Genetic Structure of Cultivated Varieties of Radicchio (Cichorium intybus L.): A Comparison between F1 Hybrids and Synthetics



Abstract
:1. Introduction
2. Results
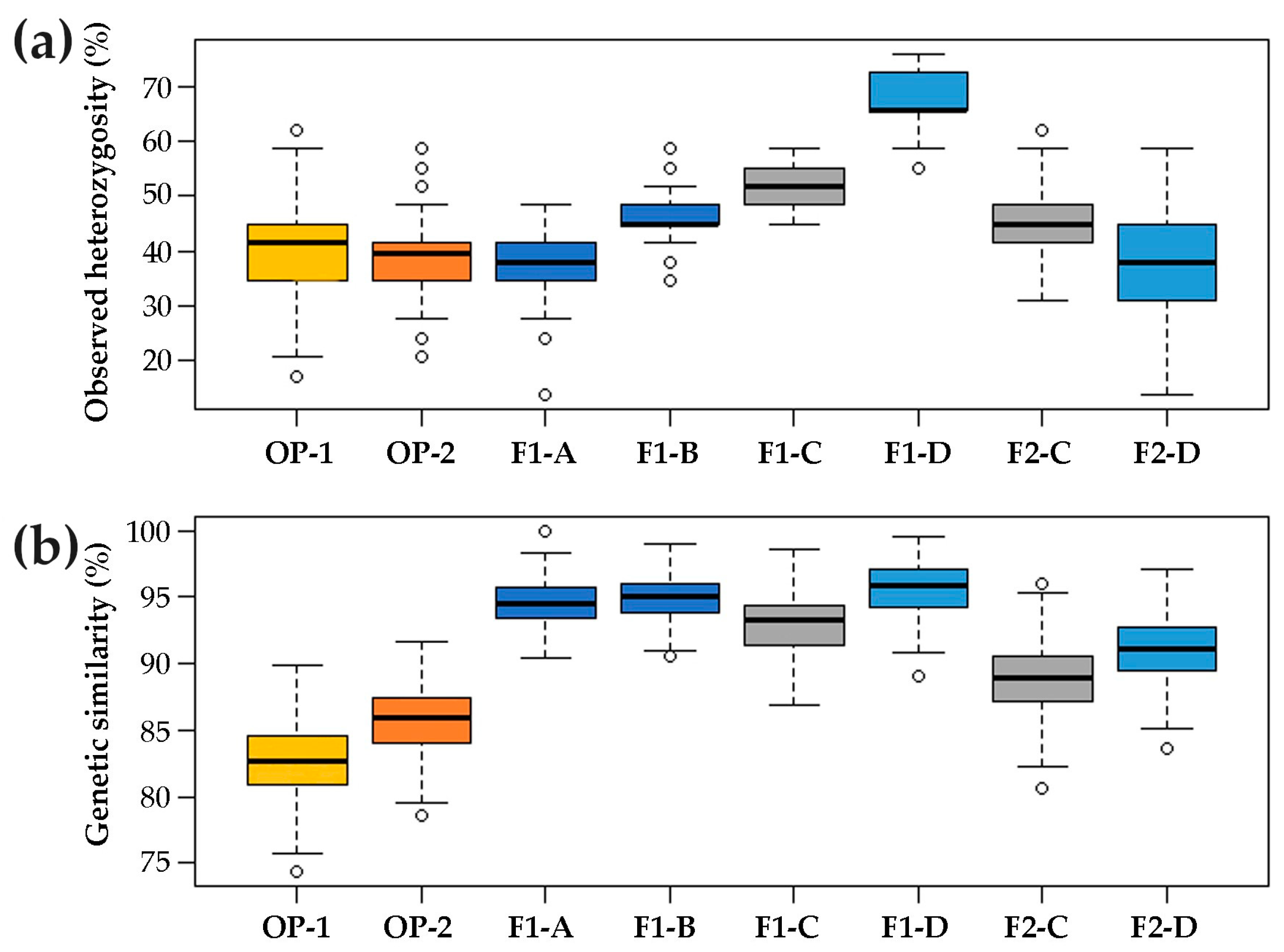
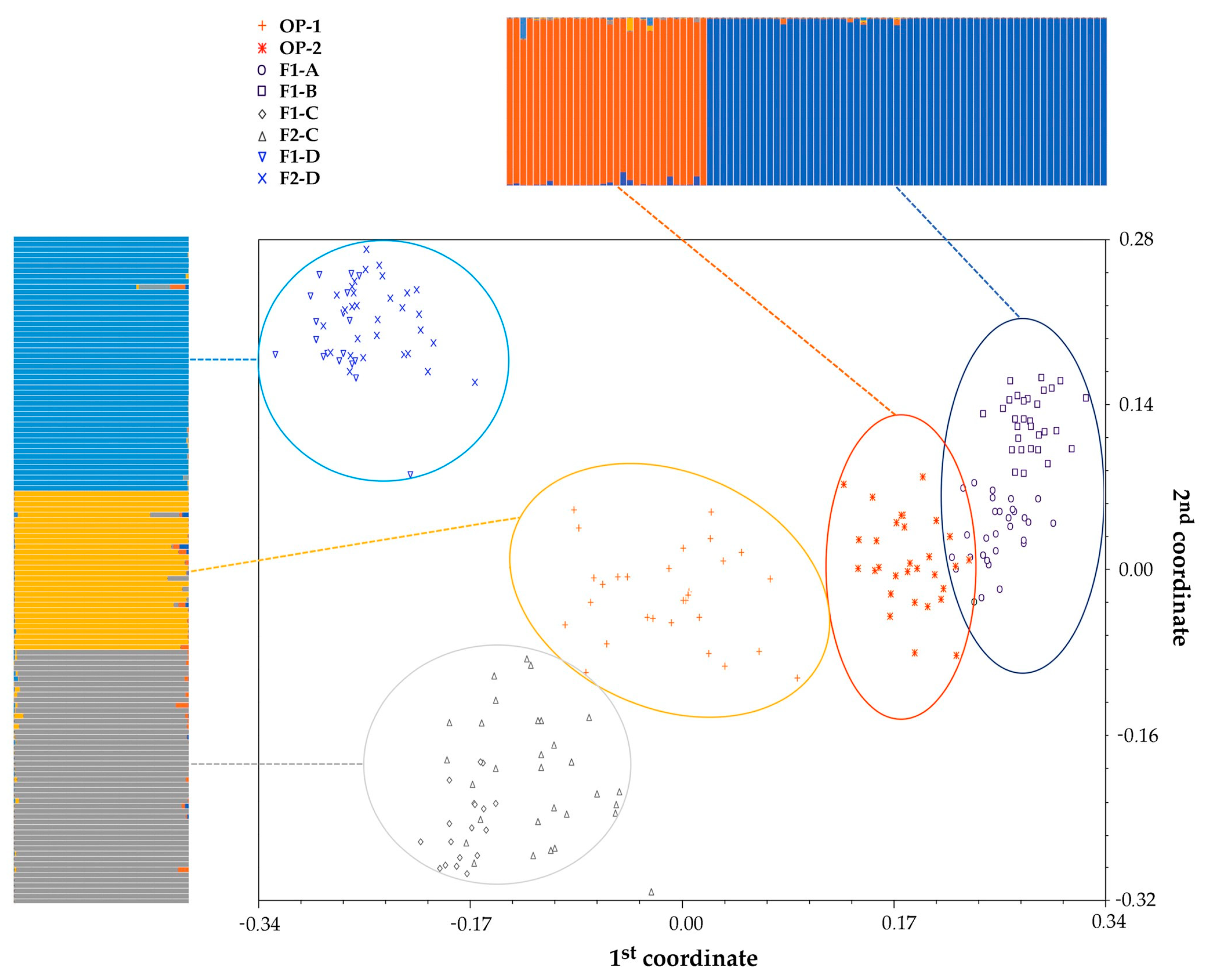
2.1. Genetic Structure of Commercial Open Pollinated (OP) Varieties, F1 Hybrids and their F2 Progenies
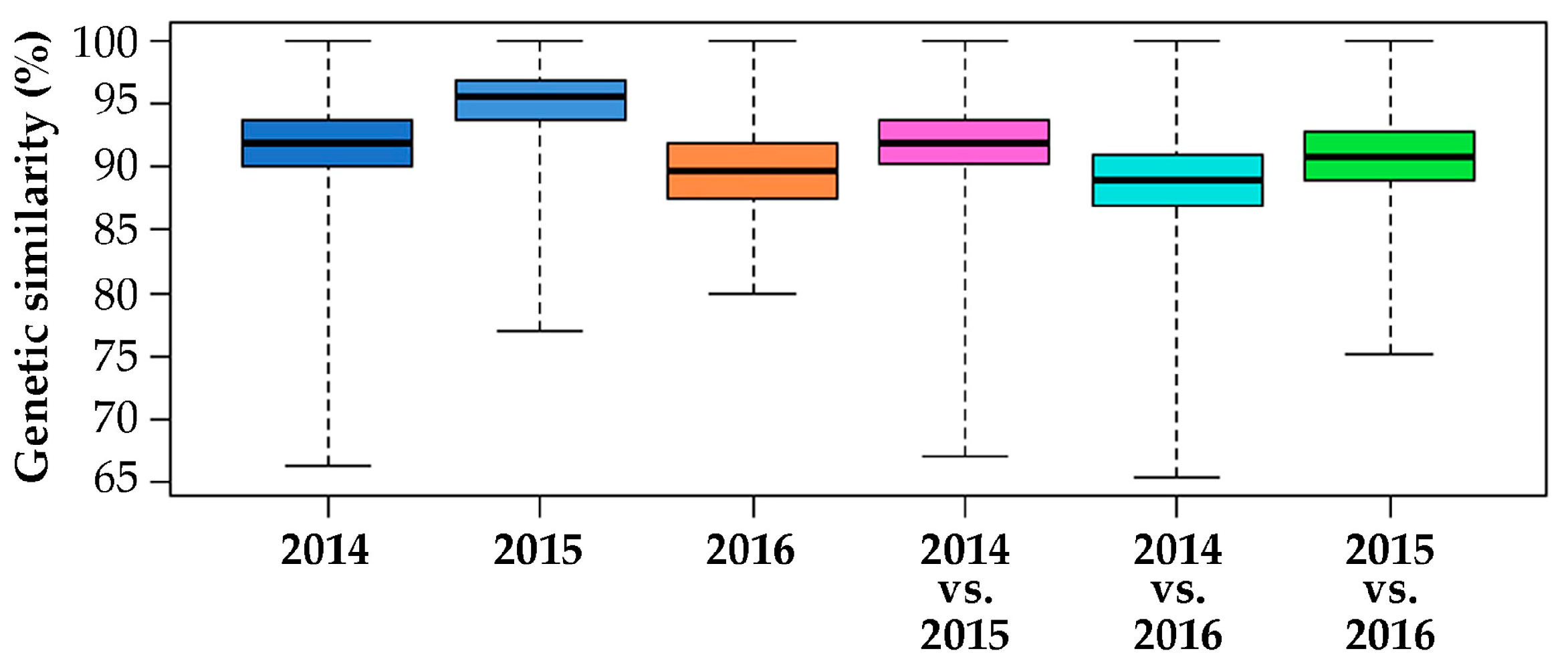
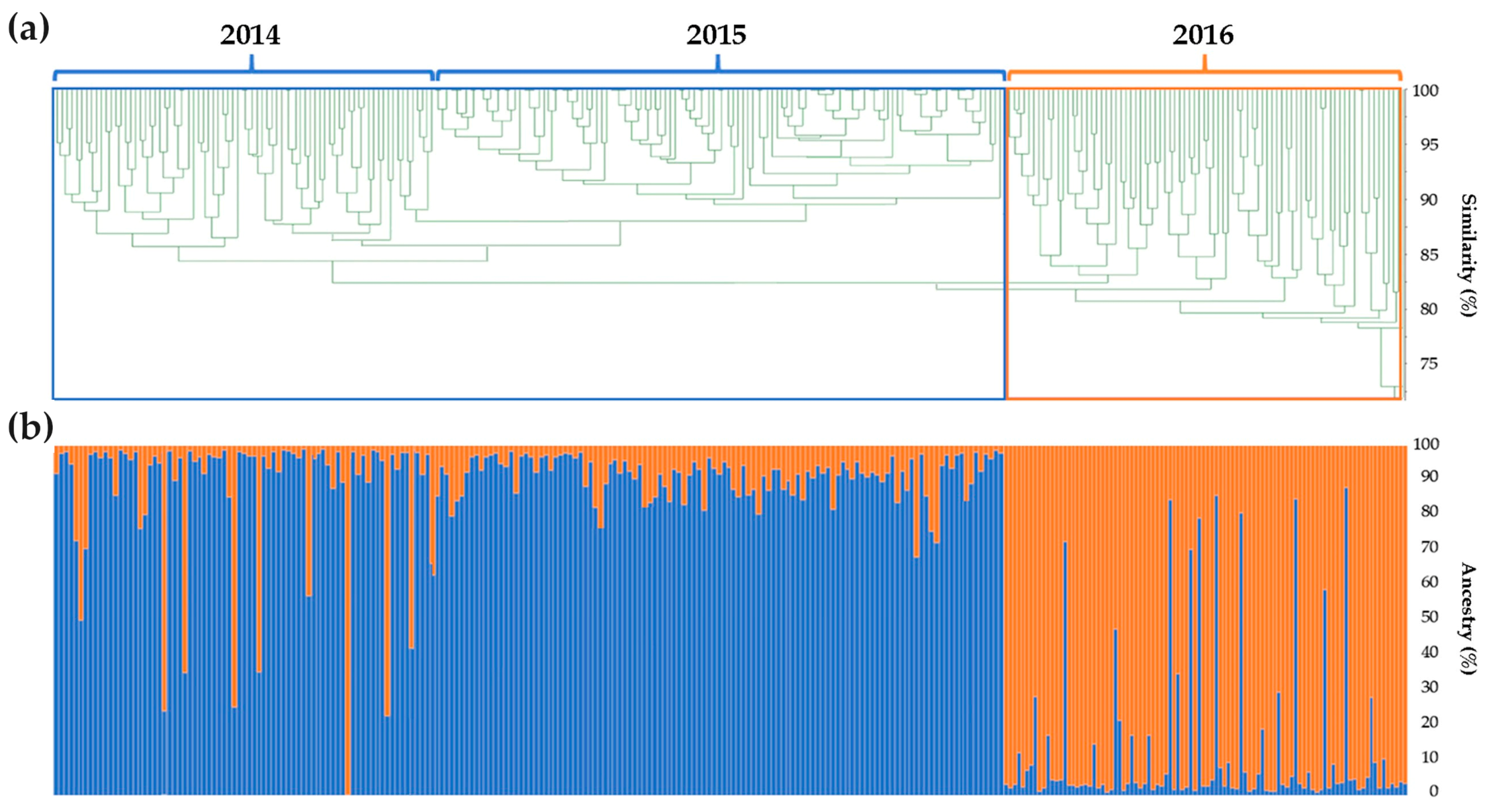
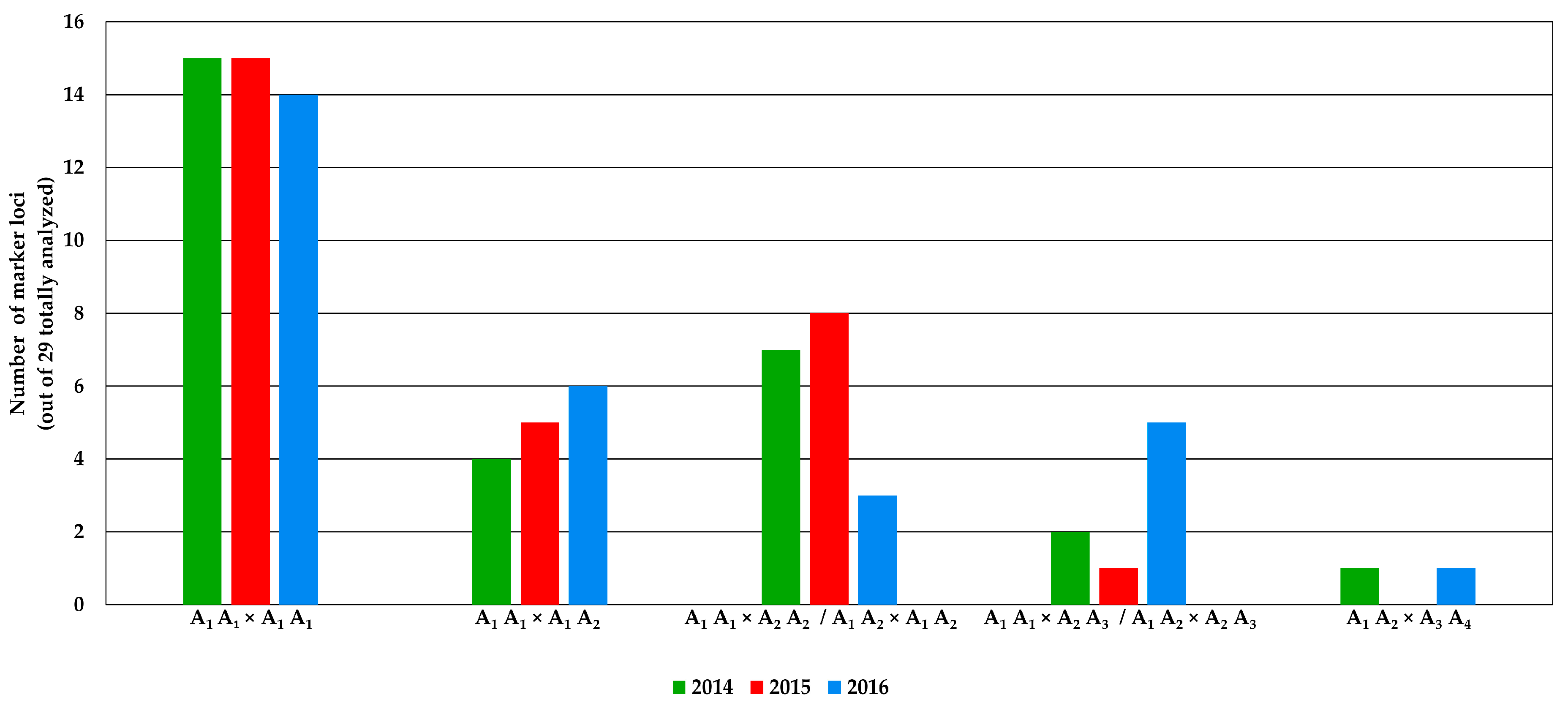
2.2. Genomic Comparison among Three Years Production of An F1 Hybrid Variety
3. Discussion
3.1. Genetic Structure of A Core Collection of OP, F1 and F2 Populations
3.2. Genomic Comparison among Three Different Production Years of A Commercial F1 Hybrid
4. Materials and Methods
4.1. Plant Materials and DNA Isolation
4.2. Genotyping by SSR Markers
4.3. Genetic Structure of Populations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Funk, V.A.; Susanna, A.; Stuessy, T.F.; Bayer, R.J. Systematics, Evolution, and Biogeography of Compositae; International Association for Plant Taxonomy, Institute of Botany, University of Vienna: Vienna, Austria, 2009; ISBN 3950175431. [Google Scholar]
