The First Genetic Map for a Psoraleoid Legume (Bituminaria bituminosa) Reveals Highly Conserved Synteny with Phaseoloid Legumes

 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. GBS Genotyping
2.2. Linkage Mapping
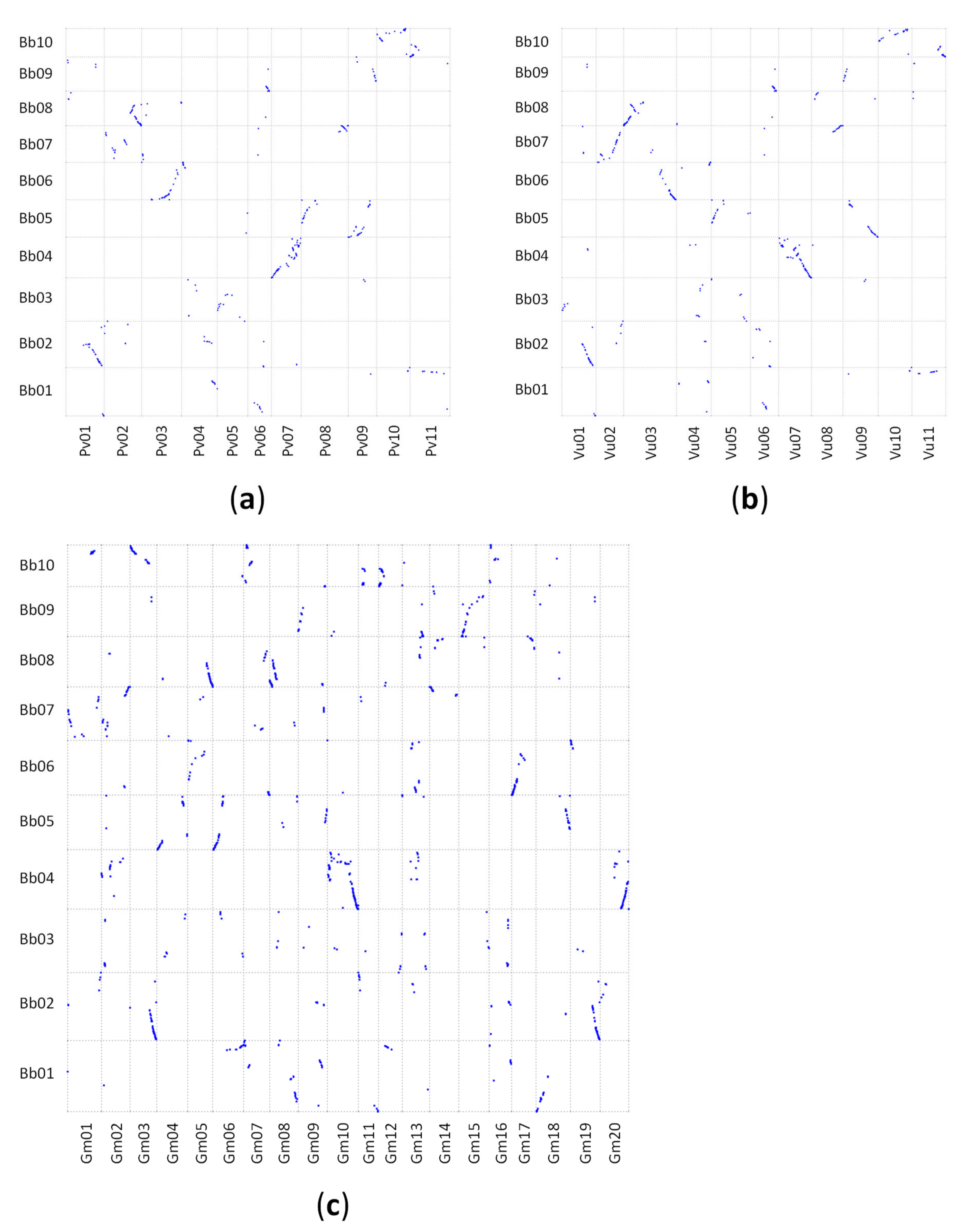
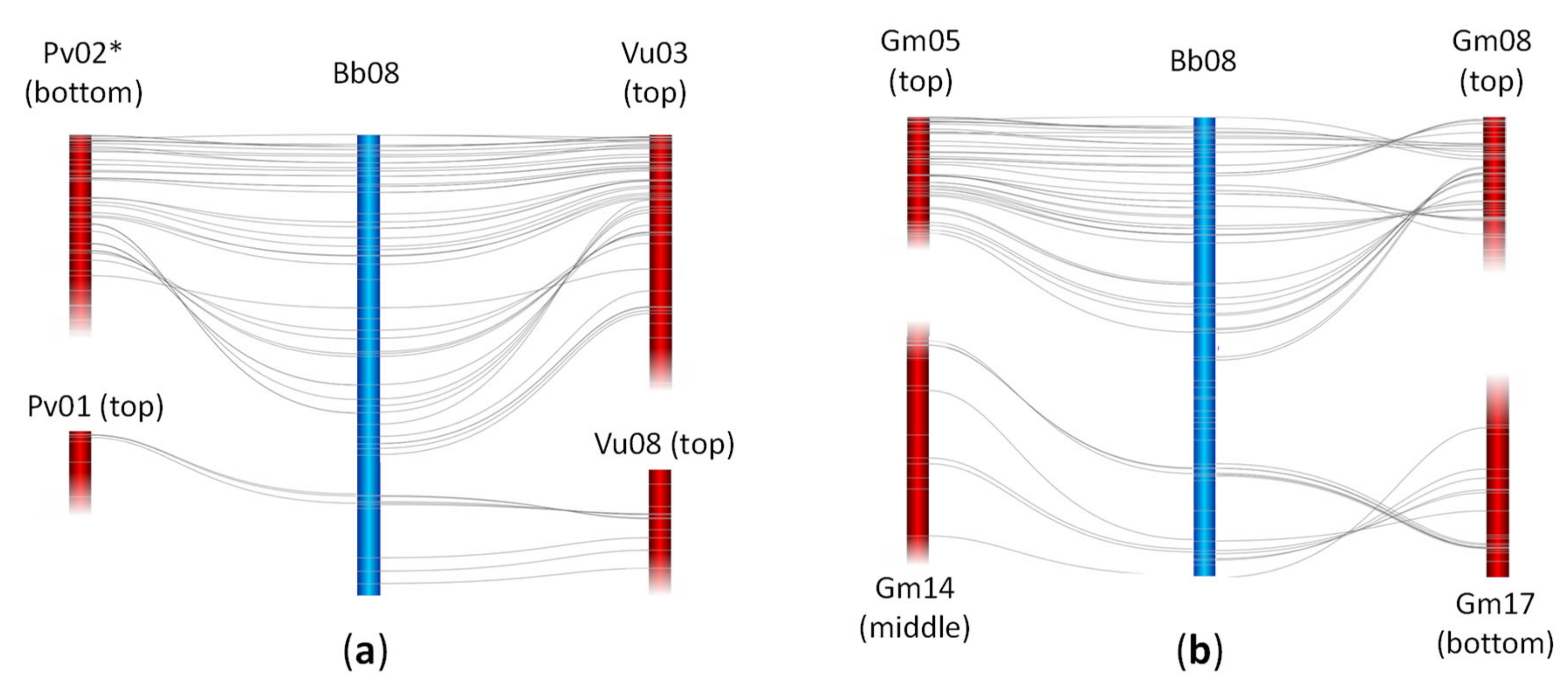
2.3. Synteny Analysis
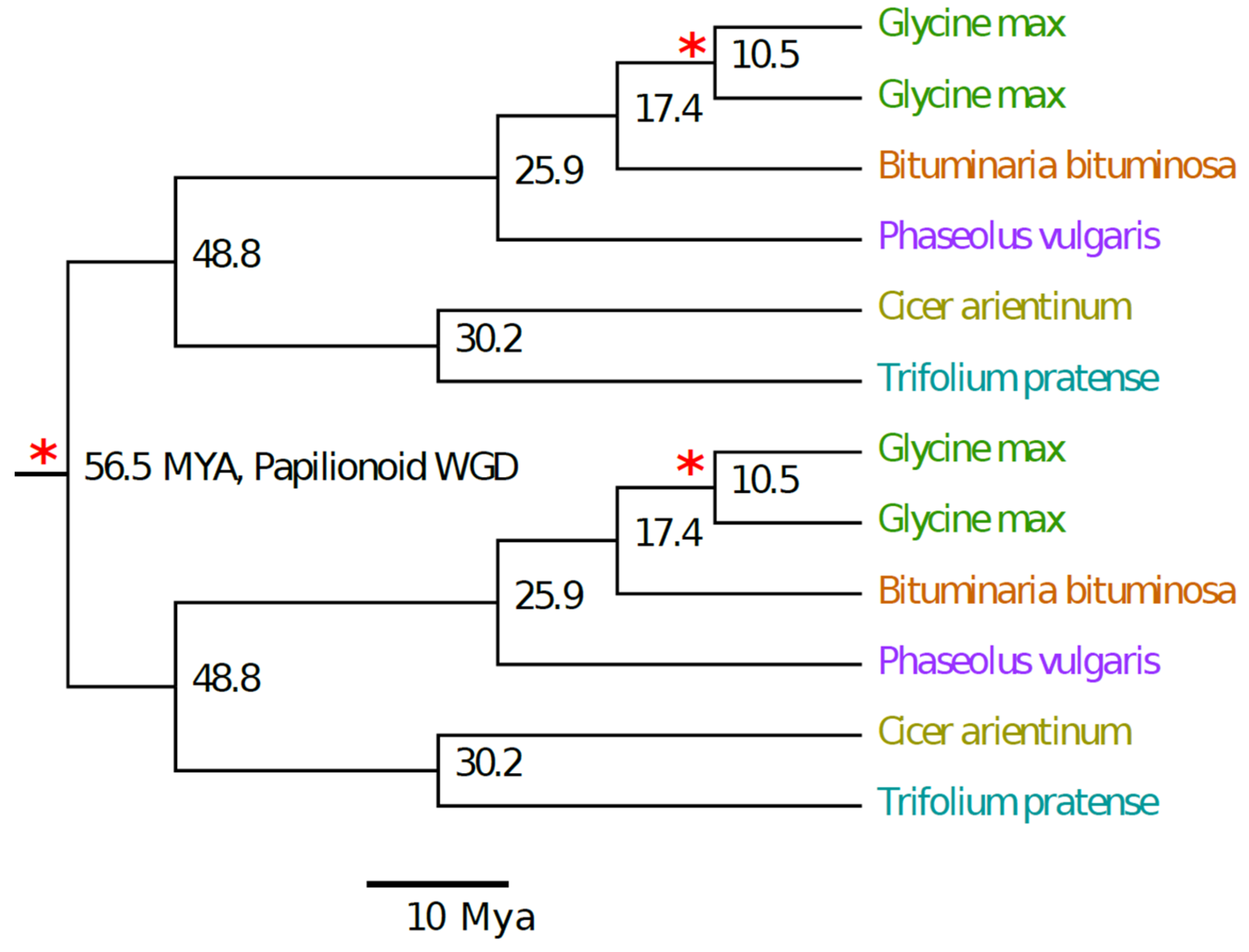
2.4. Ks Divergence Time Estimates
3. Discussion
4. Materials and Methods
4.1. Experimental Population
4.2. GBS Marker Genotyping and Bioinformatics Analysis
4.3. Linkage Mapping
4.4. Comparative Mapping of Tedera and Phaseoloid Genomes
4.5. Estimating Genome Divergence Times by Ks Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Egan, A.N.; Crandall, K.A. Incorporating gaps as phylogenetic characters across eight DNA regions: Ramifications for North American Psoraleeae (Leguminosae). Mol. Phylogenet. Evol. 2008, 46, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Real, D.; Oldham, C.M.; Nelson, M.N.; Croser, J.; Castello, M.; Verbyla, A.; Pradhan, A.; Van Burgel, A.; Mendez, P.; Correal, E.; et al. Evaluation and breeding of tedera for Mediterranean climates in southern Australia. Crop Pasture Sci. 2014, 65, 1114–1131. [Google Scholar] [CrossRef]
- Méndez, P.; Fernández, M.; Santos, A. Variedades de Bituminaria bituminosa (L.) Stirton (Leguminosae) en el archipélago canario. Pastos 1991, 2021, 157–166. [Google Scholar]
- Stirton, C.H. Tribe 11. Psoraleeae (Benth.) Rydb. (1919). In Advances in Legume Systematics Part 1; Polhill, R.M., Raven, P.H., Eds.; Royal Botanic Gardens: Kew, UK, 1981; pp. 337–343. [Google Scholar]
- Juan, A.; Monino, I.; Correal, E.; Méndez, P.; Crespo, M.B. Comparación de las tasas de fructificación de Bituminaria bituminosa (Leguminosae) bajo condiciones de cultivo en Canarias y Península Ibérica. In Pastos y Ganadería extensiva; Criado, B.G., Cuidad, A.G., Vd Aldana, B.R., Zabalgogeazcoa, I., Eds.; Sociedad Espanola para el Estudio de los Pastos: Salamanca, Spain, 2004; pp. 111–115. [Google Scholar]
