
Targeting Siderophore-Mediated Iron Uptake in M. abscessus: A New Strategy to Limit the Virulence of Non-Tuberculous Mycobacteria
 , , , , ,
, , , , ,  , , , , , and
, , , , , and
Abstract
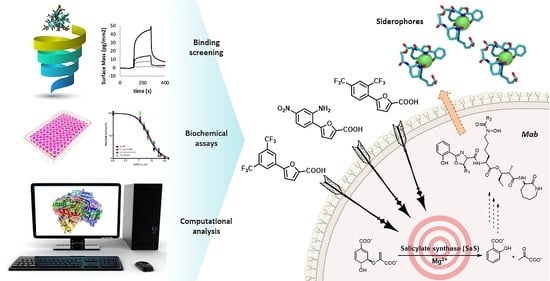
:
1. Introduction
2. Materials and Methods
2.1. Synthesis and Characterization
2.2. Mab-SaS Expression, Purification, and Characterization
2.3. Biological Tests
2.3.1. Mab-SaS Activity Dependence on Magnesium Concentration
2.3.2. Mab-SaS Inhibition Assays
2.3.3. Pan Assay Interference Compound (PAIN) Analysis
2.3.4. Siderophore Production Assay
2.3.5. Minimal Inhibitory Concentration (MIC) Evaluation by Resazurin Assay
2.4. Binding Analysis
2.5. Computational Details
2.5.1. Homology Modelling
2.5.2. Molecular Docking Simulations
2.5.3. MM-GBSA Calculations
3. Results
3.1. Biochemical Analyses
3.2. Binding Analysis
3.3. Microbiological Tests
3.4. Molecular Modelling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Faria, S.; Joao, I.; Jordao, L. General Overview on Nontuberculous Mycobacteria, Biofilms, and Human Infection. J. Pathog. 2015, 2015, 809014. [Google Scholar] [CrossRef] [PubMed]
- Sood, G.; Parrish, N. Outbreaks of nontuberculous mycobacteria. Curr. Opin. Infect. Dis. 2017, 30, 404–409. [Google Scholar] [CrossRef] [PubMed]
- To, K.; Cao, R.; Yegiazaryan, A.; Owens, J.; Venketaraman, V. General Overview of Nontuberculous Mycobacteria Opportunistic Pathogens: Mycobacterium avium and Mycobacterium abscessus. J. Clin. Med. 2020, 9, 2541. [Google Scholar] [CrossRef]
- Martiniano, S.L.; Nick, J.A.; Daley, C.L. Nontuberculous Mycobacterial Infections in Cystic Fibrosis. Clin. Chest Med. 2016, 37, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Bento, C.M.; Gomes, M.S.; Silva, T. Looking beyond Typical Treatments for Atypical Mycobacteria. Antibiotics 2020, 9, 18. [Google Scholar] [CrossRef]
- Buroni, S.; Chiarelli, L.R. Antivirulence compounds: A future direction to overcome antibiotic resistance? Future Microbiol. 2020, 15, 299–301. [Google Scholar] [CrossRef]
- Kelley, V.A.; Schorey, J.S. Mycobacterium’s arrest of phagosome maturation in macrophages requires Rab5 activity and accessibility to iron. Mol. Biol. Cell 2003, 14, 3366–3377. [Google Scholar] [CrossRef]
- Meneghetti, F.; Villa, S.; Gelain, A.; Barlocco, D.; Chiarelli, L.R.; Pasca, M.R.; Costantino, L. Iron Acquisition Pathways as Targets for Antitubercular Drugs. Curr. Med. Chem. 2016, 23, 4009–4026. [Google Scholar] [CrossRef]
- Chao, A.; Sieminski, P.J.; Owens, C.P.; Goulding, C.W. Iron Acquisition in Mycobacterium tuberculosis. Chem. Rev. 2019, 119, 1193–1220. [Google Scholar] [CrossRef]
- De Voss, J.J.; Rutter, K.; Schroeder, B.G.; Su, H.; Zhu, Y.; Barry III, C.E. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA 2000, 97, 1252–1257. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.V.; Puri, R.V.; Chauhan, P.; Kar, R.; Rohilla, A.; Khera, A.; Tyagi, A.K. Disruption of Mycobactin Biosynthesis Leads to Attenuation of Mycobacterium tuberculosis for Growth and Virulence. J. Infect. Dis. 2013, 208, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Shyam, M.; Shilkar, D.; Verma, H.; Dev, A.; Sinha, B.N.; Brucoli, F.; Bhakta, S.; Jayaprakash, V. The Mycobactin Biosynthesis Pathway: A Prospective Therapeutic Target in the Battle against Tuberculosis. J. Med. Chem. 2021, 64, 71–100. [Google Scholar] [CrossRef] [PubMed]