- Bellamy, A.; Mathieu, C.; Vedel, F.; Bannerot, H. Cytoplasmic DNAs and nuclear rDNA restriction fragment length polymorphisms in commercial witloof chicories. Theor. Appl. Genet. 1995, 91, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Barcaccia, G.; Pallottini, L.; Soattin, M.; Lazzarin, R.; Parrini, P.; Lucchin, M. Genomic DNA fingerprints as a tool for identifying cultivated types of radicchio (Cichorium intybus L.) from Veneto, Italy. Plant Breed. 2003, 122, 178–183. [Google Scholar] [CrossRef]
- Charcosset, A.; Moreau, L. Use of molecular markers for the development of new cultivars and the evaluation of genetic diversity. Euphytica 2004, 137, 81–94. [Google Scholar] [CrossRef]
- Cadalen, T.; Mörchen, M.; Blassiau, C.; Clabaut, A.; Scheer, I.; Hilbert, J.L.; Hendriks, T.; Quillet, M.C. Development of SSR markers and construction of a consensus genetic map for chicory (Cichorium intybus L.). Mol. Breed. 2010, 25, 699–722. [Google Scholar] [CrossRef]
- Gonthier, L.; Blassiau, C.; Mörchen, M.; Cadalen, T.; Poiret, M.; Hendriks, T.; Quillet, M.C. High-density genetic maps for loci involved in nuclear male sterility (NMS1) and sporophytic self-incompatibility (S-locus) in chicory (Cichorium intybus L., Asteraceae). Theor. Appl. Genet. 2013, 126, 2103–2121. [Google Scholar] [CrossRef] [PubMed]
