A Genome-Wide Survey of MATE Transporters in Brassicaceae and Unveiling Their Expression Profiles under Abiotic Stress in Rapeseed


 ,
,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Identification of MATE Transporters in Brassicaceae Species
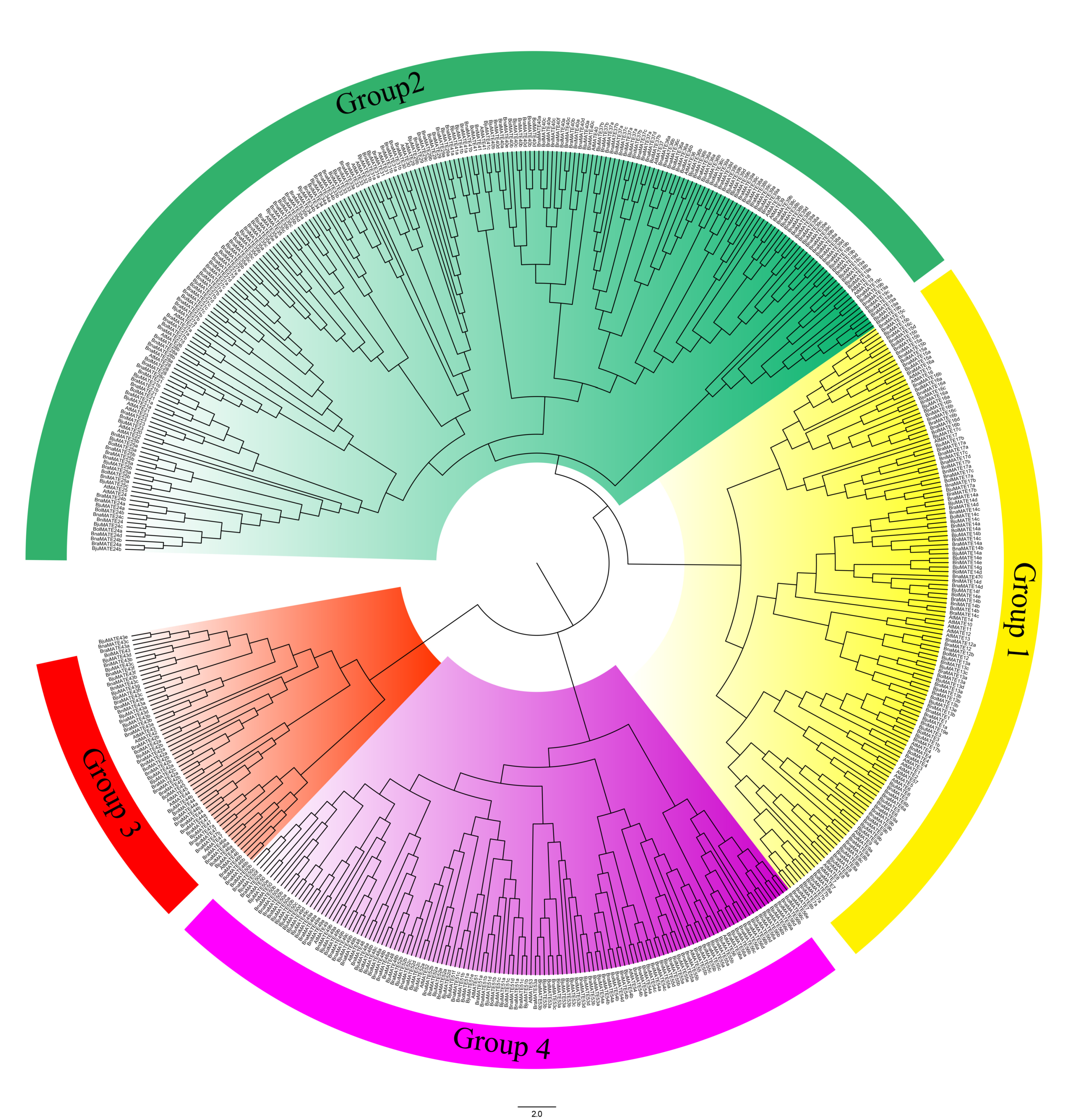
2.2. Phylogenetic Analysis of the MATE Transporters
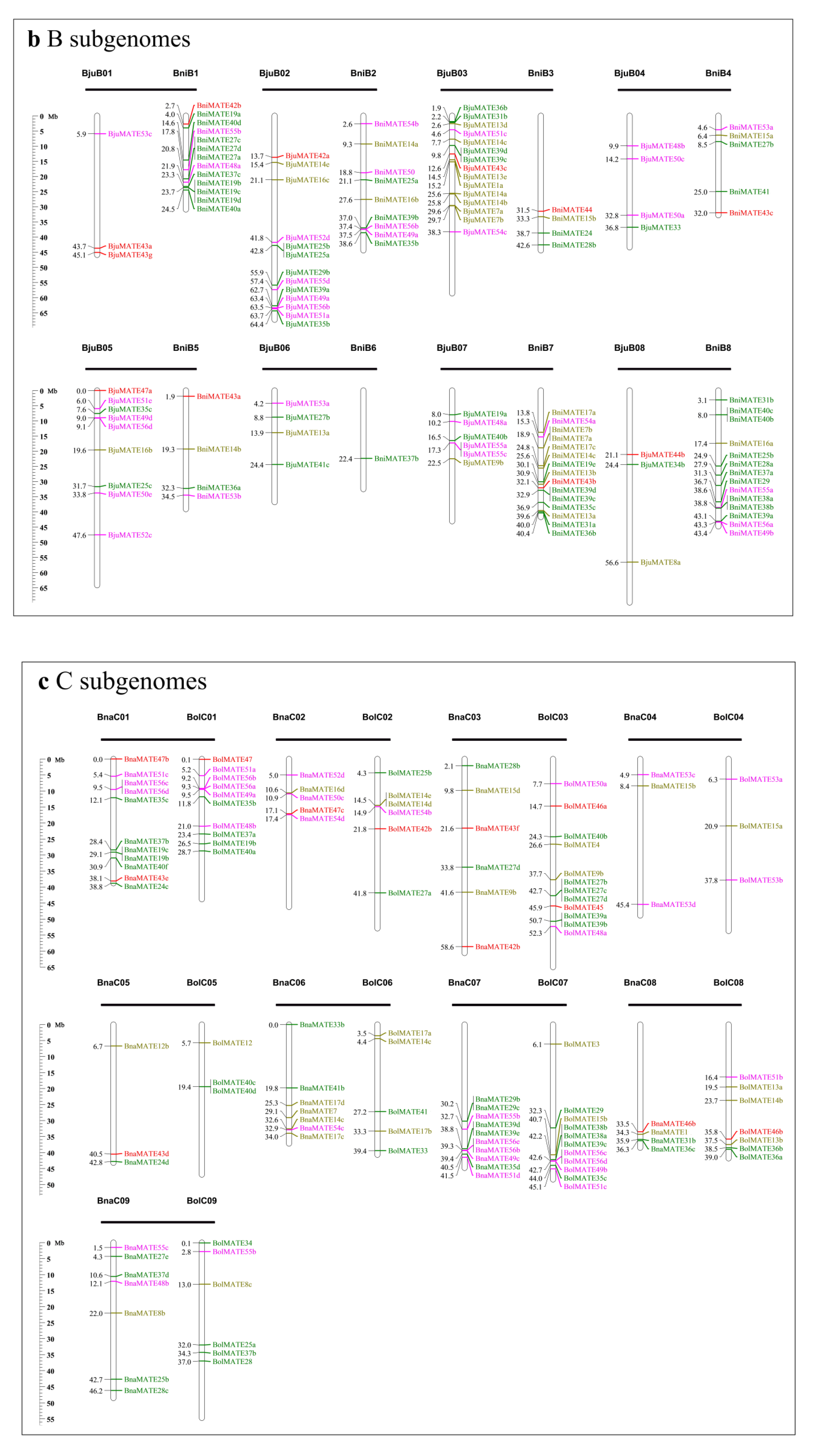
2.3. Chromosomal Location of MATE Genes in Brassicaceae Genomes
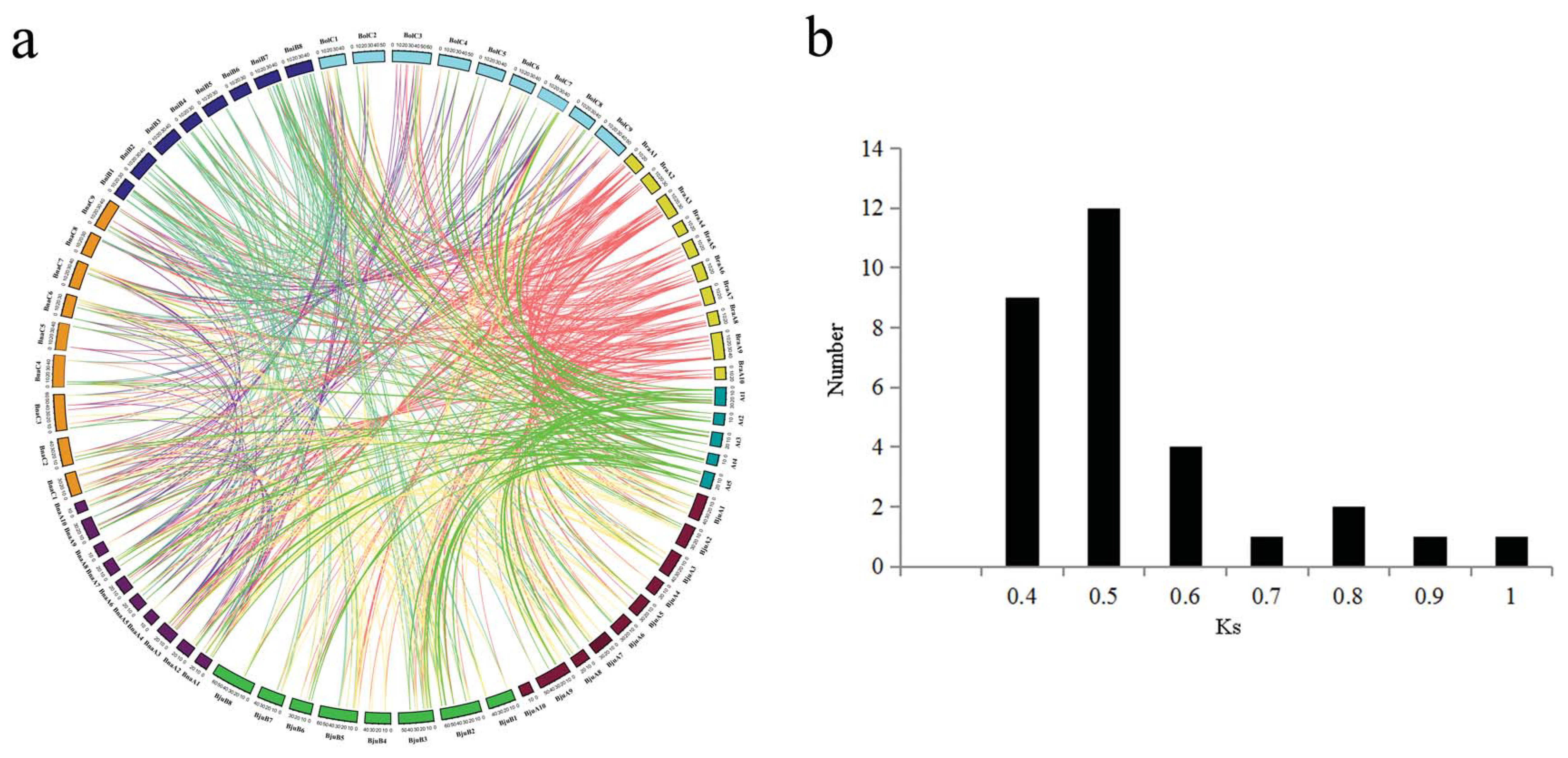
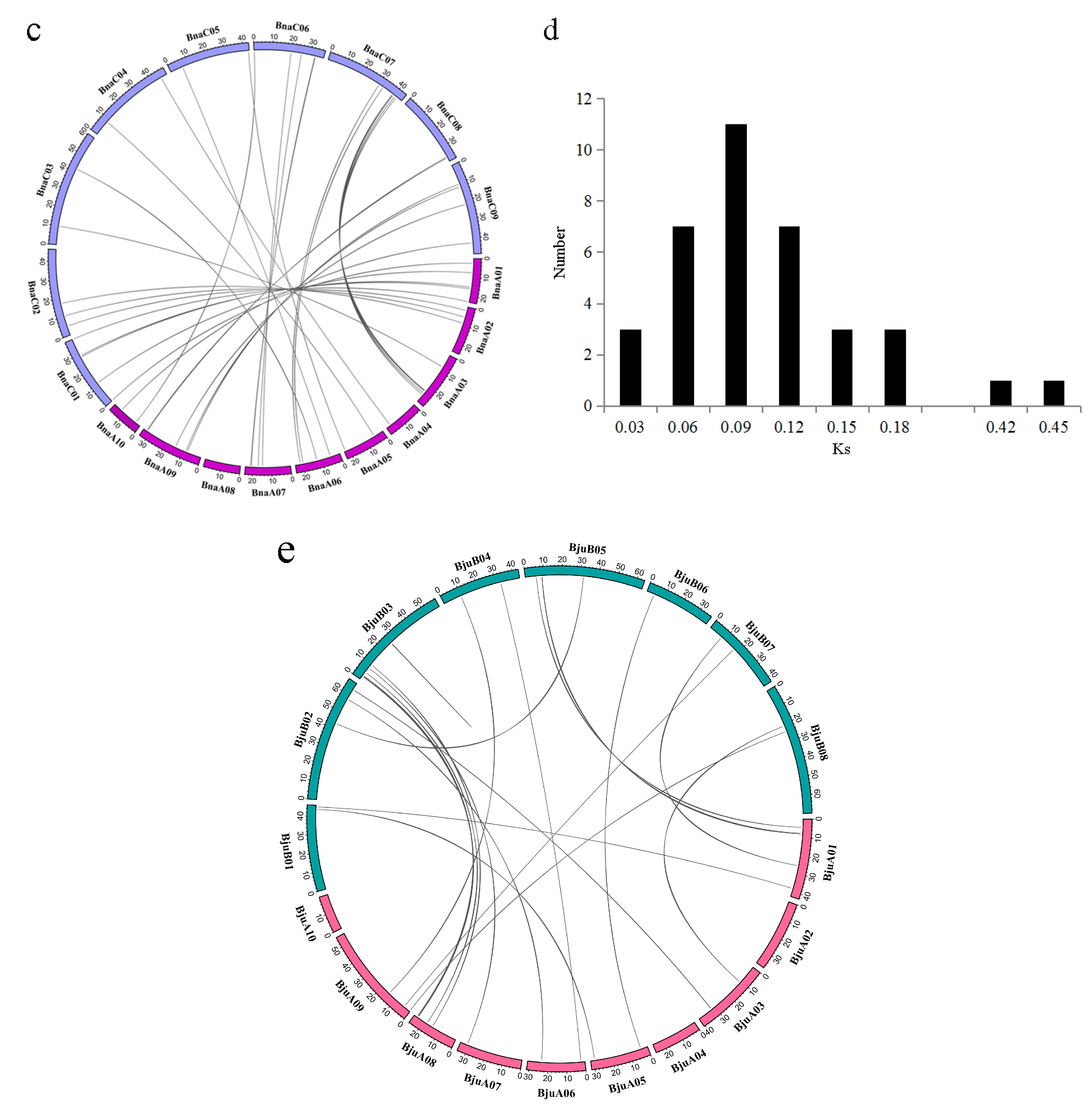
2.4. Synteny and Gene Duplication Analysis of MATE Transporters
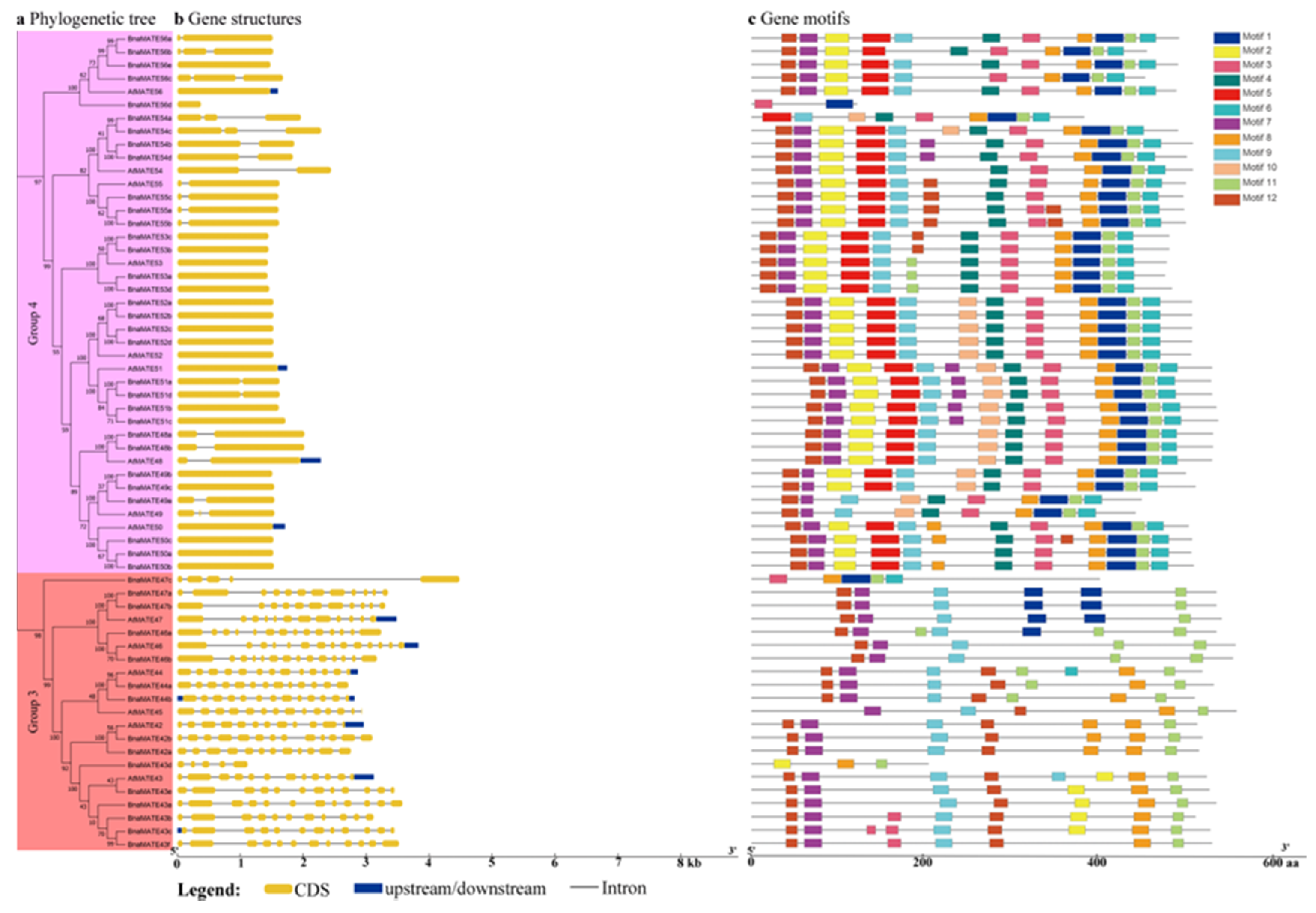
2.5. Analysis of MATE Gene Structures and Conserved Motifs between A. thaliana and B. napus
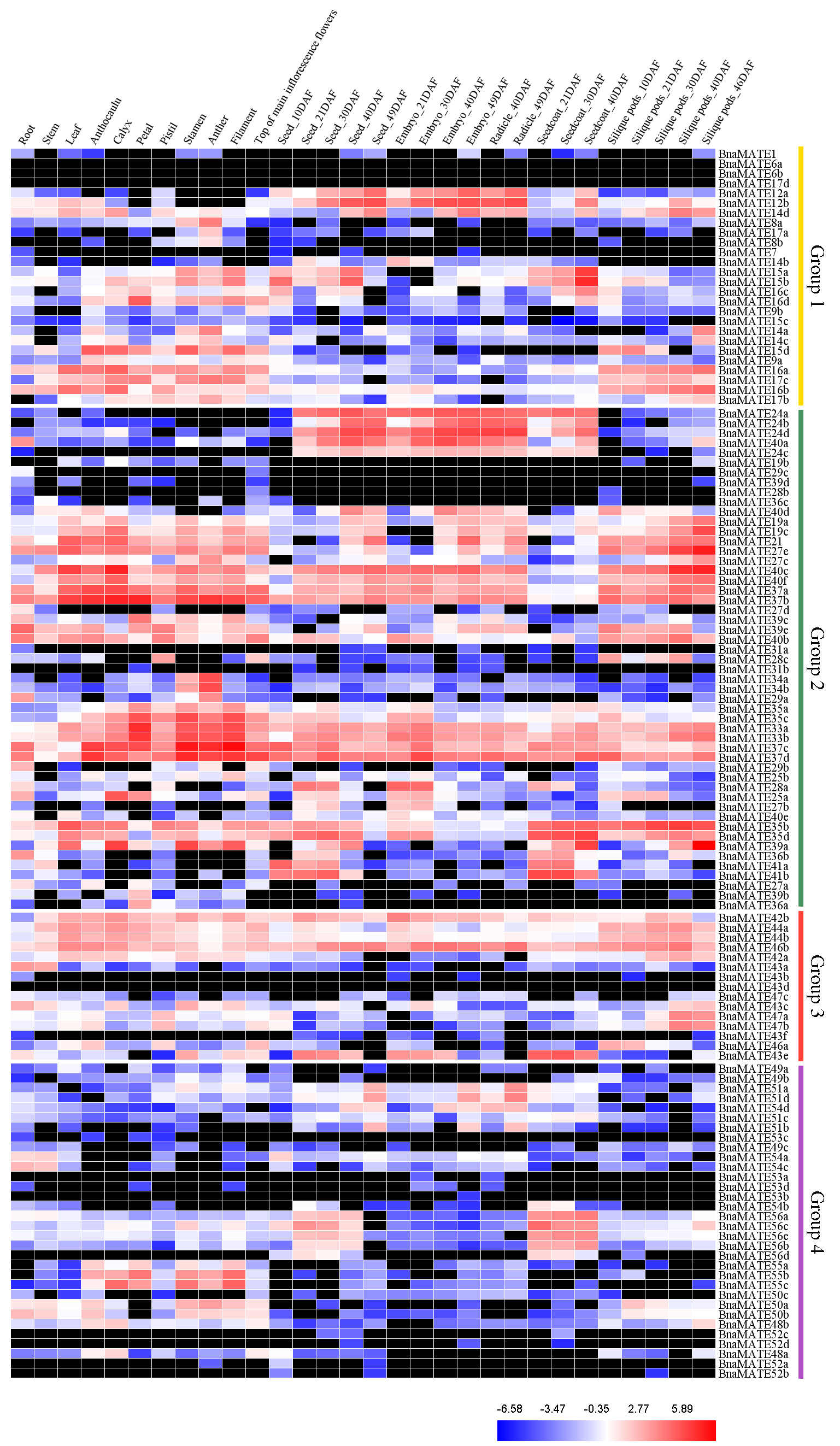
2.6. Expression Profiles of BnaMATE Genes in Various Rapeseed Tissues
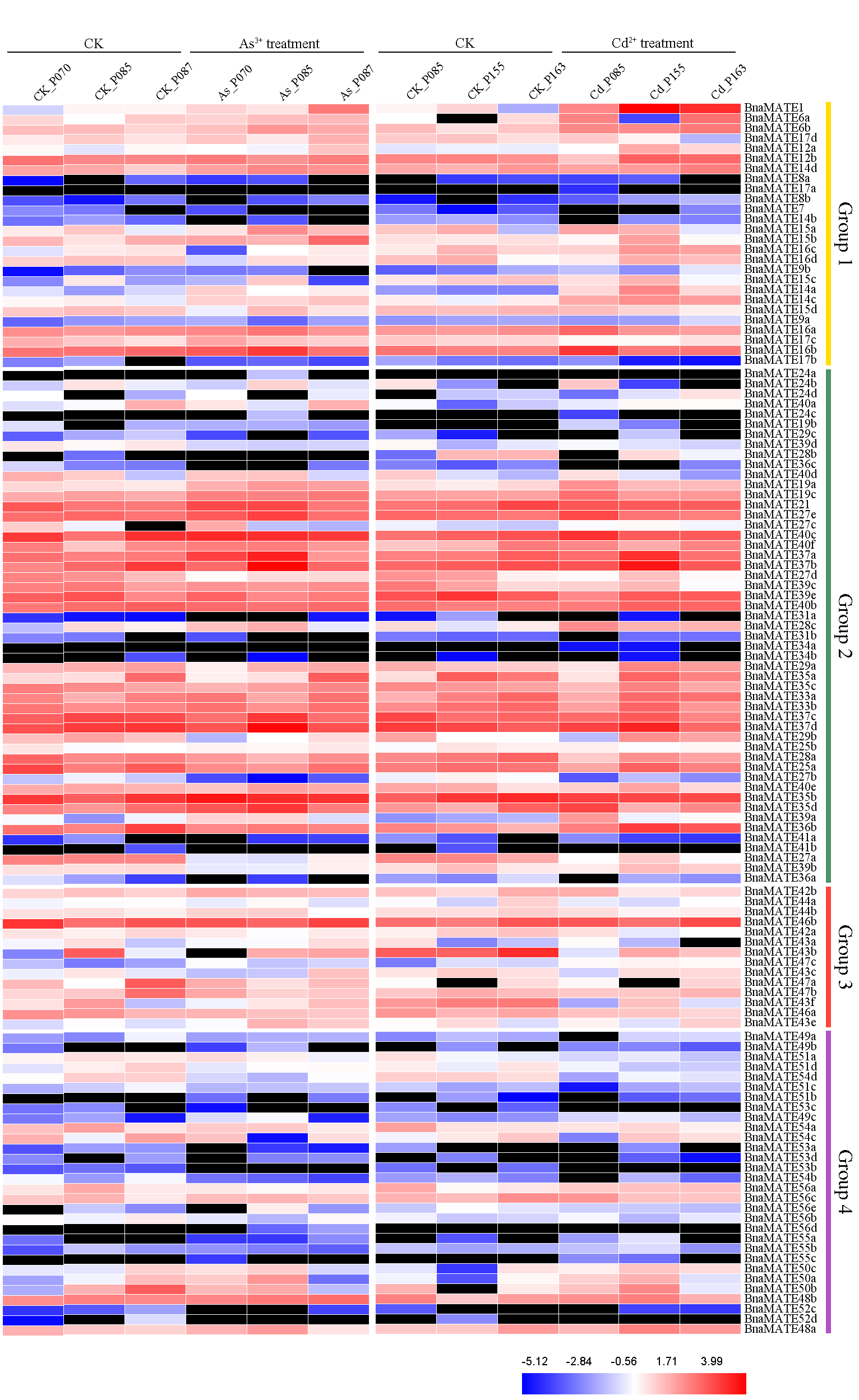
2.7. Expression Patterns of BnaMATE Genes under Heavy Metal Stress
2.8. Expression Patterns of BnaMATE under Hormone Treatments
3. Discussion
4. Materials and Methods
4.1. Identification of MATE Transporters in Brassicaceae Species
4.2. Subcellular Location Analysis of Brassicaceae MATE Transporters
4.3. Phylogenetic Analysis of Brassicaceae MATE Transporters
4.4. Chromosomal Distribution, Gene Duplication, and Evolutionary Analysis of Brassicaceae MATE Genes
4.5. Gene Structure and Conserved Motif Analysis of BnaMATE Genes
4.6. Expression Patterns of BnaMATE Genes among Tissues and Developmental stages
4.7. Transcriptome Analysis of BnaMATE Genes in Response to Abiotic Stresses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Bei, X.; Gao, J.; Li, Y.; Yan, Y.; Hu, Y. The similar and different evolutionary trends of MATE family occurred between rice and Arabidopsis thaliana. BMC Plant Biol. 2016, 16, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, Y.; Kodama, K.; Shiota, S.; Mine, T.; Kataoka, A.; Mizushima, T.; Tsuchiya, T. NorM, a Putative Multidrug Efflux Protein, of Vibrio parahaemolyticus and Its Homolog in Escherichia coli. Antimicrob. Agents Chemother. 1998, 42, 1778–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.H.; Paulsen, I.T.; Skurray, R.A. The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol. Microbiol. 1999, 31, 394–395. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.L.d.; Chaves-Silva, S.; Yang, L.; Maia, L.G.S.; Chalfun-Júnior, A.; Sinharoy, S.; Zhao, J.; Benedito, V.A. Global analysis of the MATE gene family of metabolite transporters in tomato. BMC Plant Biol. 2017, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Takanashi, K.; Shitan, N.; Yazaki, K. The multidrug and toxic compound extrusion (MATE) family in plants. Plant Biotechnol. 2014, 31, 417–430. [Google Scholar] [CrossRef] [Green Version]
