Chloroplast Genome Analysis of Two Medicinal Coelogyne spp. (Orchidaceae) Shed Light on the Genetic Information, Comparative Genomics, and Species Identification

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. Genome Sequencing and Assembly
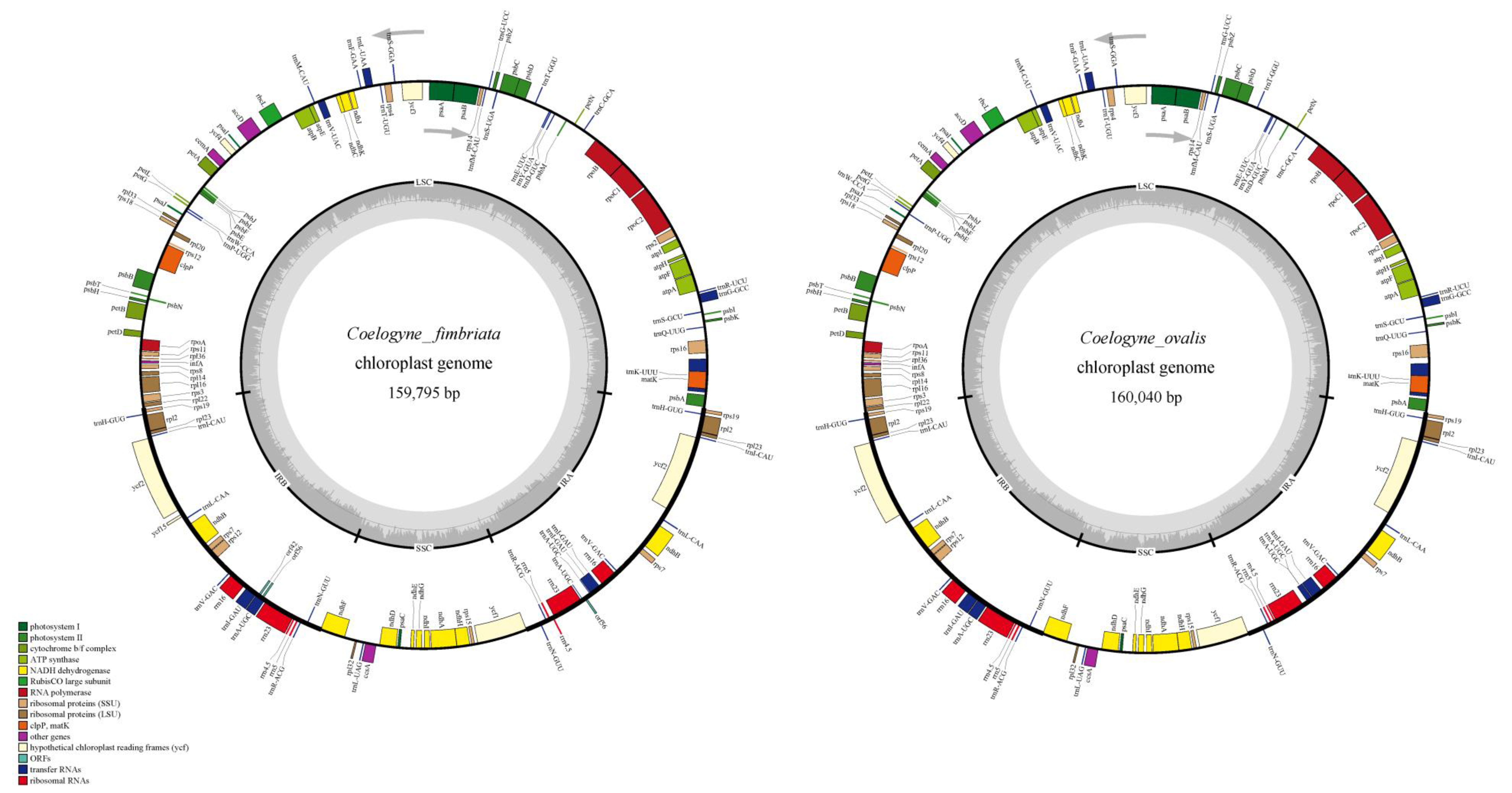
2.2. The Organization of the Coelogyne Chloroplast Genome
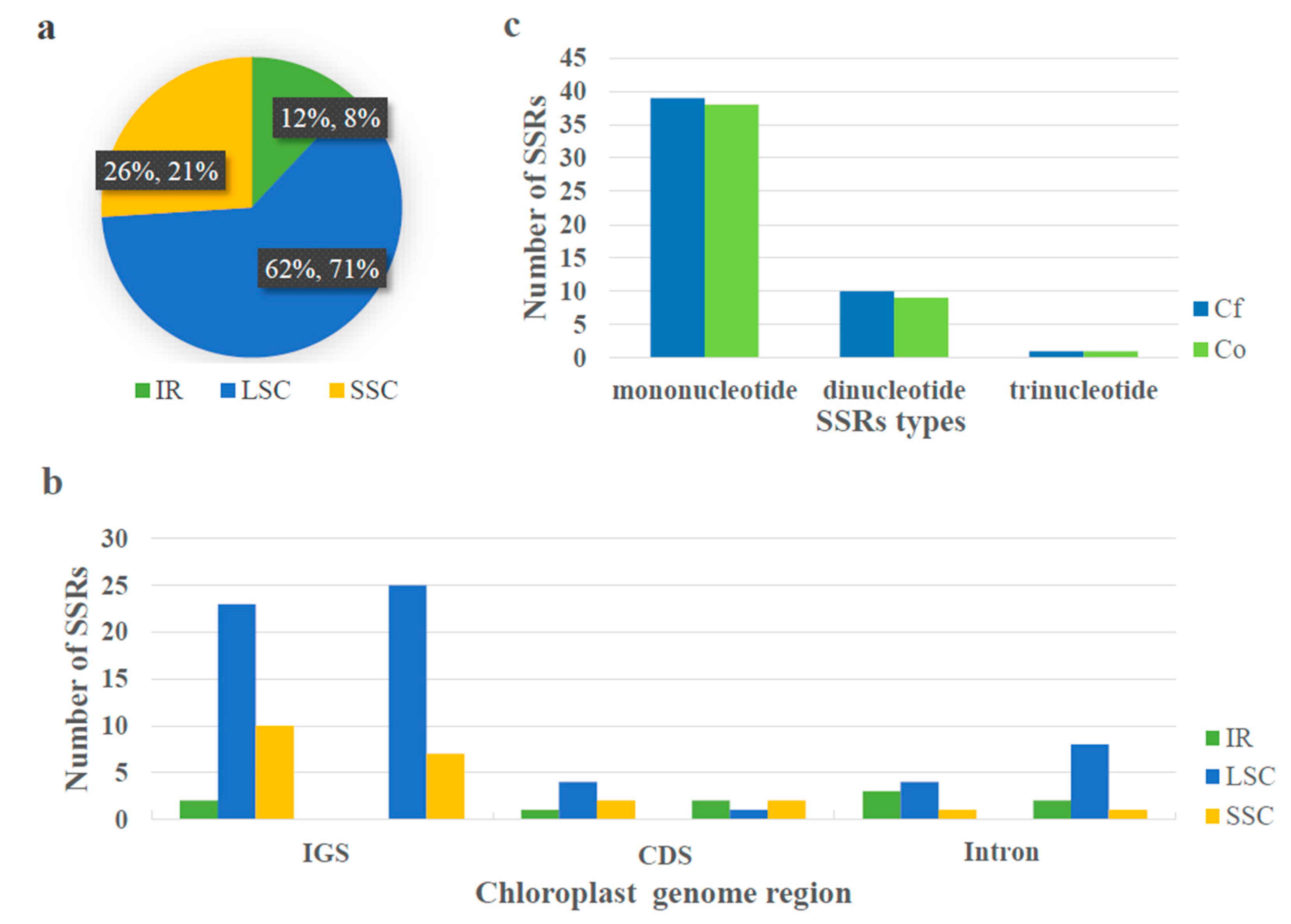
2.3. Sequence Repeats
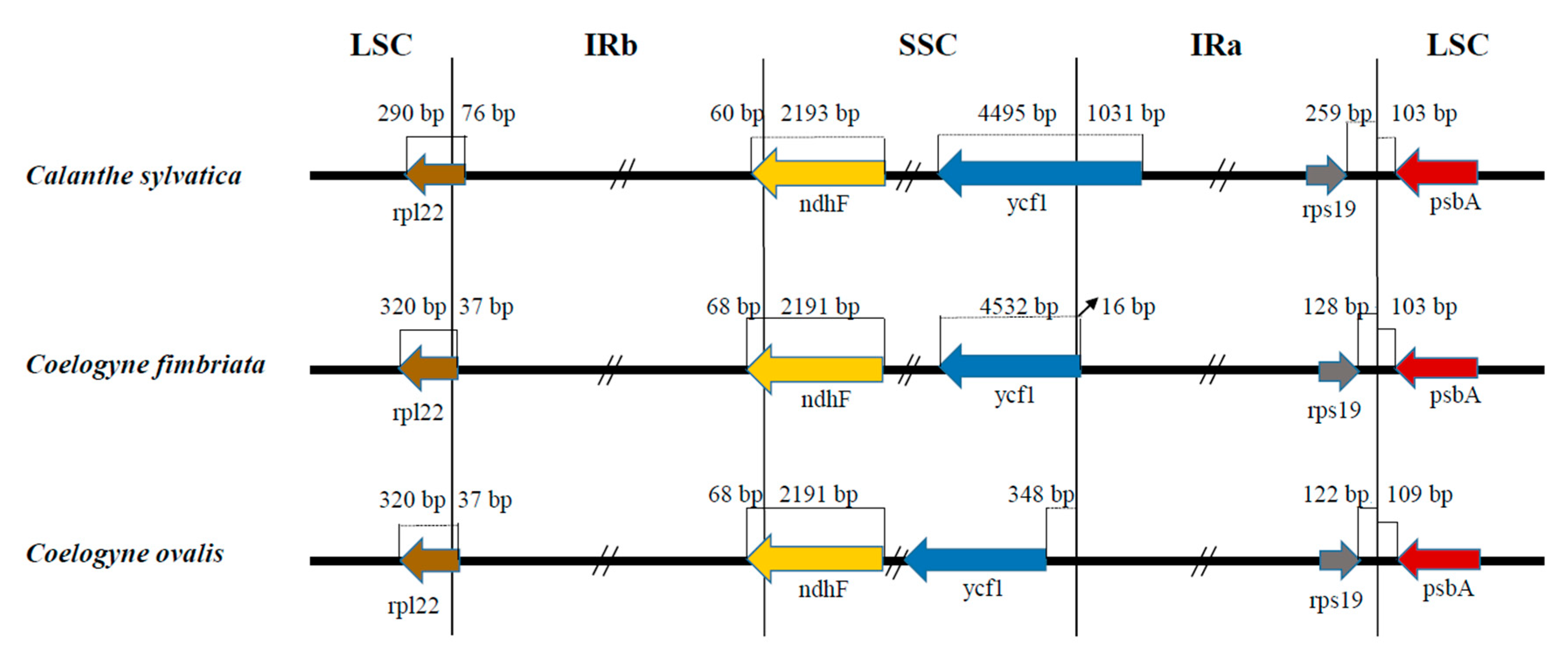
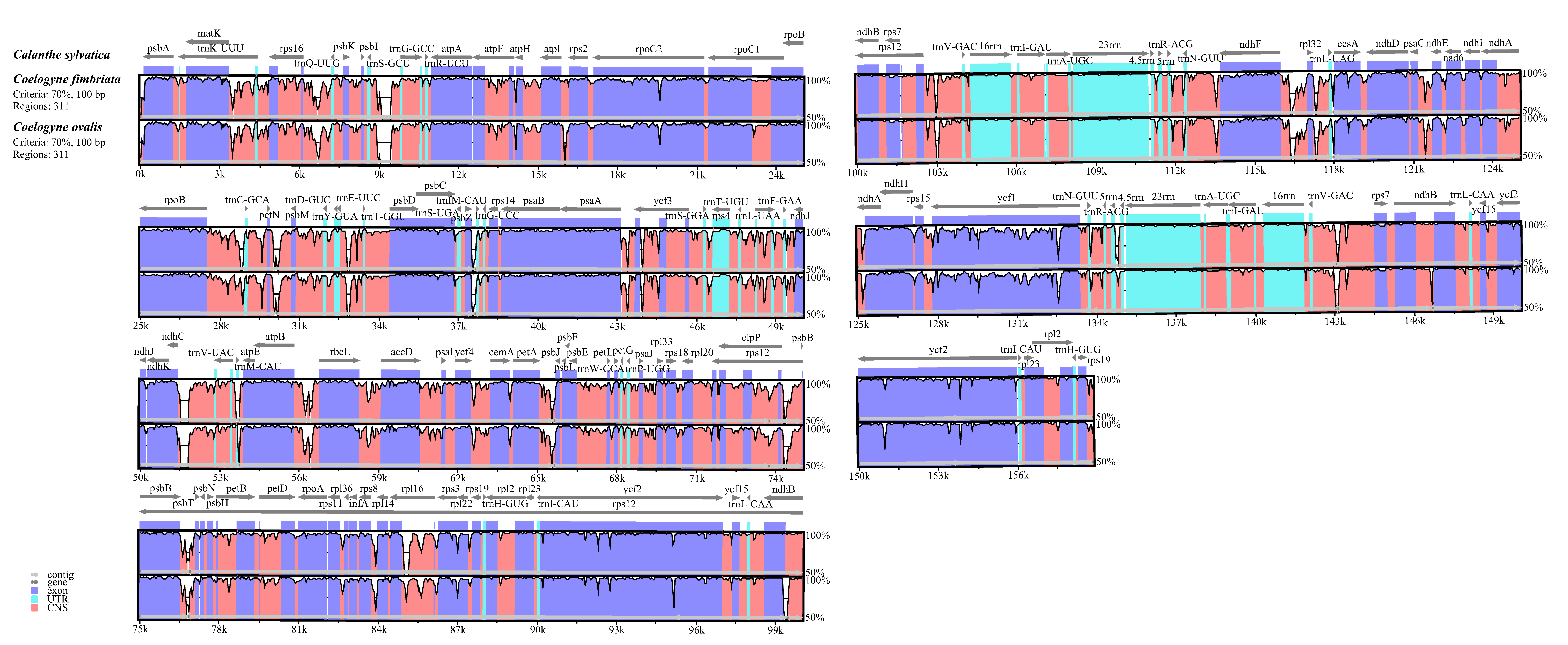
2.4. Comparative Genome Analysis
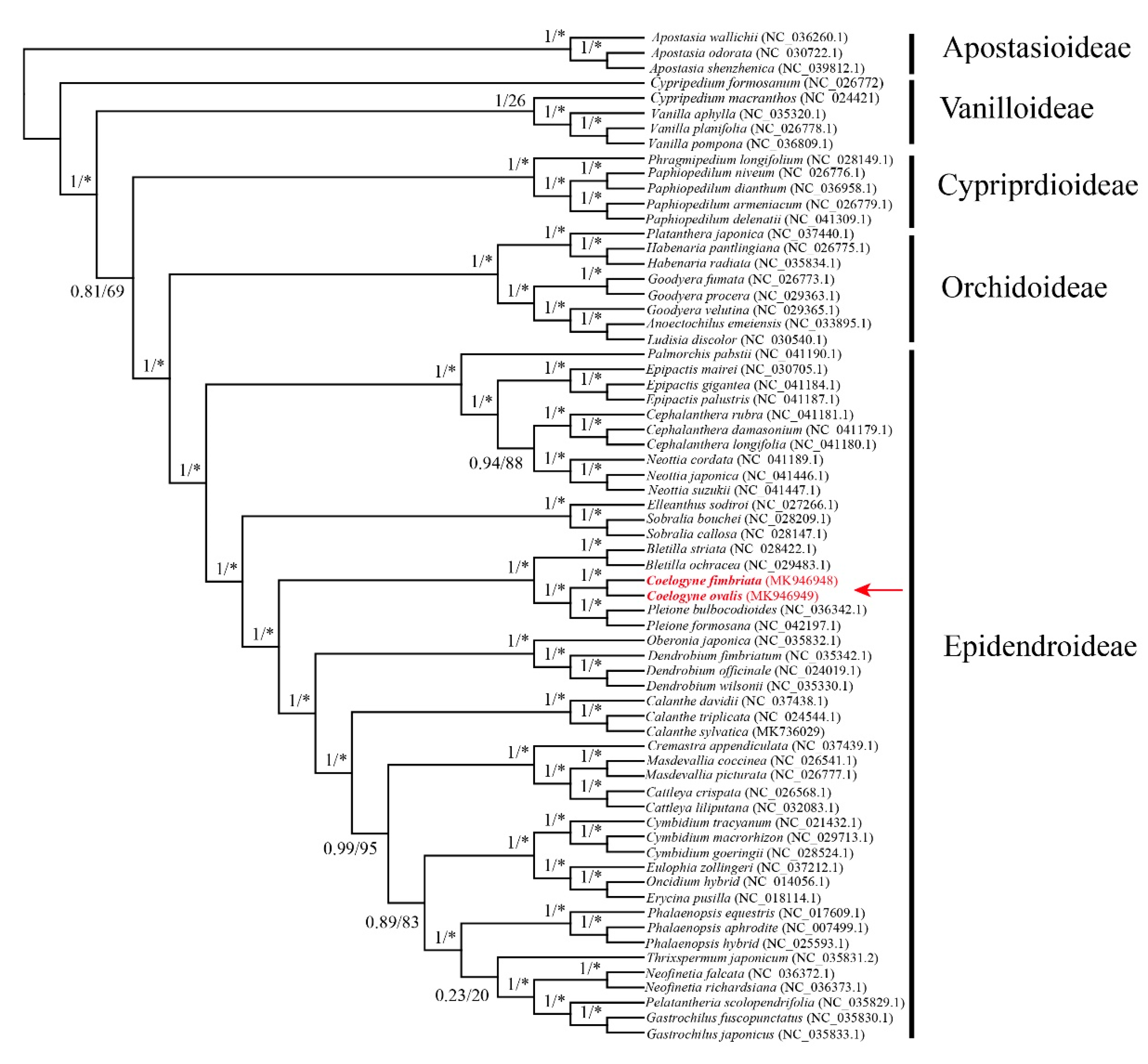
2.5. Phylogenetic Position of Coelogyne in Orchidaceae
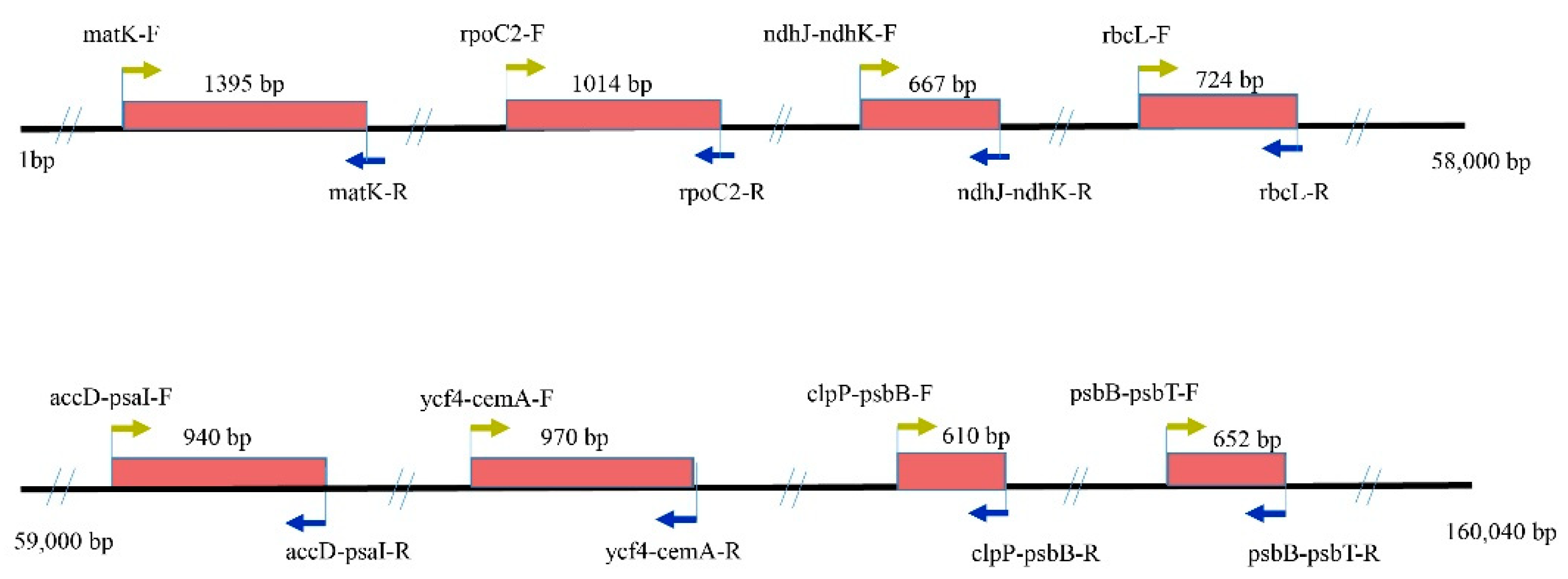
2.6. Primer Verification and Transferability
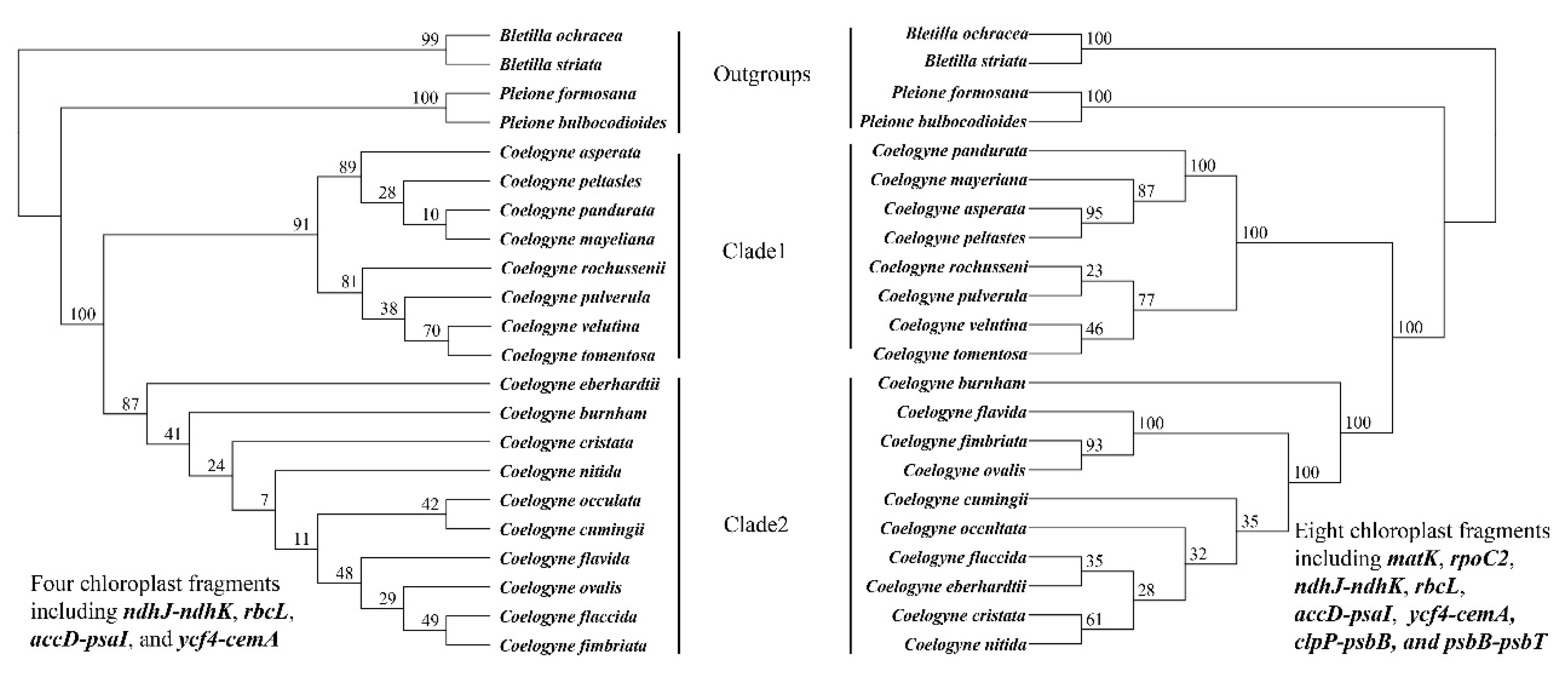
2.7. Phylogenetic Relationship within Coelogyne
3. Discussion
3.1. Coelogyne Chloroplast Genome Structure and Characterization
3.2. Phylogenetic Analysis of Inter- and Intra- Coelogyne
4. Materials and Methods
4.1. Plant Sampling and DNA Extraction
4.2. Genome Assembly and Annotation
4.3. Repeat Sequence Analysis
4.4. Comparative Genome Analysis
4.5. Phylogenetic Position of the Two Coelogyne Species
4.6. Primers Design and Verification in Other Coelogyne Species
4.7. Phylogenetic Relationship within Coelogyne
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | GenBank Accession | |||||||
|---|---|---|---|---|---|---|---|---|
| matK | rpoC2 | ndhJ-ndhK | rbcL | accD-psaI | ycf4-cemA | clpP-psbB | psbB-psbT | |
| C. rochussenii | - | - | MN512535 | MN416673 | MN512468 | MN512517 | MN512484 | - |
| C. burnham | MN400405 | MN400397 | MN512520 | MN396950 | MN512453 | MN512502 | MN512471 | MN512487 |
| C. veluting | MN416681 | MN416666 | MN512537 | MN416675 | MN512470 | MN512519 | MN512486 | MN512501 |
| C. mayeliana | MN400412 | MN400404 | MN512528 | MN400420 | MN512461 | MN512510 | - | - |
| C. peltasles | MN416679 | MN512532 | MN416670 | MN512465 | MN512514 | MN512482 | MN512498 | |
| C. cumingii | MN400407 | MN400399 | MN512522 | MN400414 | MN512455 | MN512504 | MN512473 | MN512489 |
| C. flavida | MN400409 | MN400401 | MN512525 | MN400417 | MN512458 | MN512507 | MN512476 | MN512492 |
| C. eberhardtii | MN400408 | MN400400 | MN512524 | MN400416 | MN512457 | MN512506 | MN512475 | MN512491 |
| C. cristata | - | - | MN512523 | MN400415 | MN512456 | MN512505 | MN512474 | MN512490 |
| C. tomentosa | - | MN416665 | MN512536 | MN416674 | MN512469 | MN512518 | MN512485 | MN512500 |