- Pradhan, A.; Besharat, N.; Castello, M.; Croser, J.; Real, D.; Nelson, M.N. Evidence for outcrossing in the perennial forage legume tedera (Bituminaria bituminosa). Crop Sci. 2014, 54, 2406–2412. [Google Scholar] [CrossRef]
- Real, D.; Oldham, C.M.; van Burgel, A.; Dobbe, E.; Hardy, J. Tedera proves its value as a summer and autumn feed for sheep in Mediterranean-like climates. Anim. Prod. Sci. 2018, 58, 2269–2279. [Google Scholar] [CrossRef]
- Lanza® Tedera. Available online: https://www.agric.wa.gov.au/pasture-species/lanza%C2%AE-tedera (accessed on 27 May 2020).
- Pazos-Navarro, M.; Dabauza, M.; Correal, E.; Hanson, K.; Teakle, N.; Real, D.; Nelson, M. Next generation DNA sequencing technology delivers valuable genetic markers for the genomic orphan legume species, Bituminaria bituminosa. BMC Genet. 2011, 12, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Río, J.; Díaz, L.; García-Bernal, D.; Blanquer, M.; Ortuño, A.; Correal, E.; Moraleda, J. Furanocoumarins: Biomolecules of therapeutic interest. In Studies in Natural Products Chemistry; Atta-ur-Rahman, F., Ed.; International Center for Chemical and Biological Sciences, HEJ research Institute of Chemistry, University of Karachi: Karachi, Pakistan, 2014; Volume 43, pp. 145–195. [Google Scholar]
- Innocenti, G.; Cappelletti, E.; Caporale, G. Furanocoumarin contents in the vegetative organs of cultivated Psoralea species. Int. J. Pharm. 1991, 29, 141–146. [Google Scholar]
- Martínez, S.; Correal, E.; Real, D.; Ortuño, A.; Del Río, J. Bituminaria bituminosa: A source of furanocoumarins of pharmaceutical interest. In Drug Plants I; AS Awaad, J.G.V.S., Ed.; Studium Press LLC: Houston, TX, USA, 2010; pp. 307–322. [Google Scholar]
- Morison, W.L. Psoralen ultraviolet A therapy in 2004. Photodermatol. Photoimmunol. Photomed. 2004, 20, 315–320. [Google Scholar] [CrossRef]