- Shoji, T.; Inai, K.; Yazaki, Y.; Sato, Y.; Takase, H.; Shitan, N.; Yazaki, K.; Goto, Y.; Toyooka, K.; Matsuoka, K.; et al. Multidrug and Toxic Compound Extrusion-Type Transporters Implicated in Vacuolar Sequestration of Nicotine in Tobacco Roots. Plant Physiol. 2009, 149, 708–718. [Google Scholar] [CrossRef] [Green Version]
- Debeaujon, I.; Peeters, A.; Léon-Kloosterziel, K.; Koornneef, M. The TRANSPARENT TESTA12 gene of Arabidopsis encodes a multidrug secondary transporter-like protein required for flavonoid sequestration in vacuoles of the seed coat endothelium. Plant Cell 2001, 13, 853–871. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Díaz, R.; Ryngajllo, M.; Pérez-Díaz, J.; Peña-Cortés, H.; Casaretto, J.A.; González-Villanueva, E.; Ruiz-Lara, S. VvMATE1 and VvMATE2 encode putative proanthocyanidin transporters expressed during berry development in Vitis vinifera L. Plant Cell Rep. 2014, 33, 1147–1159. [Google Scholar] [CrossRef]
- Gomez, C.; Terrier, N.; Torregrosa, L.; Vialet, S.; Fournier-Level, A.; Verriès, C.; Souquet, J.M.; Mazauric, J.P.; Klein, M.; Cheynier, V.; et al. Grapevine MATE-Type Proteins Act as Vacuolar H+-Dependent Acylated Anthocyanin Transporters. Plant Physiol. 2009, 150, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Gomez, C.; Conejero, G.; Torregrosa, L.; Cheynier, V.; Terrier, N.; Ageorges, A. In vivo grapevine anthocyanin transport involves vesicle-mediated trafficking and the contribution of anthoMATE transporters and GST. Plant J. 2011, 67, 960–970. [Google Scholar] [CrossRef]
- Sun, X.; Gilroy, E.M.; Chini, A.; Nurmberg, P.L.; Hein, I.; Lacomme, C.; Birch, P.R.J.; Hussain, A.; Yun, B.W.; Loake, G.J. ADS1 encodes a MATE-transporter that negatively regulates plant disease resistance. New Phytol. 2011, 192, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, T.; Sekine, K.T.; Hase, S.; Kanayama, Y.; Seo, S.; Ohashi, Y.; Kusano, T.; Shibata, D.; Shah, J.; Takahashi, H. Overexpression of the Arabidopsis thaliana EDS5 gene enhances resistance to viruses. Plant Biol. 2008, 10, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, Z.; Pandey, G.K.; Tsuchiya, T.; Luan, S. Functional cloning and characterization of a plant efflux carrier for multidrug and heavy metal detoxification. J. Biol. Chem. 2002, 277, 5360–5368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, J.; Yamaji, N.; Wang, H.; Mitani, N.; Murata, Y.; Sato, K.; Katsuhara, M.; Takeda, K.; Ma, J.F. An Aluminum-Activated Citrate Transporter in Barley. Plant Cell Physiol. 2007, 48, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, R.; Shi, J.; Wang, J.; Sun, Q.; Zhang, H.; Xing, Y.; Qi, Y.; Zhang, N.; Guo, Y.D. Brassica oleracea MATE Encodes a Citrate Transporter and Enhances Aluminum Tolerance in Arabidopsis thaliana. Plant Cell Physiol. 2014, 55, 1426–1436. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Wang, B.; Aryal, B.; Garcion, C.; Abou-Mansour, E.; Heck, S.; Geisler, M.; Mauch, F.; Nawrath, C.; Métraux, J.P. Export of Salicylic Acid from the Chloroplast Requires the Multidrug and Toxin Extrusion-Like Transporter EDS5. Plant Physiol. 