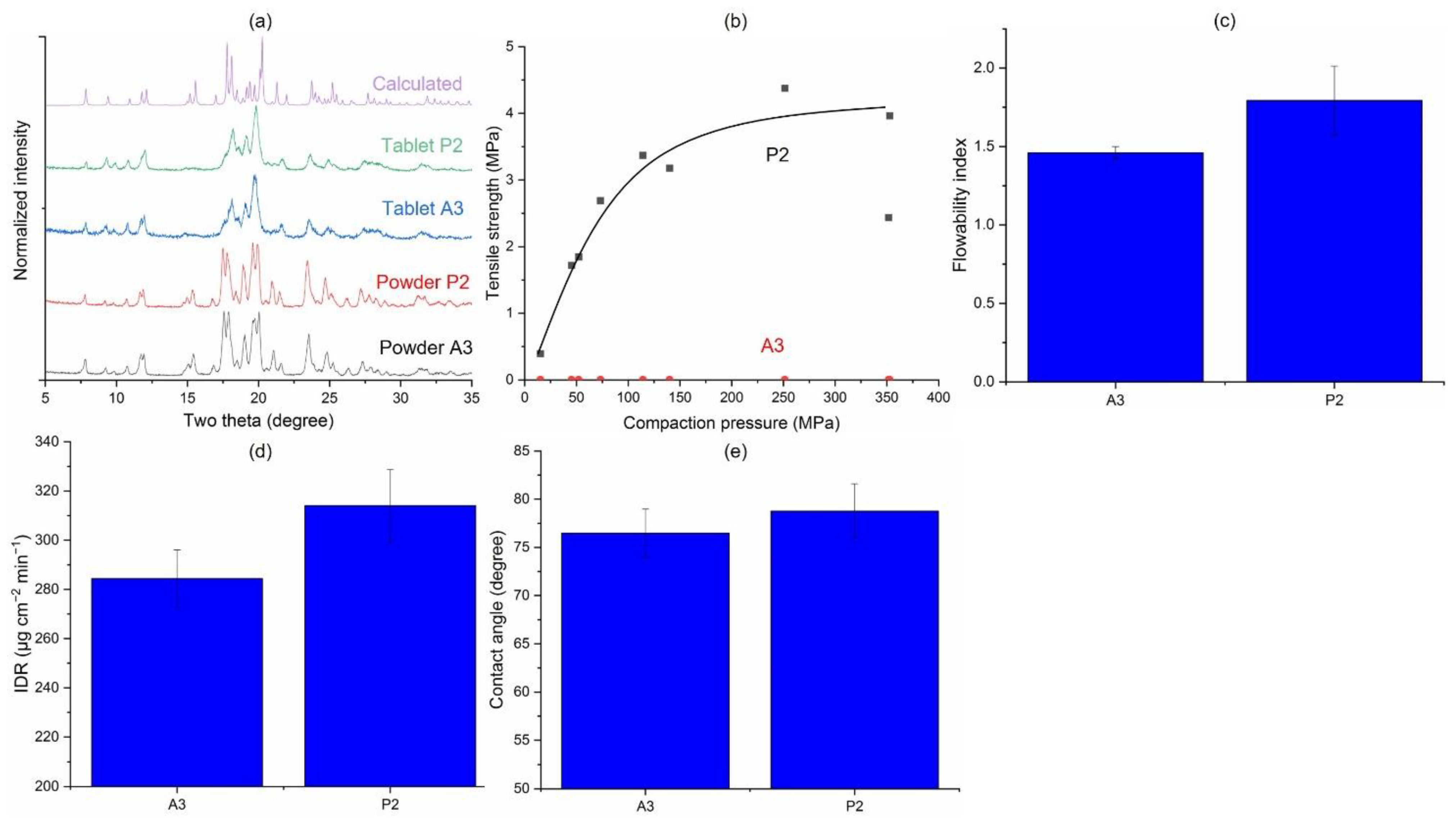
3.3.4. Jet-Milled vs. Precipitated Samples (A3 vs. P2)
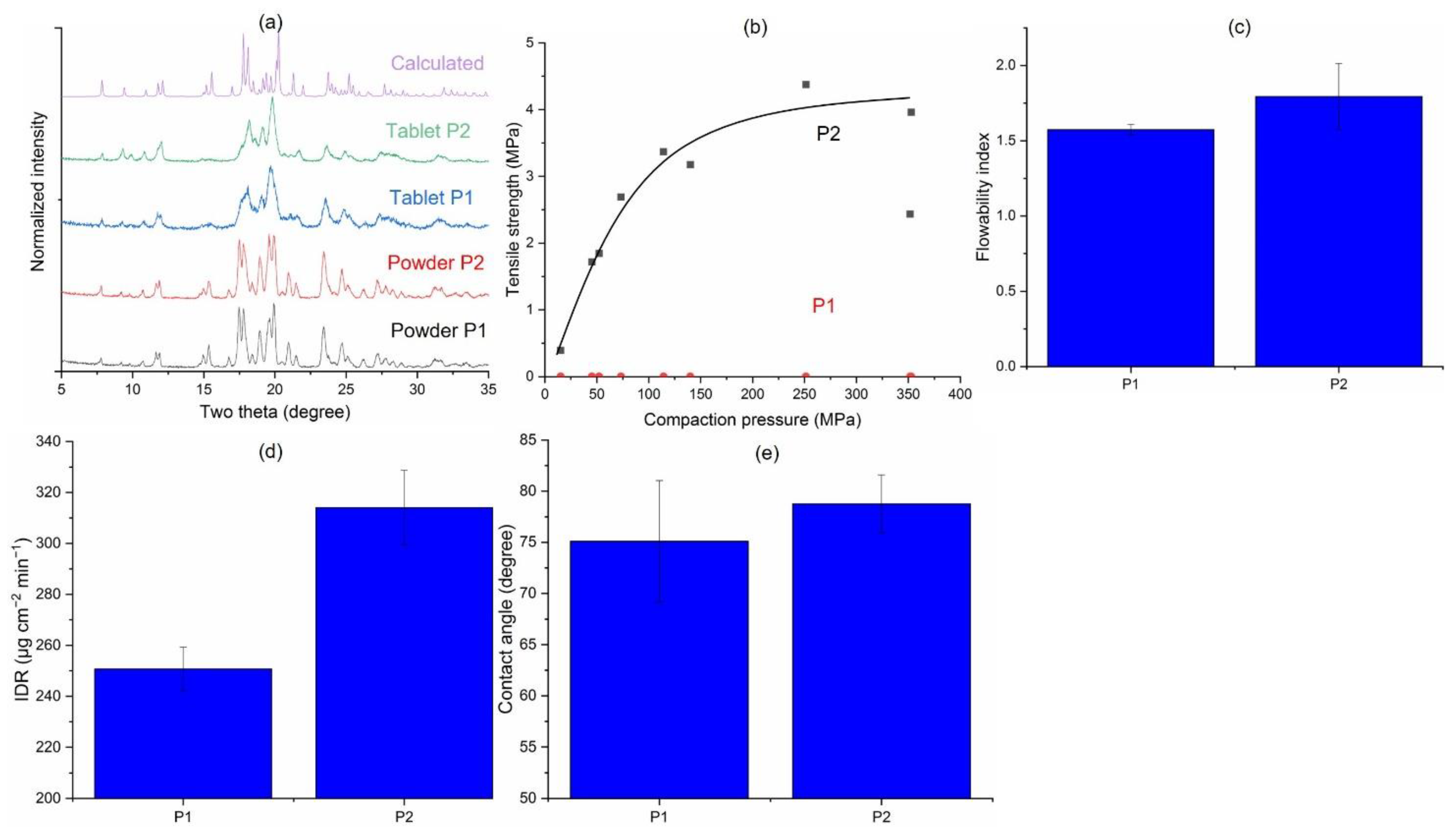
3.3.5. Effects of Solvent Systems in the Precipitation Route (P1 vs. P2)
3.3.6. Effects of Crystallinity
3.3.7. Effects of Processing Aids during Jet Milling (A3 vs. A(S) vs. A(M))
3.4. Impact on Content Uniformity in Tablets
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to Address Low Drug Solubility in Discovery and Development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Buckton, G.; Beezer, A.E. The Relationship between Particle Size and Solubility. Int. J. Pharm. 1992, 82, R7–R10. [Google Scholar] [CrossRef]
- Noyes, A.A.; Whitney, W.R. The rate of solution of solid substances in their own solutions. J. Am. Chem. Soc. 1897, 19, 930–934. [Google Scholar] [CrossRef]
- Sun, C.C. On the Mechanism of Reduced Tabletability of Granules Prepared by Roller Compaction. Int. J. Pharm. 2008, 347, 171–172. [Google Scholar] [CrossRef]
- Fu, X.; Huck, D.; Makein, L.; Armstrong, B.; Willen, U.; Freeman, T. Effect of Particle Shape and Size on Flow Properties of Lactose Powders. Particuology 2012, 10, 203–208. [Google Scholar] [CrossRef]
- Yalkowsky, S.H.; Bolton, S. Particle Size and Content Uniformity. Pharm. Res. Off. J. Am. Assoc. Pharm. Sci. 1990, 7, 962–966. [Google Scholar] [CrossRef]
- Rohrs, B.R.; Amidon, G.E.; Meury, R.H.; Secreast, P.J.; King, H.M.; Skoug, C.J. Particle Size Limits to Meet USP Content Uniformity Criteria for Tablets and Capsules. J. Pharm. Sci. 2006, 95, 1049–1059. [Google Scholar] [CrossRef]
- Jakubowska, E.; Ciepluch, N. Blend Segregation in Tablets Manufacturing and Its Effect on Drug Content Uniformity—A Review. Pharmaceutics 2021, 13, 1909. [Google Scholar] [CrossRef]