- Shikishima, Y.; Takaishi, Y.; Honda, G.; Ito, M.; Takeda, Y.; Kodzhimatov, O.K.; Ashurmetov, O.; Lee, K.-H. Chemical constituents of Prangos tschimganica; structure elucidation and absolute configuration of coumarin and furanocoumarin derivatives with anti-HIV activity. Chem. Pharm. Bull. 2001, 49, 877–880. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Takaishi, Y.; Duan, H.; Chen, B.; Honda, G.; Ito, M.; Takeda, Y.; Kodzhimatov, O.K.; Lee, K.-H. Coumarins and bicoumarin from Ferula sumbul: Anti-HIV activity and inhibition of cytokine release. Phytochemistry 2000, 53, 689–697. [Google Scholar] [CrossRef]
- Wolff, D.; Schleuning, M.; von Harsdorf, S.; Bacher, U.; Gerbitz, A.; Stadler, M.; Ayuk, F.; Kiani, A.; Schwerdtfeger, R.; Vogelsang, G.B.; et al. Consensus conference on clinical practice in chronic GVHD: Second-line treatment of chronic graft-versus-host disease. Biol. Blood Marrow Transplant. 2011, 17, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pistelli, L.; Noccioli, C.; Appendina, G.; Bianchi, F.; Sterner, O.; Ballero, M. Pterocarpans from Bituminaria morisiana and Bituminaria bituminosa. Phytochemistry 2003, 64, 595–598. [Google Scholar] [CrossRef]
- Maurich, T.; Iorio, M.; Chimenti, D.; Turchi, G. Erybraedin C and bitucarpin A, two structurally related pterocarpans purified from Bituminaria bituminosa, induced apoptosis in human colon adenocarcinoma cell lines MMR- and p53-proficient and -deficient in a dose-, time-, and structure-dependent fashion. Chem. Biol. Interact. 2006, 159, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Maurich, T.; Pistelli, L.; Turchi, G. Anti-clastogenic activity of two structurally related pterocarpans purified from Bituminaria bituminosa in cultured human lymphocytes. Mutat. Res. 2004, 561, 75–81. [Google Scholar] [CrossRef]
- Walker, D.; Moñino, I.; Correal, E. Genome size in Bituminaria bituminosa (L.) CH Stirton (Fabaceae) populations: Separation of “true” differences from environmental effects on DNA determination. Environ. Exp. Bot. 2006, 55, 258–265. [Google Scholar] [CrossRef]