2013, 162, 1815–1821. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, K.; Motomura, Y.; Yagi, Y.; Nomura, H.; Kikuchi, S.; Nakai, M.; Shiina, T. Chloroplast envelope localization of EDS5, an essential factor for salicylic acid biosynthesis in Arabidopsis thaliana. Plant Signal Behav. 2013, 8, e23603. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhu, H.; Pan, Y.; Yu, Y.; Luan, S.; Li, L. A DTX/MATE-Type Transporter Facilitates Abscisic Acid Efflux and Modulates ABA Sensitivity and Drought Tolerance in Arabidopsis. Mol. Plant 2014, 7, 1522–1532. [Google Scholar] [CrossRef] [Green Version]
- Omote, H.; Hiasa, M.; Matsumoto, T.; Otsuka, M.; Moriyama, Y. The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 2006, 27, 587–593. [Google Scholar] [CrossRef]
- Hvorup, R.N.; Winnen, B.; Chang, A.B.; Jiang, Y.; Zhou, X.F.; Saier, M.H. The multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) exporter superfamily. Eur. J. Biochem. 2003, 270, 799–813. [Google Scholar] [CrossRef]
- Tiwari, M.; Sharma, D.; Singh, M.; Tripathi, R.D.; Trivedi, P.K. Expression of OsMATE1 and OsMATE2 alters development, stress responses and pathogen susceptibility in Arabidopsis. Sci. Rep. 2014, 4, 3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wu, J.; Jiang, Y.; Jin, J.; Zhou, W.E.I.; Wang, Y.U.; Han, G.; Zhao, Y.; Cheng, B. Genomewide analysis of MATE-type gene family in maize reveals microsynteny and their expression patterns under aluminum treatment. J. Genet. 2016, 95, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shen, Z.; Chen, W.; Si, G.; Meng, Y.; Guo, N.; Sun, X.; Cai, Y.; Lin, Y.; Gao, J. Phylogenetic analysis of upland cotton MATE gene family reveals a conserved subfamily involved in transport of proanthocyanidins. Mol. Biol. Rep. 2019, 46, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Y.; Wang, W.; Gai, J.; Li, Y. Genome-wide analysis of MATE transporters and expression patterns of a subgroup of MATE genes in response to aluminum toxicity in soybean. BMC Genom. 2016, 17, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julião, M.H.M.; Silva, S.R.; Ferro, J.A.; Varani, A.M. A Genomic and Transcriptomic Overview of MATE, ABC, and MFS Transporters in Citrus sinensis Interaction with Xanthomonas citri subsp. citri. Plants 2020, 9, 794. [Google Scholar]
- Nagaharu, U.; Nagaharu, N. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. J. Japan Bot. 1935, 7, 389–452. [Google Scholar]