- Wuelfing, W.P.; El Marrouni, A.; Lipert, M.P.; Daublain, P.; Kesisoglou, F.; Converso, A.; Templeton, A.C. Dose Number as a Tool to Guide Lead Optimization for Orally Bioavailable Compounds in Drug Discovery. J. Med. Chem. 2022, 65, 1685–1694. [Google Scholar] [CrossRef]
- Kumar, R.; Thakur, A.K.; Chaudhari, P.; Banerjee, N. Particle Size Reduction Techniques of Pharmaceutical Compounds for the Enhancement of Their Dissolution Rate and Bioavailability. J. Pharm. Innov. 2021, 17, 333–352. [Google Scholar] [CrossRef]
- Kumar, D.; Worku, Z.A.; Gao, Y.; Kamaraju, V.K.; Glennon, B.; Babu, R.P.; Healy, A.M. Comparison of Wet Milling and Dry Milling Routes for Ibuprofen Pharmaceutical Crystals and Their Impact on Pharmaceutical and Biopharmaceutical Properties. Powder Technol. 2018, 330, 228–238. [Google Scholar] [CrossRef]
- Salah, N.; Habib, S.S.; Khan, Z.H.; Memic, A.; Azam, A.; Alarfaj, E.; Zahed, N.; Al-Hamedi, S. High-Energy Ball Milling Technique for ZnO Nanoparticles as Antibacterial Material. Int. J. Nanomed. 2011, 6, 863–869. [Google Scholar] [CrossRef]
- Bonakdar, T.; Ghadiri, M. Analysis of Pin Milling of Pharmaceutical Materials. Int. J. Pharm. 2018, 552, 394–400. [Google Scholar] [CrossRef]
- Yeung, C.C.; Hersey, J.A. Powder Homogenization Using a Hammer Mill. J. Pharm. Sci. 1979, 68, 721–724. [Google Scholar] [CrossRef]
- Saleem, I.; Smyth, H. Micronization of a Soft Material: Air-Jet and Micro-Ball Milling. AAPS PharmSciTech 2010, 11, 1642–1649. [Google Scholar] [CrossRef]
- Latreche, M.; Willart, J.-F.; Guerain, M.; Hédoux, A.; Danède, F. Using Milling to Explore Physical States: The Amorphous and Polymorphic Forms of Sulindac. J. Pharm. Sci. 2019, 108, 2635–2642. [Google Scholar] [CrossRef]