- Stefanović, S.; Pfeil, B.E.; Palmer, J.D.; Doyle, J.J. Relationships among phaseoloid legumes based on sequences from eight chloroplast regions. Syst. Bot. 2009, 34, 115–128. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Schmutz, J.; McClean, P.E.; Mamidi, S.; Wu, G.A.; Cannon, S.B.; Grimwood, J.; Jenkins, J.; Shu, S.; Song, Q.; Chavarro, C.; et al. A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 2014, 46, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Severin, A.J.; Woody, J.L.; Bolon, Y.-T.; Joseph, B.; Diers, B.W.; Farmer, A.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef] [Green Version]
- Bolon, Y.-T.; Haun, W.J.; Xu, W.W.; Grant, D.; Stacey, M.G.; Nelson, R.T.; Gerhardt, D.J.; Jeddeloh, J.A.; Stacey, G.; Muehlbauer, G.J.; et al. Phenotypic and genomic analyses of a fast neutron mutant population resource in soybean. Plant Physiol. 2011, 156, 240–253. [Google Scholar] [CrossRef] [Green Version]
- Leebens-Mack, J.H.; Barker, M.S.; Carpenter, E.J.; Deyholos, M.K.; Gitzendanner, M.A.; Graham, S.W.; Grosse, I.; Li, Z.; Melkonian, M.; Mirarab, S.; et al. One thousand plant transcriptomes and the phylogenomics of green plants. Nature 2019, 574, 679–685. [Google Scholar]
- Cannon, S.B.; McKain, M.R.; Harkess, A.; Nelson, M.N.; Dash, S.; Deyholos, M.K.; Peng, Y.; Joyce, B.; Stewart, C.N.; Rolf, M.; et al. Multiple polyploidy events in the early radiation of nodulating and nonnodulating legumes. Mol. Biol. Evol. 2015, 32, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Van Orsouw, N.J.; Hogers, R.C.J.; Janssen, A.; Yalcin, F.; Snoeijers, S.; Verstege, E.; Schneiders, H.; Van Der Poel, H.; Van Oeveren, J.; Verstegen, H.; et al. Complexity Reduction of Polymorphic Sequences (CRoPS™): A novel approach for large-scale polymorphism discovery in complex genomes. PLoS ONE 2007, 