- Kim, J.; Lee, J.; Choi, J.P.; Park, I.; Yang, K.; Kim, M.K.; Lee, Y.H.; Nou, I.S.; Kim, D.S.; Min, S.R.; et al. Functional innovations of three chronological mesohexaploid Brassica rapa genomes. BMC Genom. 2014, 15, 606. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Hu, F.; Dossa, K.; Wang, Z.; Ke, T. Genome-wide analysis of UDP-glycosyltransferase super family in Brassica rapa and Brassica oleracea reveals its evolutionary history and functional characterization. BMC Genom. 2017, 18, 474. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [Green Version]
- Jenczewski, E.; Chèvre, A.M.; Alix, K. Chromosomal and Gene Expression Changes in Brassica Allopolyploids. In Polyploid and Hybrid Genomics; Chen, Z.J., Birchler, J.A., Eds.; John Wiley & Sons, Inc.: Oxford, IA, USA, 2013; pp. 171–186. [Google Scholar]
- Lee, T.-H.; Tang, H.; Wang, X.; Paterson, A.H. PGDD: A database of gene and genome duplication in plants. Nucleic Acids Res. 2013, 41, D1152–D1158. [Google Scholar] [CrossRef]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Liu, Y.; Yang, X.; Tong, C.; Edwards, D.; Parkin, I.A.P.; Zhao, M.; Ma, J.; Yu, J.; Huang, S.; et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat. Commun. 2014, 5, 3930. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef]
- Perumal, S.; Koh, C.S.; Jin, L.; Buchwaldt, M.; Higgins, E.; Zheng, C.; Sankoff, D.; Robinson, S.J.; Kagale, S.; Navabi, Z.; et al. High contiguity long read assembly of Brassica nigra allows localization of active centromeres and provides insights into the ancestral Brassica genome. BioRxiv 2020, 932665. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Liu, S.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Li, P.; Hua, W.; Wang, X. BRAD, the genetics and genomics database for Brassica plants. BMC Plant. Biol. 2011, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; He, H.; He, L.F. Genome-wide analysis of the MATE gene family in potato. Mol. Biol. Rep. 2019, 46, 403–414. [Google Scholar] [CrossRef]
- Lu, P.; Magwanga, R.O.; Guo, X.; Kirungu, J.N.; Lu, H.; Cai, X.; Zhou, Z.; Wei, Y.; Wang, X.; Zhang, Z.; et al. Genome-Wide Analysis of Multidrug and Toxic Compound Extrusion (MATE) Family in Gossypium raimondii and Gossypium arboreum and Its Expression Analysis Under Salt, Cadmium, and Drought Stress. G3 Genes Genom. Genet. 2018, 8, 2483–2500. [Google Scholar]
- Wei, L.; Zhu, Y.; Liu, R.; Zhang, A.; Zhu, M.; Xu, W.; Lin, A.; Lu, K.; Li, J. Genome wide identification and comparative analysis of glutathione transferases (GST) family genes in Brassica napus. Sci. Rep. 2019, 9, 9196. [Google Scholar] [CrossRef] [Green Version]
- Cheng, F.; Wu, J.; Wang, X. Genome triplication drove the diversification of Brassica plants. Hortic. Res. 2014, 1, 14024. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Jin, J.; Guo, A.Y.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [Green Version]