| C. occulata | MN416678 | MN416661 | MN512531 | MN416669 | MN512464 | MN512513 | MN512481 | MN512497 |
| C. flaccida | MN400411 | MN400403 | MN512527 | MN400419 | MN512460 | MN512509 | MN512478 | MN512494 |
| C. pulverula | MN416680 | MN416663 | MN512534 | MN416672 | MN512467 | MN512516 | MN512483 | MN512499 |
| C. asperata | MN400406 | MN400398 | MN512521 | MN400413 | MN512454 | MN512503 | MN512472 | MN512488 |
| C. pandurata | - | MN416662 | MN512533 | MN416671 | MN512466 | MN512515 | - | - |
| C. nitida | MN416676 | MN416659 | MN512529 | MN416667 | MN512462 | MN512511 | MN512479 | MN512495 |
| C. fimbriata | MN400410 | MN400402 | MN512526 | MN400418 | MN512459 | MN512508 | MN512477 | MN512493 |
| C. ovalis | MN416677 | MN416660 | MN512530 | MN416668 | MN512463 | MN512512 | MN512480 | MN512496 |
| Species | Collector | Collection No. | Deposited Institution | n |
|---|---|---|---|---|
| C. fimbriata | Wei-Chang Huang | CS-HWC201606-2 | CSH | 1 |
| C. ovalis | Wei-Chang Huang | CS-HWC201606-5 | CSH | 1 |
| C. rochussenii | Kai Jiang | CS-JK201806-01 | CSH | 1 |
| C. burnham | Kai Jiang | CS-JK201806-02 | CSH | 1 |
| C. veluting | Kai Jiang | CS-JK201806-03 | CSH | 1 |
| C. mayeliana | Kai Jiang | CS-JK201806-04 | CSH | 1 |
| C. peltasles | Kai Jiang | CS-JK201806-05 | CSH | 1 |
| C. cumingii | Kai Jiang | CS-JK201806-06 | CSH | 1 |
| C. flavida | Kai Jiang | CS-JK201806-07 | CSH | 1 |
| C. eberhardtii | Kai Jiang | CS-JK201806-08 | CSH | 1 |
| C. cristata | Kai Jiang | CS-JK201806-09 | CSH | 1 |
| C. tomentosa | Kai Jiang | CS-JK201806-10 | CSH | 1 |
| C. occulata | Kai Jiang | CS-JK201806-11 | CSH | 1 |
| C. flaccida | Kai Jiang | CS-JK201806-12 | CSH | 1 |
| C. pulverula | Kai Jiang | CS-JK201806-13 | CSH | 1 |
| C. asperata | Kai Jiang | CS-JK201806-14 | CSH | 1 |
| C. pandurata | Kai Jiang | CS-JK201806-15 | CSH | 1 |
| C. nitida | Kai Jiang | CS-JK201806-16 | CSH | 1 |
| Model | Param | BIC | AICc | lnL | Invariant | Gamma | R |
|---|---|---|---|---|---|---|---|
| GTR + G | 140.00 | 582,962.94 | 581,155.75 | −290,437.87 | n/a | 0.94 | 1.39 |
| GTR + G + I | 141.00 | 582,977.87 | 581,157.77 | −290,437.88 | 0.00 | 0.94 | 1.39 |
| T92 + G | 134.00 | 585,265.34 | 583,535.60 | −291,633.79 | n/a | 0.93 | 1.48 |
| TN93 + G | 137.00 | 585,287.36 | 583,518.89 | −291,622.44 | n/a | 0.93 | 1.48 |
| HKY + G | 136.00 | 585,287.51 | 583,531.95 | −291,629.97 | n/a | 0.93 | 1.48 |
| T92 + G + I | 135.00 | 585,403.95 | 583,661.30 | −291,695.64 | 0.00 | 0.93 | 1.57 |