2, e1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Beissinger, T.M.; Hirsch, C.N.; Sekhon, R.S.; Foerster, J.M.; Johnson, J.M.; Muttoni, G.; Vaillancourt, B.; Buell, C.R.; Kaeppler, S.M.; De Leon, N. Marker density and read depth for genotyping populations using genotyping-by-sequencing. Genetics 2013, 193, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Sonah, H.; Bastien, M.; Iquira, E.; Tardivel, A.; Légaré, G.; Boyle, B.; Normandeau, E.; Laroche, J.; LaRose, S.; Jean, M.; et al. An improved genotyping by sequencing (GBS) approach offering increased versatility and efficiency of SNP discovery and genotyping. PLoS ONE 2013, 8, e54603. [Google Scholar] [CrossRef] [Green Version]
- Andolfatto, P.; Davison, D.; Erezyilmaz, D.; Hu, T.T.; Mast, J.; Sunayama-Morita, T.; Stern, D.L. Multiplexed shotgun genotyping for rapid and efficient genetic mapping. Genome Res. 2011, 21, 610–617. [Google Scholar] [CrossRef] [Green Version]
- Peterson, B.K.; Weber, J.N.; Kay, E.H.; Fisher, H.S.; Hoekstra, H.E. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 2012, 7, e37135. [Google Scholar] [CrossRef] [Green Version]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Meyer, E.; McKay, J.K.; Matz, M.V. 2b-RAD: A simple and flexible method for genome-wide genotyping. Nat. Methods 2012, 9, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Toonen, R.J.; Puritz, J.B.; Forsman, Z.H.; Whitney, J.L.; Fernandez-Silva, I.; Andrews, K.R.; Bird, C.E. ezRAD: A simplified method for genomic genotyping in non-model organisms. PeerJ 2013, 1, e203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Liu, D.; Zhang, X.; Li, W.; Liu, H.; Hong, W.; Jiang, C.; Guan, N.; Ma, C.; Zeng, H.; et al. SLAF-seq: An efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS ONE 2013, 8, e58700. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.; Herendeen, P.; Wojchiechowski, M. Evolutionary rates analysis of Leguminosae implicates a rapid diversification of lineages during the Tertiary. Syst. Biol. 2005, 54, 575–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Huang, W.; Cannon, S.B. Reconstruction of ancestral genome reveals chromosome evolution history for selected legume species. New Phytol. 