- Fan, K.; Wang, M.; Miao, Y.; Ni, M.; Bibi, N.; Yuan, S.; Li, F.; Wang, X. Molecular Evolution and Expansion Analysis of the NAC Transcription Factor in Zea mays. PLoS ONE 2014, 9, e111837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.S.; Raes, J. Duplication and Divergence: The Evolution of New Genes and Old Ideas. Annu. Rev. Genet. 2004, 38, 615–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Town, C.D.; Cheung, F.; Maiti, R.; Crabtree, J.; Haas, B.J.; Wortman, J.R.; Hine, E.E.; Althoff, R.; Arbogast, T.S.; Tallon, L.J.; et al. Comparative Genomics of Brassica oleracea and Arabidopsis thaliana Reveal Gene Loss, Fragmentation, and Dispersal after Polyploidy. Plant Cell 2006, 18, 1348–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinf. 2006, 4, 259–263. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Fan, H.J.; Ling, H.Q. Genome-wide identification and characterization of the bHLH gene family in tomato. BMC Genom. 2015, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Diener, A.C.; Gaxiola, R.A.; Fink, G.R. Arabidopsis ALF5, a multidrug efflux transporter gene family member, confers resistance to toxins. Plant Cell 2001, 13, 1625–1637. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.P.; Wilkins, C.; Demidchik, V.; Davies, J.M.; Glover, B.J. An Arabidopsis flavonoid transporter is required for anther dehiscence and pollen development. J. Exp. Bot. 2010, 61, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhao, F.; Tang, R.; Yu, Y.; Song, J.; Wang, Y.; Li, L.; Luan, S. Two tonoplast MATE proteins function as turgor-regulating chloride channels in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, E2036–E2045. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.; Lei, B.; Huang, H.; Li, J.; Yin, J.; Tang, Z.; Wang, R.; Chen, L. TRANSPARENT TESTA 12 genes from Brassica napus and parental species: Cloning, evolution, and differential involvement in yellow seed trait. Mol. Genet. Genom. 2009, 281, 109–123. [Google Scholar] [CrossRef]
- Marinova, K.; Pourcel, L.; Weder, B.; Schwarz, M.; Barron, D.; Routaboul, J.M.; Debeaujon, I.; Klein, M. The Arabidopsis MATE Transporter TT12 Acts as a Vacuolar Flavonoid/H+-Antiporter Active in Proanthocyanidin-Accumulating Cells of the Seed Coat. Plant Cell 2007, 19, 2023–2038. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Luo, X.; Shaff, J.; Liang, C.; Jia, X.; Li, Z.; Magalhaes, J.; Kochian, L.V. A promoter-swap strategy between the AtALMT and AtMATE genes increased Arabidopsis aluminum resistance andimproved carbon-use efficiency for aluminum resistance. Plant J. 2012, 71, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Green, L.S.; Rogers, E.E. FRD3 Controls Iron Localization in Arabidopsis. Plant Physiol. 2004, 136, 2523–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durrett, T.P.; Gassmann, W.; Rogers, E.E. The FRD3-Mediated Efflux of Citrate into the Root Vasculature Is Necessary for Efficient Iron Translocation. Plant Physiol. 