| TN93 + G + I | 138.00 | 585,425.88 | 583,644.51 | −291,684.25 | 0.00 | 0.92 | 1.57 |
| HKY + G + I | 137.00 | 585,426.37 | 583,657.91 | −291,691.95 | 0.00 | 0.93 | 1.57 |
| GTR + I | 140.00 | 587,343.22 | 585,536.03 | −292,628.01 | 0.31 | n/a | 1.37 |
| T92 + I | 134.00 | 589,665.50 | 587,935.76 | −293,833.87 | 0.31 | n/a | 1.34 |
| HKY + I | 136.00 | 589,688.10 | 587,932.55 | −293,830.27 | 0.31 | n/a | 1.34 |
| TN93 + I | 137.00 | 589,690.94 | 587,922.48 | −293,824.23 | 0.31 | n/a | 1.34 |
| K2 + G | 133.00 | 591,575.20 | 589,858.37 | −294,796.18 | n/a | 0.84 | 1.56 |
| K2 + G + I | 134.00 | 591,756.46 | 590,026.72 | −294,879.35 | 0.00 | 0.83 | 1.65 |
| GTR | 139.00 | 592,361.70 | 590,567.41 | −295,144.70 | n/a | n/a | 1.33 |
| T92 | 133.00 | 594,707.63 | 592,990.80 | −296,362.39 | n/a | n/a | 1.32 |
| HKY | 135.00 | 594,729.93 | 592,987.28 | −296,358.63 | n/a | n/a | 1.32 |
| TN93 | 136.00 | 594,733.26 | 592,977.71 | −296,352.85 | n/a | n/a | 1.32 |
| K2 + I | 133.00 | 596,345.39 | 594,628.56 | −297,181.27 | 0.33 | n/a | 1.45 |
| JC + G | 132.00 | 600,630.85 | 598,926.93 | −299,331.46 | n/a | 0.87 | 0.50 |
| JC + G + I | 133.00 | 600,645.77 | 598,928.94 | −299,331.46 | 0.00 | 0.87 | 0.50 |
| K2 | 132.00 | 602,173.44 | 600,469.52 | −300,102.75 | n/a | n/a | 1.39 |
| JC + I | 132.00 | 605,419.13 | 603,715.21 | −301,725.60 | 0.32 | n/a | 0.50 |
| JC | 131.00 | 610,996.30 | 609,305.29 | −304,521.64 | n/a | n/a | 0.50 |


References
- Dyall, S.D.; Brown, M.T.; Johnson, P.J. Ancient invasions: From endosymbionts to organelles. Science 2004, 304, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chumley, T.W.; Palmer, J.D.; Mower, J.P.; Fourcade, H.M.; Calie, P.J.; Boore, J.L.; Jansen, R.K. The complete chloroplast genome sequence of Pelargonium × hortorum: Organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 2006, 23, 2175–2190. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.D.; Jansen, R.K.; Michaels, H.J.; Chase, M.W.; Manhart, J.R. Chloroplast DNA variation and plant phylogeny. Ann. Mo. Bot. Gard. 1988, 1180–1206. [Google Scholar] [CrossRef]
- Corriveau, J.L.; Goff, L.J.; Coleman, A.W. Plastid DNA is not detectable in the male gametes and pollen tubes of an angiosperm (Antirrhinum majus) that is maternal for plastid inheritance. Curr. Genet. 1990, 17, 439–444. [Google Scholar] [CrossRef]