- Muys, C.; Thienpont, C.N.; Dauchot, N.; Maudoux, O.; Draye, X.; Cutsem, P.V. Integration of AFLPs, SSRs and SNPs markers into a new genetic map of industrial chicory (Cichorium intybus L. var. sativum). Plant Breed. 2014, 133, 130–137. [Google Scholar] [CrossRef]
- Palumbo, F.; Qi, P.; Batista Pinto, V.; Devos, K.M.; Barcaccia, G. Construction of the first SNP-based linkage map using genotyping-by-sequencing and mapping of the male-sterility gene in leaf chicory. Front. Plant Sci. 2019, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Galla, G.; Ghedina, A.; Tiozzo, S.C.; Barcaccia, G. Toward a First High-quality Genome Draft for Marker-assisted Breeding in Leaf Chicory, Radicchio (Cichorium intybus L.). Plant Genom. 2016, 1, 65–87. [Google Scholar] [CrossRef]
- Ghedina, A.; Galla, G.; Cadalen, T.; Hilbert, J.L.; Caenazzo, S.T.; Barcaccia, G. A method for genotyping elite breeding stocks of leaf chicory (Cichorium intybus L.) by assaying mapped microsatellite marker loci Genetics. BMC Res. Notes 2015, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.D.; Singh, A.K. Marker-Assisted Plant Breeding: Principles and Practices; Springer: New Delhi, India, 2015; ISBN 9788132223160. [Google Scholar]