2007, 144, 197–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrath, C.; Heck, S.; Parinthawong, N.; Métraux, J.P. EDS5, an Essential Component of Salicylic Acid–Dependent Signaling for Disease Resistance in Arabidopsis, Is a Member of the MATE Transporter Family. Plant Cell 2002, 14, 275–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, P.J.; Park, J.; Park, M.J.; Kim, Y.S.; Kim, S.G.; Jung, J.H.; Park, C.M. A Golgi-localized MATE transporter mediates iron homoeostasis under osmotic stress in Arabidopsis. Biochem. J. 2012, 442, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Ingram, G.C.; Boisnard-Lorig, C.; Dumas, C.; Rogowsky, P.M. Expression patterns of genes encoding HD-ZipIV homeo domain proteins define specific domains in maize embryos and meristems. Plant J. 2000, 22, 401–414. [Google Scholar] [CrossRef]
- Gallie, D.R.; Geisler-Lee, J.; Chen, J.; Jolley, B. Tissue-specific expression of the ethylene biosynthetic machinery regulates root growth in maize. Plant Mol. Biol. 2009, 69, 195–211. [Google Scholar] [CrossRef]
- Tian, W.; Hou, C.; Ren, Z.; Pan, Y.; Jia, J.; Zhang, H.; Bai, F.; Zhang, P.; Zhu, H.; He, Y.; et al. A molecular pathway for CO2 response in Arabidopsis guard cells. Nat. Commun. 2015, 6, 6057. [Google Scholar] [CrossRef] [Green Version]
- Yokosho, K.; Yamaji, N.; Fujii-Kashino, M.; Ma, J.F. Functional Analysis of a MATE Gene OsFRDL2 Revealed its Involvement in Al-Induced Secretion of Citrate, but a Lower Contribution to Al Tolerance in Rice. Plant Cell Physiol. 2016, 57, 976–985. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Magalhaes, J.V.; Shaff, J.; Kochian, L.V. Aluminum-activated citrate and malate transporters from the MATE and ALMT families function independently to confer Arabidopsis aluminum tolerance. Plant J. 2009, 57, 389–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hou, Q.; Li, P.; Yang, L.; Sun, X.; Benedito, V.A.; Wen, J.; Chen, B.; Mysore, K.S.; Zhao, J. Diverse functions of multidrug and toxin extrusion (MATE) transporters in citric acid efflux and metal homeostasis in Medicago truncatula. Plant J. 2017, 90, 79–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, S.; Chen, G.; Wang, Y.; Huang, Y.; Marchant, D.B.; Wang, Y.; Yang, Q.; Dai, F.; Hills, A.; Franks, P.J.; et al. Evolutionary Conservation of ABA Signaling for Stomatal Closure. Plant Physiol. 2017, 174, 732–747. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Mao, Y.; Zhang, W.; Lai, D.; Wang, Q.; Shen, W. Reactive Oxygen Species-Dependent Nitric Oxide Production Contributes to Hydrogen-Promoted Stomatal Closure in Arabidopsis. Plant Physiol. 2014, 165, 759–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Reiser, L.; Subramaniam, S.; Li, D.; Huala, E. Using the Arabidopsis information resource (TAIR) to find information about Arabidopsis genes. Curr. Protoc. Bioinform. 2017, 60, 1.11.1–1.11.45. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3398–3402. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [Green Version]