- Chen, X.Q.; Clayton, D. Coelogyne Lindley. In Flora of China; Wu, Z.Y., Raven, P.H., Eds.; 25; Science Press, Beijing & Missouri Botanical Garden Press: St. Louis, MO, USA, 2009; pp. 315–325. [Google Scholar]
- Cheng, J.; Shi, J.; Shangguan, F.Z.; Dafni, A.; Deng, Z.H.; Luo, Y.B. The pollination of a self-incompatible, food-mimic orchid, Coelogyne fimbriata (Orchidaceae), by female Vespula wasps. Ann. Bot. 2009, 104, 565–571. [Google Scholar] [CrossRef]
- Satake, M.; Ijung, L. Flowers in Myanmar (Part II): Wild orchid and medicinal orchid. Aroma Res. 2004, 5, 83–89. [Google Scholar]
- Pramanick, D.D. Pharmacognostic studies on the pseudobulb of Coelogyne cristata Lindl.(Orchidaceae)-An epiphytic orchid of ethno-medicinal importance. J. Pharmacogn. Phytochem. 2016, 5, 120. [Google Scholar]
- Singh, N.; Kumaria, S. A Combinational Phytomolecular-Mediated Assessment in Micropropagated Plantlets of Coelogyne ovalis Lindl.: A Horticultural and Medicinal Orchid. Proc. Natl. Acad. Sci. India Sect. B Biol. Sci. 2020, 90, 455–466. [Google Scholar] [CrossRef]
- Teoh, E.S. Medicinal orchids of Asia; Springer: Cham, Switzerland, 2016. [Google Scholar]
- Wu, X.R. A Concise Edition of Medicinal Plants in China; Guangdong Higher Education Publication House: Guangdong, China, 1994. (In Chinese) [Google Scholar]
- Gravendeel, B.; Chase, M.W.; de Vogel, E.F.; Roos, M.C.; Mes, T.H.; Bachmann, K. Molecular phylogeny of Coelogyne (Epidendroideae; Orchidaceae) based on plastid RFLPs, matK, and nuclear ribosomal ITS sequences: Evidence for polyphyly. Am. J. Bot. 2001, 88, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.S.; Chen, J.J.; Chiu, C.C.; Hsiao, H.C.; Yang, C.J.; Jin, X.H.; Leebens-Mack, J.; de Pamphilis, C.W.; Huang, Y.T.; Yang, L.H.; et al. Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some orchids. Plant J. 2017, 90, 994–1006. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, J.; Shimada, H.; Whittier, R.; Ishibashi, T.; Sakamoto, M.; Mori, M.; Kondo, C.; Honji, Y.; Sun, C.R.; Meng, B.Y. The complete sequence of the rice (Oryza sativa) chloroplast genome: Intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. MGG 1989, 217, 185–194. [Google Scholar] [CrossRef]
- Maier, R.M.; Neckermann, K.; Igloi, G.L.; Kössel, H. Complete sequence of the maize chloroplast genome: Gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J. Mol. Biol. 1995, 251, 614–628. [Google Scholar] [CrossRef]