- Van Stallen, N.; Vandenbussche, B.; Verdoodt, V.; De Proft, M. Construction of a genetic linkage map for witloof (Cichorium intybus L. var. foliosum Hegi). Plant Breed. 2003, 122, 521–526. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Brumlop, S.; Finckh, M.R. Applications and Potentials of Marker Assisted Selection (MAS) in Plant Breeding; Bundesamt für Naturschutz: Bonn, Germany, 2011; Volume 298, ISBN 9783896240330. [Google Scholar]
- Palumbo, F.; Galla, G.; Martínez-Bello, L.; Barcaccia, G. Venetian local corn (Zea mays L.) germplasm: Disclosing the genetic anatomy of old landraces suited for typical cornmeal mush production. Diversity 2017, 9, 32. [Google Scholar] [CrossRef]
- Barcaccia, G.; Ghedina, A.; Lucchin, M. Current Advances in Genomics and Breeding of Leaf Chicory (Cichorium intybus L.). Agriculture 2016, 6, 50. [Google Scholar] [CrossRef]
- Barcaccia, G.; Tiozzo, C.S. New Male Sterile Chicorium spp. Mutant, Parts or Derivatives, Where Male Sterility is Due to a Nuclear Recessive Mutation Linked to a Polymorphic Genetic Marker, Useful for Producing Mutant F1 Hybrids of Chicorium spp. EU Patent No. WO2012163389-A1, 6 December 2012. [Google Scholar]
- Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GenALEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Yeh, F.; Boyle, T. Population genetic analysis of co-dominant and dominant markers and quantitative traits. Belgian J. Bot. 1997, 130, 129–157. [Google Scholar] [CrossRef]
- Rohlf, F.J. NTSYS-pc: Microcomputer Programs for Numerical Taxonomy and Multivariate Analysis; American Statistician: New York, NY, USA, 1987; Volume 41, ISBN 0925031313. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession ID | No. of Individuals | Population Type | Varietal Cycles (d) |
|---|---|---|---|
| OP-1 | 30 | OP | 70 |
| OP-2 | 30 | OP | 110 |
| F1-A | 30 | F1 | 70 |
| F1-B | 30 | F1 | 110 |
| F1-C | 18 | F1 | 100 |
| F1-D | 18 | F1 | 70 |
| F2-C | 30 | F2 | 100 |
| F2-D | 30 | F2 | 70 |
| ID | OPs | F1s | F2s | |||
|---|---|---|---|---|---|---|
| Na | Ne | Na | Ne | Na | Ne | |
| Mean | 4.5 | 2.6 | 2.3 | 1.8 | 2.8 | 1.9 |
| St. Dev. | 2.4 | 1.2 | 0.9 | 0.6 | 0.9 | 0.5 |
| ID | LG | Motif | Primer Sequence and Tail | Multiplex | |
|---|---|---|---|---|---|
| M1.1 | 1 | (GA)40 | F | [PAN3]CCAACGGATACCAAGGTGTT | 1 |
| R | AACCGCACGGGTTCTATG | ||||