- Emanuelsson, O.; Brunak, S.; von Heijne, G.; Nielsen, H. Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2007, 2, 953–971. [Google Scholar] [CrossRef]
- Boden, M.; Hawkins, J. Prediction of subcellular localization using sequence-biased recurrent networks. Bioinformatics 2005, 21, 2279–2286. [Google Scholar] [CrossRef] [Green Version]
- Horton, P.; Park, K.J.; Obayashi, T.; Fujita, N.; Harada, H.; Adams-Collier, C.J.; Nakai, K. WoLF PSORT: Protein localization predictor. Nucleic Acids Res. 2007, 35, W585–W587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Zuckerkandl, E.; Pauling, L. Evolutionary divergence and convergence in proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: New York, NY, USA, 1965; pp. 97–166. [Google Scholar]
- Voorrips, R.E. MapChart: Software for the graphical presentation of linkage maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant. 2020, 13, 1192–1204. [Google Scholar] [CrossRef]
- Nei, M.; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 1986, 3, 418–426. [Google Scholar]
- Koch, M.A.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Deng, W.; Wang, Y.; Liu, Z.; Cheng, H.; Xue, Y. HemI: A Toolkit for Illustrating Heatmaps. PLoS ONE 2014, 9, e111988. [Google Scholar]
- Wan, Y.; Wang, Z.; Xia, J.; Shen, S.; Guan, M.; Zhu, M.; Qiao, C.; Sun, F.; Liang, Y.; Li, J.; et al. Genome-Wide Analysis of Phosphorus Transporter Genes in Brassica and Their Roles in Heavy Metal Stress Tolerance. Int. J. Mol. Sci. 2020, 21, 2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, P.; Wen, J.; Yang, J.; Ke, Y.; Wang, M.; Liu, M.; Ran, F.; Wu, Y.; Li, P.; Li, J.; et al. Genome-wide survey and expression analyses of the GRAS gene family in Brassica napus reveals their roles in root development and stress response. Planta 2019, 250, 1051–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, Y.Z.; Wu, Y.W.; Zhou, H.J.; Chen, P.; Wang, M.M.; Liu, M.M.; Li, P.F.; Yang, J.; Li, J.N.; Du, H. Genome-wide survey of the bHLH super gene family in Brassica napus. BMC Plant Biol. 2020, 20, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Family | A. thaliana | B. napus | B. oleracea | B. rapa | B. juncea | B. nigra |
|---|---|---|---|---|---|---|---|
| Group 1 | MATE1 | AtMATE1 | 1 | / | 1 | 2 | / |
| MATE1 | AtMATE2 | / | / | / | / | / | |
| MATE1 | AtMATE3 | / | 1 | / | 1 | / | |
| MATE1 | AtMATE4 | / | 1 | 1 | / | 1 | |
| MATE1 | AtMATE5 | / | / | / | 1 | 1 | |
| MATE1 | AtMATE6 | 2 | / | 1 | 1 | / | |
| MATE1 | AtMATE7 | 1 | / | 1 | 2 | 2 | |
| MATE1 | AtMATE8 | 2 | 3 | 1 | 2 | 1 | |
| MATE1 | AtMATE9 | 2 | 2 | 2 | 2 | 1 | |
| MATE1 | AtMATE10 | / | / | / | / | / | |
| MATE1 | AtMATE11 | / | / | / | / | / | |
| MATE1 | AtMATE12 | 2 | 1 | 1 | / | / | |
| MATE1 | AtMATE13 | / | 2 | 2 | 5 | 3 | |
| MATE1 | AtMATE14 | 4 | 5 | 4 | 7 | 5 | |