- Gantt, J.S.; Baldauf, S.L.; Calie, P.J.; Weeden, N.F.; Palmer, J.D. Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO J. 1991, 10, 3073–3078. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F.; Massenet, O.; Dome, A.M.; Briat, J.F.; Mache, R. Expression of the rpl23, rpl2 and rps19 genes in spinach chloroplasts. Nucleic Acids Res. 1988, 16, 2461–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagano, Y.; Matsuno, R.; Sasaki, Y. Sequence and transcriptional analysis of the gene cluster trnQ-zfpA-psaI-ORF231-petA in pea chloroplasts. Curr. Genet. 1991, 20, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.H.; Chan, M.T.; Liao, D.C.; Hsu, C.T.; Lee, Y.W.; Daniell, H.; Duvall, M.R.; Lin, C.S. Complete chloroplast genome of Oncidium Gower Ramsey and evaluation of molecular markers for identification and breeding in Oncidiinae. BMC Plant Biol. 2010, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roma, L.; Cozzolino, S.; Schlüter, P.M.; Scopece, G.; Cafasso, D. The complete plastid genomes of Ophrys iricolor and O. sphegodes (Orchidaceae) and comparative analyses with other orchids. PLoS ONE 2018, 13, e0204174. [Google Scholar] [CrossRef] [Green Version]
- Pan, I.C.; Liao, D.C.; Wu, F.H.; Daniell, H.; Singh, N.D.; Chang, C.; Shih, M.C.; Chan, M.T.; Lin, C.S. Complete chloroplast genome sequence of an orchid model plant candidate: Erycina pusilla apply in tropical Oncidium breeding. PLoS ONE 2012, 7, e34738. [Google Scholar] [CrossRef]
- Yang, J.B.; Tang, M.; Li, H.T.; Zhang, Z.R.; Li, D.Z. Complete chloroplast genome of the genus Cymbidium: Lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 2013, 13, 84. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, C.; Wang, Z.Z. The complete chloroplast genome of the Dendrobium strongylanthum (Orchidaceae: Epidendroideae). Mitochondrial DNA Part A 2016, 27, 3048–3049. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Kim, J.S.; Moore, M.J.; Neubig, K.M.; Williams, N.H.; Whitten, W.M.; Kim, J.H. Seven new complete plastome sequences reveal rampant independent loss of the ndh gene family across orchids and associated instability of the inverted repeat/small single-copy region boundaries. PLoS ONE 2015, 10, e0142215. [Google Scholar] [CrossRef] [PubMed]
- Cavalier-Smith, T. Chloroplast evolution: Secondary symbiogenesis and multiple losses. Curr. Biol. 2002, 12, R62–R64. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Tang, P.; Li, Z.; Li, D.; Liu, Y.; Huang, H. The first complete chloroplast genome sequences in Actinidiaceae: Genome structure and comparative analysis. PLoS ONE 2015, 10, e0129347. [Google Scholar] [CrossRef] [PubMed]