2019, 223, 2090–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catchen, J.; Hohenlohe, P.A.; Bassham, S.; Amores, A.; Cresko, W.A. Stacks: An analysis tool set for population genomics. Mol. Ecol. 2013, 22, 3124–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mester, D.; Ronin, Y.; Minkov, D.; Nevo, E.; Korol, A. Constructing large-scale genetic maps using an evolutionary strategy algorithm. Genetics 2003, 165, 2269–2282. [Google Scholar] [PubMed]
- Nelson, M.N.; Moolhuijzen, P.M.; Boersma, J.G.; Chudy, M.; Lesniewska, K.; Bellgard, M.I.; Oliver, R.P.; Święcicki, W.; Wolko, B.; A Cowling, W.; et al. Aligning a new reference genetic map of Lupinus angustifolius with the genome sequence of the model legume, Lotus japonicus. DNA Res. 2010, 17, 73–83. [Google Scholar] [CrossRef] [Green Version]
- Kroc, M.; Koczyk, G.; Święcicki, W.; Kilian, A.; Nelson, M.N. New evidence of ancestral polyploidy in the Genistoid legume Lupinus angustifolius L. (narrow-leafed lupin). Theor. Appl. Genet. 2014, 127, 1237–1249. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vega, J.J.; Ayling, S.; Hegarty, M.; Kudrna, D.; Goicoechea, J.L.; Ergon, Å.; Rognli, O.A.; Jones, C.; Swain, M.; Geurts, R.; et al. Red clover (Trifolium pratense L.) draft genome provides a platform for trait improvement. Sci. Rep. 2015, 5, 17394. [Google Scholar] [CrossRef]
- Varshney, R.K.; Song, C.; Saxena, R.K.; Azam, S.; Yu, S.; Sharpe, A.G.; Cannon, S.; Baek, J.; Rosen, B.D.; Tar’An, B.; et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 2013, 31, 240–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H. GitHub. Available online: https://github.com/tanghaibao/bio-pipeline/blob/master/synonymous_calculation/synonymous_calc.py (accessed on 20 June 2017).