- Shyam, M.; Shilkar, D.; Rakshit, G.; Jayaprakash, V. Approaches for targeting the mycobactin biosynthesis pathway for novel anti-tubercular drug discovery: Where we stand. Expert Opin. Drug Discov. 2022, 17, 699–715. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, L.R.; Mori, M.; Barlocco, D.; Beretta, G.; Gelain, A.; Pini, E.; Porcino, M.; Mori, G.; Stelitano, G.; Costantino, L.; et al. Discovery and Development of Novel Salicylate Synthase (MbtI) Furanic Inhibitors as Antitubercular Agents. Eur. J. Med. Chem. 2018, 155, 754–763. [Google Scholar] [CrossRef]
- Pini, E.; Poli, G.; Tuccinardi, T.; Chiarelli, L.; Mori, M.; Gelain, A.; Costantino, L.; Villa, S.; Meneghetti, F.; Barlocco, D. New Chromane-Based Derivatives as Inhibitors of Mycobacterium tuberculosis Salicylate Synthase (MbtI): Preliminary Biological Evaluation and Molecular Modeling Studies. Molecules 2018, 23, 1506. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, M.; Beretta, G.; Gelain, A.; Pini, E.; Sammartino, J.C.; Stelitano, G.; Barlocco, D.; Costantino, L.; Lapillo, M.; et al. New Insight into Structure-Activity of Furan-based Salicylate Synthase (MbtI) Inhibitors as Potential Antitubercular Agents. J. Enzyme Inhib. Med. Chem. 2019, 34, 823–828. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Stelitano, G.; Gelain, A.; Pini, E.; Chiarelli, L.R.; Sammartino, J.C.; Poli, G.; Tuccinardi, T.; Beretta, G.; Porta, A.; et al. Shedding X-ray Light on the Role of Magnesium in the Activity of M. tuberculosis Salicylate Synthase (MbtI) for Drug Design. J. Med. Chem. 2020, 63, 7066–7080. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Chiarelli, L.R.; Cazzaniga, G.; Gelain, A.; Barlocco, D.; Pini, E.; Meneghetti, F.; Villa, S. Synthesis, Characterization, and Biological Evaluation of New Derivatives Targeting MbtI as Antitubercular Agents. Pharmaceuticals 2021, 14, 155. [Google Scholar] [CrossRef]
- Mori, M.; Stelitano, G.; Griego, A.; Chiarelli, L.R.; Cazzaniga, G.; Gelain, A.; Pini, E.; Camera, M.; Canzano, P.; Fumagalli, A.; et al. Synthesis and Assessment of the In Vitro and Ex Vivo Activity of Salicylate Synthase (Mbti) Inhibitors as New Candidates for the Treatment of Mycobacterial Infections. Pharmaceuticals 2022, 15, 992. [Google Scholar] [CrossRef]
- Schwyn, B.; Neilands, J.B. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 1987, 160, 47–56. [Google Scholar] [CrossRef]
- Creanza, T.M.; Delre, P.; Ancona, N.; Lentini, G.; Saviano, M.; Mangiatordi, G.F. Structure-Based Prediction of hERG-Related Cardiotoxicity: A Benchmark Study. J. Chem. Inf. Model. 2021, 61, 4758–4770. [Google Scholar] [CrossRef]
- Schrödinger Release 2022-2: Prime; Schrödinger LLC: New York, NY, USA, 2022.
- Schrödinger Release 2022-2: Protein Preparation Wizard; Schrödinger LLC: New York, NY, USA, 2022.
- Epik; Schrödinger LLC: New York, NY, USA, 2021.