- Small, R.L.; Ryburn, J.A.; Cronn, R.C.; Seelanan, T.; Wendel, J.F. The tortoise and the hare: Choosing between noncoding plastome and nuclear Adh sequences for phylogeny reconstruction in a recently diverged plant group. Am. J. Bot. 1998, 85, 1301–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef]
- Jakobsson, M.; Säll, T.; Lind-Halldén, C.; Halldén, C. Evolution of chloroplast mononucleotide microsatellites in Arabidopsis thaliana. Theor. Appl. Genet. 2007, 114, 223. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, H.L. Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 2004, 11, 247–261. [Google Scholar] [CrossRef]
- Vu, H.T.; Tran, N.; Nguyen, T.-D.; Vu, Q.L.; Bui, M.H.; Le, M.T.; Le, L. Complete chloroplast genome of Paphiopedilum delenatii and phylogenetic relationships among Orchidaceae. Plants 2020, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Jo, S.; Cheon, S.H.; Joo, M.J.; Hong, J.R.; Kwak, M.; Kim, K.J. Plastome evolution and phylogeny of Orchidaceae, with 24 new sequences. Front. Plant Sci. 2020, 11, 22. [Google Scholar] [CrossRef]
- Shi, C.; Hu, N.; Huang, H.; Gao, J.; Zhao, Y.J.; Gao, L.Z. An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS ONE 2012, 7, e31468. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, R. Kmernator: An MPI Toolkit for Large Scale Genomic Analysis. 2014. Available online: https://github.com/JGI-Bioinformatics/Kmernator (accessed on 8 October 2020).
- Miao, L.Y.; Hu, C.; Huang, W.C.; Jiang, K. Chloroplast genome structure and phylogenetic position of Calanthe sylvatica (Thou.) Lindl. (Orchidaceae). Mitochondrial DNA Part B 2019, 4, 2625–2626. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stephan, G.; Pascal, L.; Ralph, B. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Vaidya, G.; Lohman, D.J.; Meier, R. SequenceMatrix: Concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 2011, 27, 171–180. [Google Scholar] [CrossRef]







| Characteristics and Parameters | C. fimbriata | C. ovalis |
|---|---|---|
| Raw reads (bp) | 3,142,569 | 3,763,406 |
| Clean reads (bp) | 3,041,719 | 3,624,370 |
| Average read length (bp) | 300 | 300 |
| Number of contigs | 1 | 1 |
| Total length of contigs (bp) | 159,795 | 160,040 |
| N50 length of contigs (bp) | 159,795 | 160,040 |
| Total cp genome size (bp) | 159,795 | 160,040 |
| LSC length (bp) | 87,606 | 87,759 |