- Elisei, E.; Willart, J.-F.; Danède, F.; Siepmann, J.; Siepmann, F.; Descamps, M. Crystalline Polymorphism Emerging From a Milling-Induced Amorphous Form: The Case of Chlorhexidine Dihydrochloride. J. Pharm. Sci. 2018, 107, 121–126. [Google Scholar] [CrossRef]
- Chikhalia, V.; Forbes, R.T.; Storey, R.A.; Ticehurst, M. The Effect of Crystal Morphology and Mill Type on Milling Induced Crystal Disorder. Eur. J. Pharm. Sci. 2006, 27, 19–26. [Google Scholar] [CrossRef]
- Thorat, A.A.; Dalvi, S.V. Liquid Antisolvent Precipitation and Stabilization of Nanoparticles of Poorly Water Soluble Drugs in Aqueous Suspensions: Recent Developments and Future Perspective. Chem. Eng. J. 2012, 181–182, 1–34. [Google Scholar] [CrossRef]
- Modi, S.R.; Dantuluri, A.K.R.; Puri, V.; Pawar, Y.B.; Nandekar, P.; Sangamwar, A.T.; Perumalla, S.R.; Sun, C.C.; Bansal, A.K. Impact of Crystal Habit on Biopharmaceutical Performance of Celecoxib. Cryst. Growth Des. 2013, 13, 2824–2832. [Google Scholar] [CrossRef]
- Modi, S.R.; Dantuluri, A.K.R.; Perumalla, S.R.; Sun, C.C.; Bansal, A.K. Effect of Crystal Habit on Intrinsic Dissolution Behavior of Celecoxib Due to Differential Wettability. Cryst. Growth Des. 2014, 14, 5283–5292. [Google Scholar] [CrossRef]
- Chattoraj, S.; Sun, C.C. Crystal and Particle Engineering Strategies for Improving Powder Compression and Flow Properties to Enable Continuous Tablet Manufacturing by Direct Compression. J. Pharm. Sci. 2018, 107, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Xu, H.; Wang, C.; Kang, H.; Haynes, C.L.; Mahanthappa, M.K.; Sun, C.C. Novel Quasi-Emulsion Solvent Diffusion-Based Spherical Cocrystallization Strategy for Simultaneously Improving the Manufacturability and Dissolution of Indomethacin. Cryst. Growth Des. 2020, 20, 6752–6762. [Google Scholar] [CrossRef]