- Impact; Schrödinger LLC: New York, NY, USA, 2021.
- Schrödinger Release 2022-2: LigPrep; Schrödinger LLC: New York, NY, USA, 2022.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2022-2: Glide; Schrödinger LLC: New York, NY, USA, 2022.
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Nissink, J.W.M.; Strasser, J.M.; Francis, S.; Higgins, L.; Zhou, H.; Zhang, Z.; Walters, M.A. PAINS in the Assay: Chemical Mechanisms of Assay Interference and Promiscuous Enzymatic Inhibition Observed during a Sulfhydryl-Scavenging HTS. J. Med. Chem. 2015, 58, 2091–2113. [Google Scholar] [CrossRef] [PubMed]
- Jankovics, H.; Kovacs, B.; Saftics, A.; Gerecsei, T.; Tóth, É.; Szekacs, I.; Vonderviszt, F.; Horvath, R. Grating-coupled interferometry reveals binding kinetics and affinities of Ni ions to genetically engineered protein layers. Sci. Rep. 2020, 10, 22253. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.C.; Da Costa, P.M.; Sousa, E.; Resende, D.I.S.P. Emerging Target-Directed Approaches for the Treatment and Diagnosis of Microbial Infections. J. Med. Chem. 2023, 66, 32–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hendrickson, R.C.; Meikle, V.; Lefkowitz, E.J.; Ioerger, T.R.; Niederweis, M. Comprehensive analysis of iron utilization by Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008337. [Google Scholar] [CrossRef]
- Parida, A.; Mohanty, A.; Raut, R.K.; Padhy, I.; Behera, R.K. Modification of 4-Fold and B-Pores in Bacterioferritin from Mycobacterium tuberculosis Reveals Their Role in Fe2+ Entry and Oxidoreductase Activity. Inorg. Chem. 2022, 62, 178–191. [Google Scholar] [CrossRef]
- Patidar, A.; Malhotra, H.; Chaudhary, S.; Kumar, M.; Dilawari, R.; Chaubey, G.K.; Dhiman, A.; Modanwal, R.; Talukdar, S.; Raje, C.I.; et al. Host glyceraldehyde-3-phosphate dehydrogenase-mediated iron acquisition is hijacked by intraphagosomal Mycobacterium tuberculosis. Cell. Mol. Life Sci. 2022, 79, 62. [Google Scholar] [CrossRef]
- Zhang, L.; Kent, J.E.; Whitaker, M.; Young, D.C.; Herrmann, D.; Aleshin, A.E.; Ko, Y.H.; Cingolani, G.; Saad, J.S.; Moody, D.B.; et al. A periplasmic cinched protein is required for siderophore secretion and virulence of Mycobacterium tuberculosis. Nat. Commun. 2022, 13, 2255. [Google Scholar] [CrossRef]
- Arnold, F.M.; Weber, M.S.; Gonda, I.; Gallenito, M.J.; Adenau, S.; Egloff, P.; Zimmermann, I.; Hutter, C.A.J.; Hürlimann, L.M.; Peters, E.E.; et al. The ABC exporter IrtAB imports and reduces mycobacterial siderophores. Nature 2020, 580, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Ingale, P.; Lad, B.; Kabra, R.; Singh, S. Dissecting druggability of ABC transporter proteins in Mycobacterium species through network modeling. J. Biomol. Struct. Dyn. 2021, 40, 8365–8374. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.; Koduru, T.N.; Kumar, N.; Salimi, S.; Desai, K.; Prabhu, N.P.; Sritharan, M. Iron uptake and transport by the carboxymycobactin-mycobactin siderophore machinery of Mycobacterium tuberculosis is dependent on the iron-regulated protein HupB. BioMetals 2021, 34, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.; Wells, G.; Bhakta, S.; Johnson, J.; Guzman, J.; Parish, T.; Prentice, R.A.; Brucoli, F. Integrated Target-Based and Phenotypic Screening Approaches for the Identification of Anti-Tubercular Agents That Bind to the Mycobacterial Adenylating Enzyme MbtA. ChemMedChem 2019, 14, 1735–1741. [Google Scholar] [CrossRef] [PubMed]