| SSC length (bp) | 18,839 | 18,851 |
| IR length (bp) | 26,675 | 26,715 |
| Total CDS length (bp) | 79,891 | 78,258 |
| Total tRNA length (bp) | 2865 | 2911 |
| Total rRNA length (bp) | 9038 | 9041 |
| Total GC content (%) | 37.39 | 37.35 |
| GC content for LSC (%) | 35.30 | 35.20 |
| GC content for SSC (%) | 30.50 | 30.40 |
| GC content for IR (%) | 43.30 | 43.30 |
| Total number of genes | 136 | 133 |
| Protein-coding genes | 90 | 87 |
| rRNAs genes | 38 | 38 |
| tRNAs genes | 8 | 8 |
| Duplicated genes | 17 | 17 |
| Categories of Genes | Groups of Genes | Name of Genes |
|---|---|---|
| RNA genes | Ribosomal RNAs | rrn5a, rrn4.5a, rrn16a, rrn23a |
| Transfer RNAs | trnK-UUUb, trnQ-UUG, trnS-GCU, trnG-GCCb, trnR-UCU, trnC-GCA, trnD-GUC, trnA-UGC, trnY-GUA, trnE-UUC, trnF-GAA, trnT-GGU, trnS-UGA, trnG-UCC, trnfM-CAU, trnS-GGA, trnT-UGU, trnL-UAAb, trnF-GAA, trnV-UACb, trnM-CAU, trnW-CCA, trnP-UGG, trnH-GUGa, trnI-CAUa, trnL-CAAa, trnV-GACa, trnI-GAUa,b, trnA-UGCa,b, trnR-ACGa, trnN-GUUa, trnL-UAG, trnS-GCU | |
| Transcription- and translation-related genes | Small subunit of ribosome | rps2, rps3, rps4, rps7a, rps8, rps11, rps12c, rps14, rps15, rps16b, rps18, rps19a |
| Large subunit of ribosome | rpl2a,b, rpl14, rpl16b, rpl20, rpl22, rpl23a, rpl32, rpl33, rpl36 | |
| Transcription | rpoA, rpoB, rpoC1b, rpoC2 | |
| Translation initiation factor | infA | |
| Photosynthesis-related genes | NADH dehydrogenase | ndhAb, ndhBa,b, ndhC, ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK |
| Photosystem I | psaA, psaB, psaC, psaI, psaJ | |
| Photosystem II | psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbK, psbL, psbJ, psbN, psbT, psbZ, psbM | |
| RubisCO large subunit | rbcL | |
| Cytochrome b/f complex | petA, petBb, petD, petG, petL, petN | |
| ATP synthase | atpA, atpB, atpE, atpFb, atpH, atpI | |
| Cytochrome c synthesis | ccsA | |
| Others | RNA processing | matK |
| Carbon metabolism | cemA | |
| Fatty acid synthesis | accD | |
| Proteolysis | clpPc | |
| Genes of unknown function | Conserved reading frames | ycf1, ycf2a, ycf4, ycf3c, ycf15, ycf68d |
| Gene | Location | Nucleotides in Base Pairs | ||||
|---|---|---|---|---|---|---|
| Exon I | Intron I | Exon II | Intron II | Exon III | ||
| atpF | LSC | 144/144 | 965/964 | 411/411 | ||
| clpP | LSC | 69/69 | 963/950 | 291/291 | 675/673 | 252/252 |
| ndhA | SSC | 552/552 | 1235/1235 | 540/540 | ||
| ndhB | IR | 777/777 | 701/710 | 756/756 | ||