| MATE1 | AtMATE15 | 4 | 2 | 1 | 2 | 2 | |
| MATE1 | AtMATE16 | 4 | 2 | 3 | 3 | 2 | |
| MATE1 | AtMATE17 | 4 | 2 | 2 | 3 | 3 | |
| MATE1 | AtMATE57 | / | / | / | / | / | |
| Group 2 | MATE2 | AtMATE18 | / | / | / | 3 | / |
| MATE2 | AtMATE19 | 3 | 2 | 2 | 2 | 5 | |
| MATE2 | AtMATE20 | / | / | / | / | / | |
| MATE2 | AtMATE21 | 1 | 1 | 2 | 2 | 1 | |
| MATE2 | AtMATE22 | / | / | / | / | / | |
| MATE2 | AtMATE23 | / | / | 1 | 1 | 1 | |
| MATE2 | AtMATE24 | 4 | 2 | 2 | 3 | 1 | |
| MATE2 | AtMATE25 | 2 | 2 | 2 | 3 | 2 | |
| MATE2 | AtMATE26 | / | / | / | / | / | |
| MATE2 | AtMATE27 | 5 | 5 | 4 | 2 | 4 | |
| MATE2 | AtMATE28 | 3 | 1 | 1 | / | 2 | |
| MATE2 | AtMATE29 | 3 | 1 | 1 | 2 | 1 | |
| MATE2 | AtMATE30 | / | / | 1 | / | / | |
| MATE2 | AtMATE31 | 2 | / | 1 | 2 | 2 | |
| MATE2 | AtMATE32 | / | / | / | / | / | |
| MATE2 | AtMATE33 | 2 | 1 | 1 | 1 | 1 | |
| MATE2 | AtMATE34 | 2 | 1 | / | 2 | 1 | |
| MATE2 | AtMATE35 | 4 | 3 | 3 | 5 | 3 | |
| MATE2 | AtMATE36 | 3 | 3 | 2 | 2 | 2 | |
| MATE2 | AtMATE37 | 4 | 2 | 2 | 2 | 3 | |
| MATE2 | AtMATE38 | / | 2 | 4 | 2 | 2 | |
| MATE2 | AtMATE39 | 5 | 3 | 2 | 4 | 4 | |
| MATE2 | AtMATE40 | 6 | 4 | 5 | 3 | 4 | |
| MATE2 | AtMATE41 | 2 | 1 | 1 | 3 | 1 | |
| Group 3 | MATE3 | AtMATE42 | 2 | 2 | 2 | 3 | 2 |
| MATE3 | AtMATE43 | 6 | 1 | 3 | 8 | 3 | |
| MATE3 | AtMATE44 | 2 | / | 1 | 2 | 1 | |
| MATE3 | AtMATE45 | / | 1 | 1 | / | / | |
| MATE3 | AtMATE46 | 2 | 2 | 2 | 1 | / | |
| MATE3 | AtMATE47 | 3 | 1 | 1 | 1 | / | |
| Group 4 | MATE4 | AtMATE48 | 2 | 2 | 1 | 3 | 1 |
| MATE4 | AtMATE49 | 3 | 2 | 2 | 4 | 2 | |
| MATE4 | AtMATE50 | 3 | 2 | 2 | 6 | 1 | |
| MATE4 | AtMATE51 | 4 | 3 | 3 | 6 | / | |
| MATE4 | AtMATE52 | 4 | / | 2 | 5 | / | |
| MATE4 | AtMATE53 | 4 | 2 | 2 | 4 | 2 | |
| MATE4 | AtMATE54 | 4 | 2 | 2 | 3 | 2 | |
| MATE4 | AtMATE55 | 3 | 2 | 2 | 5 | 2 | |
| MATE4 | AtMATE56 | 5 | 4 | 2 | 4 | 2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, C.; Yang, J.; Wan, Y.; Xiang, S.; Guan, M.; Du, H.; Tang, Z.; Lu, K.; Li, J.; Qu, C. A Genome-Wide Survey of MATE Transporters in Brassicaceae and Unveiling Their Expression Profiles under Abiotic Stress in Rapeseed. Plants 2020, 9, 1072. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9091072
Qiao C, Yang J, Wan Y, Xiang S, Guan M, Du H, Tang Z, Lu K, Li J, Qu C. A Genome-Wide Survey of MATE Transporters in Brassicaceae and Unveiling Their Expression Profiles under Abiotic Stress in Rapeseed. Plants. 2020; 9(9):1072. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9091072
Chicago/Turabian StyleQiao, Cailin, Jing Yang, Yuanyuan Wan, Sirou Xiang, Mingwei Guan, Hai Du, Zhanglin Tang, Kun Lu, Jiana Li, and Cunmin Qu. 2020. "A Genome-Wide Survey of MATE Transporters in Brassicaceae and Unveiling Their Expression Profiles under Abiotic Stress in Rapeseed" Plants 9, no. 9: 1072. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9091072