| M2.4 | 2 | (GA)25 | F | [M13]CCGCTCTCTCATCACTCCTC | 1 |
| R | GCTCGAAAATCGGCTACAAC | ||||
| M2.5 | 2 | (CT)5CC(CT)13 TT(CT)5 | F | [PAN1]GTGCCGGTCTTCAGGTTACA | 1 |
| R | CGCCTACCGATTACGATTGA | ||||
| M3.7 | 3 | (CT)22 | F | TTCGAGTCTTGCCTTAATTGTT | 1 |
| R | [PAN1]CAGACGACCTTACGGCAACT | ||||
| M4.10a | 4 | (CT)22 | F | [PAN2]CATCACCTTCACGAAAAGCA | 1 |
| R | CGAAGACCATCCATCACCA | ||||
| M4.10b | 4 | (CT)21CATA(CA)5CT(CA)5 | F | [M13] CCATTATTGGGCAGCA | 1 |
| R | CACCAACGAACTCCTTACAAAG | ||||
| M4.11a | 4 | (CT)12N5(CA)11 | F | [PAN3] GAAGGAACCTATGAACCAACCACTCA | 1 |
| R | GTTTTGAGCCTGAGCCAGA | ||||
| M5.15 | 5 | (CT)11N7(CAA)5 | F | AGCACGACTCTGCTGTCTTTT | 1 |
| R | [PAN1]CGAGCCATGTTAGGGTTTGT | ||||
| M8.22 | 8 | (CA)5AA(CA)9 | F | [PAN1]TCGTCATCAGAAACAAAGCAA | 1 |
| R | CAAAGAAGGCACTCTTGTCG | ||||
| M9.26 | 9 | (GATA)3N19(GA)9 | F | [PAN2]CCTACACTCGGCCACCTACT | 1 |
| R | TCGACGGTATAACAACACCTG | ||||
| M3.8 | 3 | (CT)16 | F | [PAN1]AGGAAGCGGTGTCATCTGT | 2 |
| R | CGCCCACATATTCATTCTCA | ||||
| M6.16 | 6 | (CT)12TT(CT)15TT(CT)2TT(CT)4 | F | [PAN1]TATTGCATTGTTGTTCCTTG | 2 |
| R | TATTTAGAAGAGGGAAATAGATG | ||||
| M7.19 | 7 | (CT)18 | F | ATGTCGGAGCAAAATCGTTC | 2 |
| R | [PAN1]CATGTTCCCGCTCATGAATA | ||||