| petB | LSC | 6/6 | 739/736 | 642/642 | ||
| rpl16 | LSC | 9/9 | 1007/1248 | 399/399 | ||
| rpl2 | IR | 387/387 | 663/663 | 432/432 | ||
| rpoC1 | LSC | 435/435 | 766/778 | 1617/1617 | ||
| rps12a | LSC | 126/126 | - | 232/232 | 549/549 | 26/26 |
| rps16 | LSC | 40/40 | 894/893 | 248/248 | ||
| ycf3 | LSC | 126/126 | 721/723 | 228/228 | 672/672 | 152/152 |
| trnG-GCC | LSC | 23/23 | 700/700 | 47/47 | ||
| trnI-GAU | IR | 42/42 | 948/948 | 35/35 | ||
| trnK-UUU | LSC | 37/37 | 2915/2917 | 26/26 | ||
| trnL-UAA | LSC | 35/35 | 574/574 | 50/50 | ||
| trnV-UAC | LSC | 39/39 | 577/577 | 35/35 | ||
| Locus | Primer Sequence (5′-3′) | Location | Product Length (bp) | Annealing Temperature/Tm (°C) |
|---|---|---|---|---|
| matK | F: CACCAGATCATTGATACGGA | CDS | 1395 | 55 |
| R: CCTGTGGAAATTCTCGGTTA | ||||
| rpoC2 | F: TATTGTCCATGCCTCTTCAC | CDS | 1014 | 55 |
| R: CATTTTTCTGGAGAGGTGGA | ||||
| ndhJ-ndhK | F: CCTATCCAACTTTCAGGCAT | IGS | 667 | 55 |
| R: ATCACAAGTTTGACCTTCGA | ||||
| rbcL | F: TCGAGTAGACCTTGTTGTTG | IGS | 724 | 55 |
| R: CGGCACAAAATAAGAAACGA | ||||
| accD-psaI | F: TGTTTTCTTTGGGGACATCA | IGS | 940 | 55 |
| R: CGGAAAGGCCACATATCATA | ||||
| ycf4-cemA | F: TGAGAATTTGACTCCACGAG | IGS | 970 | 55 |
| R: ATTTCGGATTGCCTGGTATT | ||||
| clpP-psbB | F: ACACCAATGGGCATTAAGAT | IGS | 610 | 55 |
| R: ACCTGTTCGGTAGATTTTGT | ||||
| psbB-psbN | F: ATGCTCAAGTGGAATTTGGA | IGS | 652 | 55 |
| R: GAACTTTAGGTGGTTCTCGA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, K.; Miao, L.-Y.; Wang, Z.-W.; Ni, Z.-Y.; Hu, C.; Zeng, X.-H.; Huang, W.-C. Chloroplast Genome Analysis of Two Medicinal Coelogyne spp. (Orchidaceae) Shed Light on the Genetic Information, Comparative Genomics, and Species Identification. Plants 2020, 9, 1332. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9101332
Jiang K, Miao L-Y, Wang Z-W, Ni Z-Y, Hu C, Zeng X-H, Huang W-C. Chloroplast Genome Analysis of Two Medicinal Coelogyne spp. (Orchidaceae) Shed Light on the Genetic Information, Comparative Genomics, and Species Identification. Plants. 2020; 9(10):1332. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9101332
Chicago/Turabian StyleJiang, Kai, Li-Yuan Miao, Zheng-Wei Wang, Zi-Yi Ni, Chao Hu, Xin-Hua Zeng, and Wei-Chang Huang. 2020. "Chloroplast Genome Analysis of Two Medicinal Coelogyne spp. (Orchidaceae) Shed Light on the Genetic Information, Comparative Genomics, and Species Identification" Plants 9, no. 10: 1332. https://0-doi-org.brum.beds.ac.uk/10.3390/plants9101332