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; López, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP-phylogeny inference package (version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Linkage Group | Skeleton 1 | Redundant 2 | Attached 3 | Total | Length (cM) | Mean Interval Size 4 | Max Interval Size 4 |
|---|---|---|---|---|---|---|---|
| Bb01 | 77 | 62 | 13 | 152 | 113.7 | 1.50 | 11.2 |
| Bb02 | 103 | 155 | 6 | 264 | 109.3 | 1.07 | 4.6 |
| Bb03 | 55 | 70 | 19 | 144 | 102.1 | 1.89 | 10.2 |
| Bb04 | 107 | 181 | 10 | 298 | 95.3 | 0.90 | 7.0 |
| Bb05 | 87 | 109 | 12 | 208 | 88.2 | 1.03 | 7.6 |
| Bb06 | 72 | 116 | 21 | 209 | 88.2 | 1.24 | 13.1 |
| Bb07 | 69 | 117 | 22 | 208 | 85.9 | 1.26 | 5.2 |
| Bb08 | 81 | 136 | 8 | 225 | 81.7 | 1.02 | 4.6 |
| Bb09 | 54 | 54 | 19 | 127 | 80 | 1.51 | 7.3 |
| Bb10 | 66 | 136 | 5 | 207 | 66.9 | 1.03 | 6.1 |
| Total | 771 | 1136 | 135 | 2042 | 911.3 | 1.20 | 13.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nelson, M.N.; Jabbari, J.S.; Turakulov, R.; Pradhan, A.; Pazos-Navarro, M.; Stai, J.S.; Cannon, S.B.; Real, D. The First Genetic Map for a Psoraleoid Legume (Bituminaria bituminosa) Reveals Highly Conserved Synteny with Phaseoloid Legumes. Plants 2020, 9, 973. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9080973
Nelson MN, Jabbari JS, Turakulov R, Pradhan A, Pazos-Navarro M, Stai JS, Cannon SB, Real D. The First Genetic Map for a Psoraleoid Legume (Bituminaria bituminosa) Reveals Highly Conserved Synteny with Phaseoloid Legumes. Plants. 2020; 9(8):973. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9080973
Chicago/Turabian StyleNelson, Matthew N., Jafar S. Jabbari, Rust Turakulov, Aneeta Pradhan, Maria Pazos-Navarro, Jacob S. Stai, Steven B. Cannon, and Daniel Real. 2020. "The First Genetic Map for a Psoraleoid Legume (Bituminaria bituminosa) Reveals Highly Conserved Synteny with Phaseoloid Legumes" Plants 9, no. 8: 973. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9080973