- Shyam, M.; Verma, H.; Bhattacharje, G.; Mukherjee, P.; Singh, S.; Kamilya, S.; Jalani, P.; Das, S.; Dasgupta, A.; Mondal, A.; et al. Mycobactin Analogues with Excellent Pharmacokinetic Profile Demonstrate Potent Antitubercular Specific Activity and Exceptional Efflux Pump Inhibition. J. Med. Chem. 2022, 65, 234–256. [Google Scholar] [CrossRef]
- McQueen, C.F.; Groves, J.T. Toxicity of the iron siderophore mycobactin J in mouse macrophages: Evidence for a hypoxia response. J. Inorg. Biochem. 2022, 227, 111669. [Google Scholar] [CrossRef]
- Foreman, M.; Kolodkin-Gal, I.; Barkan, D. A Pivotal Role for Mycobactin/ mbtE in Growth and Adaptation of Mycobacterium abscessus. Microbiol. Spectr. 2022, 10, e02623-22. [Google Scholar] [CrossRef]
- de Oliveira, F.M.; Corrêa, V.L.R.; Corrêa, A.F.; da Costa, A.C.; Procopio, V.O.; Junqueira-Kipnis, A.P.; Kipnis, A. The mycma_1113 Gene from Mycobacterium abscessus subsp. massiliense is Related to Siderophore Synthesis. Indian J. Microbiol. 2019, 59, 180–187. [Google Scholar] [CrossRef]
- Bythrow, G.V.; Farhat, M.F.; Levendosky, K.; Mohandas, P.; Germain, G.A.; Yoo, B.; Quadri, L.E.N. Mycobacterium abscessus Mutants with a Compromised Functional Link between the Type VII ESX-3 System and an Iron Uptake Mechanism Reliant on an Unusual Mycobactin Siderophore. Pathogens 2022, 11, 953. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Structure | %RA * | IC50 (μM) |
|---|---|---|---|
| 1 |  | 1.2 ± 2.8 | 5.3 ± 1.5 |
| 2 |  | 2.3 ± 0.7 | 23.6 ± 1.8 |
| 3 |  | 15.6 ± 2.5 | 29.5 ± 1.6 |
| Code | kon (M−1s−1) | koff (s−1) | KD (uM) | χ2 |
|---|---|---|---|---|
| 1 | 1.54 ± 0.05 × 101 | 9.77 ± 0.12 × 10−5 | 6.11 ± 0.16 | 0.86 ± 0.05 |
| 2 | 2.44 ± 0.62 × 102 | 3.03 ± 0.09 × 10−3 | 12.65 ± 3.32 | 0.53 ± 0.08 |
| 3 | 1.00 ± 0.05 × 102 | 6.75 ± 0.61 × 10−3 | 67.50 ± 9.51 | 0.91 ± 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mori, M.; Stelitano, G.; Cazzaniga, G.; Gelain, A.; Tresoldi, A.; Cocorullo, M.; Roversi, M.; Chiarelli, L.R.; Tomaiuolo, M.; Delre, P.; et al. Targeting Siderophore-Mediated Iron Uptake in M. abscessus: A New Strategy to Limit the Virulence of Non-Tuberculous Mycobacteria. Pharmaceutics 2023, 15, 502. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15020502
Mori M, Stelitano G, Cazzaniga G, Gelain A, Tresoldi A, Cocorullo M, Roversi M, Chiarelli LR, Tomaiuolo M, Delre P, et al. Targeting Siderophore-Mediated Iron Uptake in M. abscessus: A New Strategy to Limit the Virulence of Non-Tuberculous Mycobacteria. Pharmaceutics. 2023; 15(2):502. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15020502
Chicago/Turabian StyleMori, Matteo, Giovanni Stelitano, Giulia Cazzaniga, Arianna Gelain, Andrea Tresoldi, Mario Cocorullo, Martina Roversi, Laurent R. Chiarelli, Martina Tomaiuolo, Pietro Delre, and et al. 2023. "Targeting Siderophore-Mediated Iron Uptake in M. abscessus: A New Strategy to Limit the Virulence of Non-Tuberculous Mycobacteria" Pharmaceutics 15, no. 2: 502. https://0-doi-org.brum.beds.ac.uk/10.3390/pharmaceutics15020502