| M1.2 | 7 | (CT)19 | F | CCGGCAGAATTTTTAGGG | 2 |
| R | [PAN3]CAGGTCATAGGTCCATGTGAAA | ||||
| M1.3 | 1 | (CT)17 | F | [PAN3]TGGAGAAAAATGAAGCAC | 2 |
| R | GAATGAGTGAGAGAATGATAGGG | ||||
| M5.13 | 5 | (CT)23 | F | [M13]AGGCATAAAGAGGTGTGG | 2 |
| R | TCAAACATGAAAACCGCTC | ||||
| M6.17 | 6 | (CA)8(CT)18 | F | CGTGTCCAAACGCAAACATTAT | 2 |
| R | [PAN2]GCACAATTTTCCTACCACTTATCC | ||||
| M5.14 | 5 | (TC)11 | F | [M13]AAAGTCACACATCGCATTTCCT | 2 |
| R | GTAGCAGCAGCAGCCATCTT | ||||
| M4.11b | 4 | (TG)5CG(TG)7 | F | [M13]GCCATTCCTTTCAAGAGCAG | 2 |
| R | AACCCAAAACCGCAACAATA | ||||
| M4.12 | 4 | (CT)8TT(CT)5CC(CT)3TT(CT)7 | F | GGCATCGGGATAGAAAAACA | 2 |
| R | [PAN2]TCAATGCCTCAACAGAAATCC | ||||
| M3.9 | 3 | (CA)12 | F | CTGCTATGGACAGTTCCAGT | 3 |
| R | [PAN3]CAATTCAGTTGTGATAGACGC | ||||
| M7.20 | 7 | (CT)31 | F | [PAN2]ACACTCACTCACACTCCGTAA | 3 |
| R | GTCATGATGGCGTAAAAGTC | ||||
| M8.23 | 8 | (CA)11(CT)9 | F | TGTAGACACACAAAATGCACA | 3 |
| R | [M13]ACCGGTTGAAAACATGAAAT | ||||
| M8.24 | 8 | (TC)16(CA)13 | F | [PAN2]GGTCCGTAGACTGCAGACTTTT | 3 |
| R | CACCGTCCCACTTTTTAGG | ||||
| M9.25 | 9 | (CA)11 | F | [M13]GTGTGGGTGTTTGAAGAGC | 3 |
| R | TCAAGAACATCAACGCGTAA | ||||
| M7.21 | 1 | (CT)13 | F | GGACACCGAGCTGGAGAA | 3 |