- Bartkowiak, T.; Berglund, J.; Brown, C.A. Multiscale Characterizations of Surface Anisotropies. Materials 2020, 13, 3028. [Google Scholar] [CrossRef]
- Jain, T.; Sheokand, S.; Modi, S.R.; Ugale, B.; Yadav, R.N.; Kumar, N.; Nagaraja, C.M.; Bansal, A.K. Effect of Differential Surface Anisotropy on Performance of Two Plate Shaped Crystals of Aspirin Form I. Eur. J. Pharm. Sci. 2017, 99, 318–327. [Google Scholar] [CrossRef]
- Dirba, I.; Li, J.; Sepehri-Amin, H.; Ohkubo, T.; Schrefl, T.; Hono, K. Anisotropic, Single-Crystalline SmFe12-Based Microparticles with High Roundness Fabricated by Jet-Milling. J. Alloys Compd. 2019, 804, 155–162. [Google Scholar] [CrossRef]
- Gauthier, J.Y.; Chauret, N.; Cromlish, W.; Desmarais, S.; Duong, L.T.; Falgueyret, J.-P.; Kimmel, D.B.; Lamontagne, S.; Léger, S.; LeRiche, T.; et al. The Discovery of Odanacatib (MK-0822), a Selective Inhibitor of Cathepsin K. Bioorg. Med. Chem. Lett. 2008, 18, 923–928. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Sun, C.; Grant, D.J.W. Influence of Crystal Structure on the Tableting Properties of Sulfamerazine Polymorphs. Pharm. Res. 2001, 18, 274–280. [Google Scholar] [CrossRef]
- Standard Shear Test Method for Bulk Solids Using the Schulze Ring Shear Tester. Available online: https://www.astm.org/d6773-08.html (accessed on 4 June 2022).
- Shi, L.; Feng, Y.; Sun, C.C. Origin of Profound Changes in Powder Properties during Wetting and Nucleation Stages of High-Shear Wet Granulation of Microcrystalline Cellulose. Powder Technol. 2011, 208, 663–668. [Google Scholar] [CrossRef]
- Wang, C.; Perumalla, S.R.; Lu, R.; Fang, J.; Sun, C.C. Sweet Berberine. Cryst. Growth Des. 2016, 16, 933–939. [Google Scholar] [CrossRef]
- United States Pharmacopeia Convention. USP 37 NF 32, USP General Chapter <905> Uniformity of Dosage Units.