| R | [PAN1]TTCCACTTTCGGGAGTTACC | ||||
| M9.27 | 9 | (GA)10TAAA(GA)5 | F | GCTAAAAGAAGTGCAAGGAGA | 3 |
| R | [PAN1]TGTTCTTTCAAGTGCCAA | ||||
| M6.18 | 6 | (CT)16 | F | [PAN3]CTCAACGAATGCTTTGGACA | 3 |
| R | CCTCGCGGTAGCTTATTGTT | ||||
| M2.6 | 2 | (CT)26 | F | GGAGCAGGTAGAGTCCCATC | 3 |
| R | [PAN1]CGTTTGAAAATTTATACCAAAATG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patella, A.; Scariolo, F.; Palumbo, F.; Barcaccia, G. Genetic Structure of Cultivated Varieties of Radicchio (Cichorium intybus L.): A Comparison between F1 Hybrids and Synthetics. Plants 2019, 8, 213. https://0-doi-org.brum.beds.ac.uk/10.3390/plants8070213
Patella A, Scariolo F, Palumbo F, Barcaccia G. Genetic Structure of Cultivated Varieties of Radicchio (Cichorium intybus L.): A Comparison between F1 Hybrids and Synthetics. Plants. 2019; 8(7):213. https://0-doi-org.brum.beds.ac.uk/10.3390/plants8070213
Chicago/Turabian StylePatella, Alice, Francesco Scariolo, Fabio Palumbo, and Gianni Barcaccia. 2019. "Genetic Structure of Cultivated Varieties of Radicchio (Cichorium intybus L.): A Comparison between F1 Hybrids and Synthetics" Plants 8, no. 7: 213. https://0-doi-org.brum.beds.ac.uk/10.3390/plants8070213