- Keselman, H.J.; Rogan, J.C. The Tukey Multiple Comparison Test: 1953–1976. Psychol. Bull. 1977, 84, 1050–1056. [Google Scholar] [CrossRef]
- Watanabe, S.; Akahori, T. Determination of Crystallinity in Cellulose Fibers by X-Ray Diffraction. J. Soc. Chem. Ind. Jpn. 1969, 72, 1565–1572. [Google Scholar] [CrossRef]
- Surana, R.; Suryanarayanan, R. Quantitation of Crystallinity in Substantially Amorphous Pharmaceuticals and Study of Crystallization Kinetics by X-Ray Powder Diffractometry. Powder Diffr. 2000, 15, 2–6. [Google Scholar] [CrossRef]
- Vreeman, G.; Sun, C.C. Air Entrapment during Tablet Compression—Diagnosis, Impact on Tableting Performance, and Mitigation Strategies. Int. J. Pharm. 2022, 615, 121514. [Google Scholar] [CrossRef]
- Mazel, V.; Busignies, V.; Diarra, H.; Tchoreloff, P. Lamination of Pharmaceutical Tablets Due to Air Entrapment: Direct Visualization and Influence of the Compact Thickness. Int. J. Pharm. 2015, 478, 702–704. [Google Scholar] [CrossRef]
- Schulze, D. Powders and Bulk Solids: Behavior, Characterization, Storage and Flow; Springer International Publishing: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Hou, H.; Sun, C.C. Quantifying Effects of Particulate Properties on Powder Flow Properties Using a Ring Shear Tester. J. Pharm. Sci. 2008, 97, 4030–4039. [Google Scholar] [CrossRef]
- Bell, T.A. Challenges in the Scale-up of Particulate Processes—An Industrial Perspective. Powder Technol. 2005, 150, 60–71. [Google Scholar] [CrossRef]
- Paliwal, R.; Babu, R.J.; Palakurthi, S. Nanomedicine Scale-up Technologies: Feasibilities and Challenges. AAPS PharmSciTech 2014, 15, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Ramakers, L.A.I.; McGinty, J.; Beckmann, W.; Levilain, G.; Lee, M.; Wheatcroft, H.; Houson, I.; Sefcik, J. Investigation of Metastable Zones and Induction Times in Glycine Crystallization across Three Different Antisolvents. Cryst. Growth Des. 2020, 20, 4935–4944. [Google Scholar] [CrossRef]
- Marchese, P.; Celli, A.; Fiorini, M.; Gabaldi, M. Effects of Annealing on Crystallinity and Phase Behaviour of PET/PC Block Copolymers. Eur. Polym. J. 2003, 39, 1081–1089. [Google Scholar] [CrossRef]
- Simmons, H.; Tiwary, P.; Colwell, J.E.; Kontopoulou, M. Improvements in the Crystallinity and Mechanical Properties of PLA by Nucleation and Annealing. Polym. Degrad. Stab. 2019, 166, 248–257. [Google Scholar] [CrossRef]
- Horio, T.; Yasuda, M.; Matsusaka, S. Measurement of Flowability of Lubricated Powders by the Vibrating Tube Method. Drug Dev. Ind. Pharm. 2013, 39, 1063–1069. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Particle Size Distribution (µm) | Span | SSA | |||
|---|---|---|---|---|---|---|
| d10 | d50 | d90 | Mv | (m2/g) | ||
| A | 10.2 | 21.8 | 49.2 | 26.6 | 2.4 | 0.03 |
| A1 | 1.4 | 3.4 | 6.8 | 3.9 | 1.6 | 0.06 |
| A2 | 1.0 | 3.2 | 8.6 | 4.6 | 2.4 | 0.02 |
| A2 (t = 3 wks) | 1.2 | 3.5 | 8.5 | 4.5 | 2.1 | N/R |
| A2 (t = 5 wks) | 1.2 | 3.4 | 8.0 | 4.2 | 2.0 | N/R |
| A3 | 2.2 | 4.9 | 7.8 | 5.0 | 1.1 | 0.02 |
| A(S) | 0.8 | 1.9 | 3.9 | 2.2 | 1.6 | 0.02 |
| A(M) | 1.1 | 2.6 | 4.8 | 2.8 | 1.4 | 0.02 |
| B | 4.1 | 15.8 | 102.5 | 40.9 | 10.7 | 0.11 |
| B1 | 0.7 | 1.6 | 2.9 | 1.7 | 1.4 | 0.03 |
| P1 | 0.8 | 2.7 | 8.2 | 3.9 | 2.7 | 0.03 |
| P2 | 2.6 | 4.9 | 9.9 | 5.7 | 1.5 | 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Solomos, M.; Axnanda, S.; Chen, C.; Figus, M.; Schenck, L.; Sun, C.C. Varied Bulk Powder Properties of Micro-Sized API within Size Specifications as a Result of Particle Engineering Methods. Pharmaceutics 2022, 14, 1901. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14091901
Wang Z, Solomos M, Axnanda S, Chen C, Figus M, Schenck L, Sun CC. Varied Bulk Powder Properties of Micro-Sized API within Size Specifications as a Result of Particle Engineering Methods. Pharmaceutics. 2022; 14(9):1901. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14091901
Chicago/Turabian StyleWang, Zijian, Marina Solomos, Stephanus Axnanda, Chienhung Chen, Margaret Figus, Luke Schenck, and Changquan Calvin Sun. 2022. "Varied Bulk Powder Properties of Micro-Sized API within Size Specifications as a Result of Particle Engineering Methods" Pharmaceutics 14, no. 9: 1901. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics14091901