Anionic Polymer Brushes for Biomimetic Calcium Phosphate Mineralization—A Surface with Application Potential in Biomaterials

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation
2.2. Sample Nomenclature
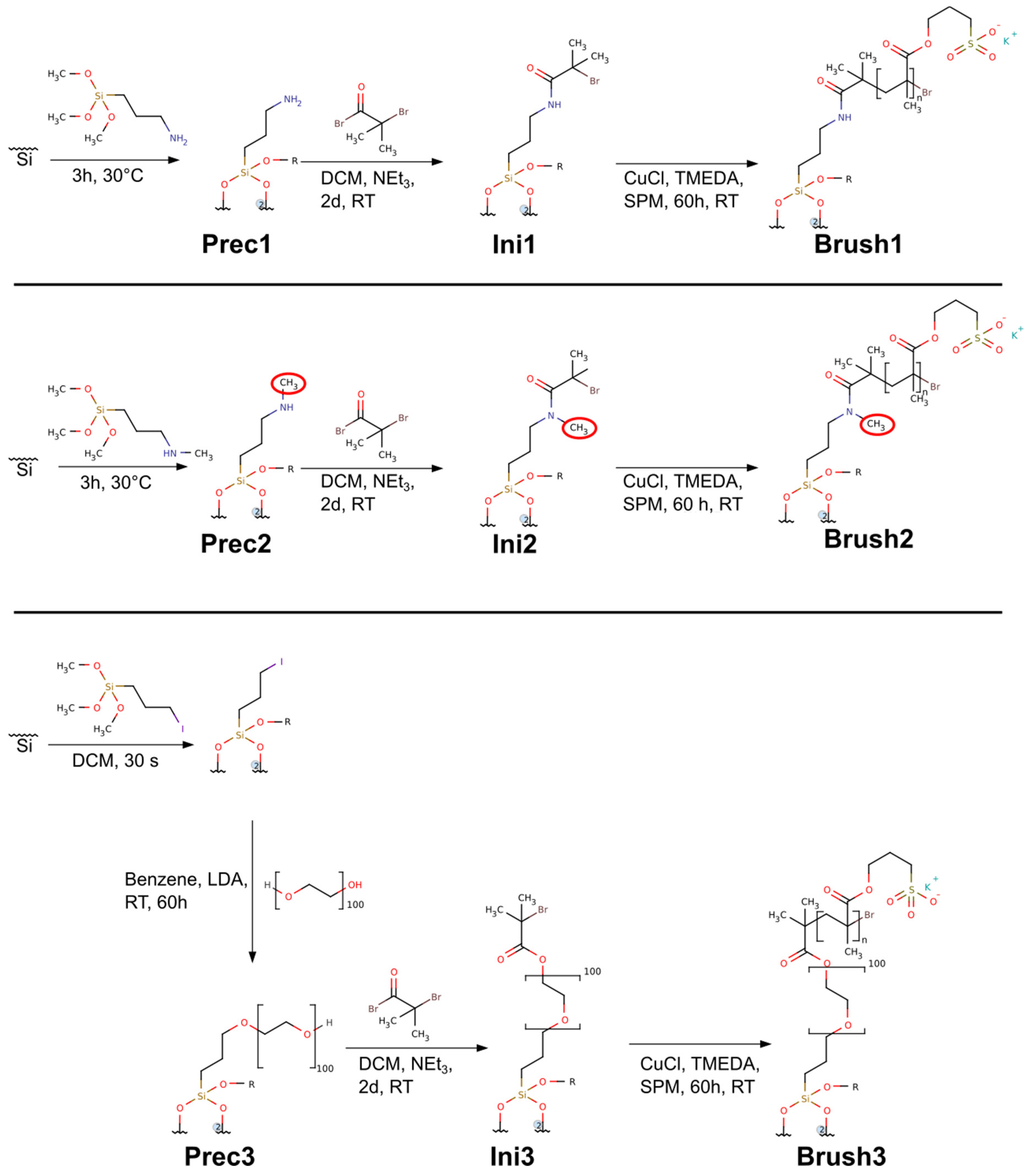
2.3. Precursors—Prec1, Prec2, Prec3
2.4. ATRP Initiators—Ini1, Ini2, Ini3
2.5. Brushes—Brush1, Brush2, Brush3
2.6. Mineralization—Min1, Min2, Min3
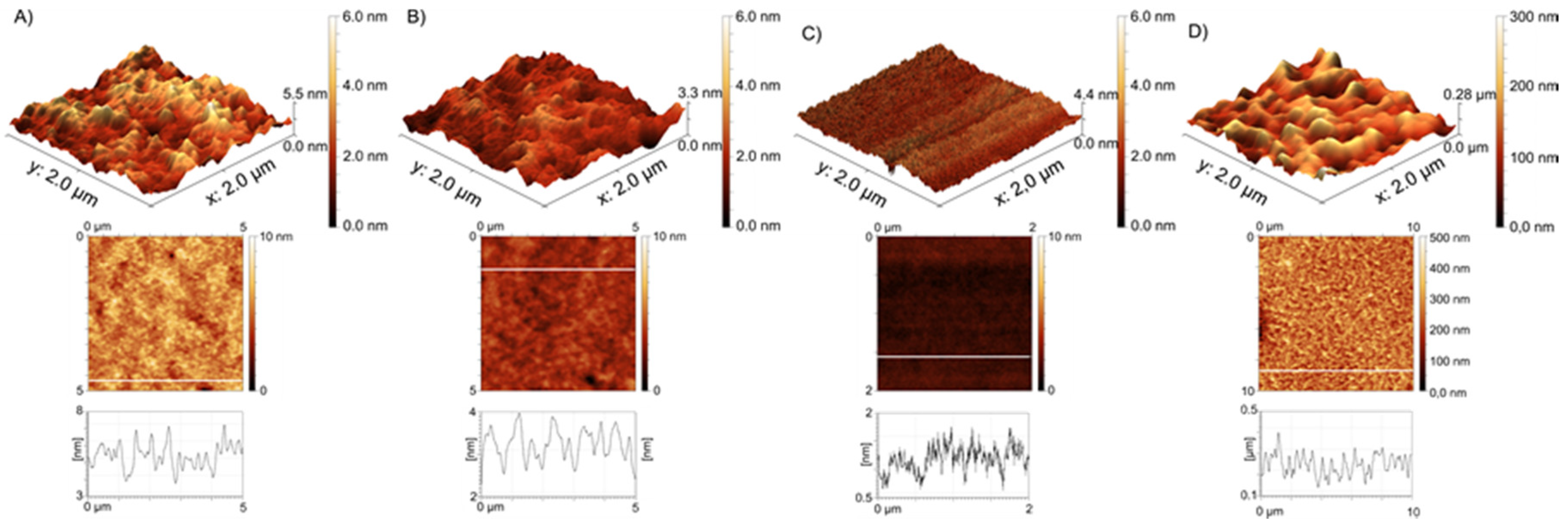
2.7. Atomic Force Microscopy (AFM)
2.8. Contact Angle Measurements
2.9. Scanning Electron Microscopy (SEM)
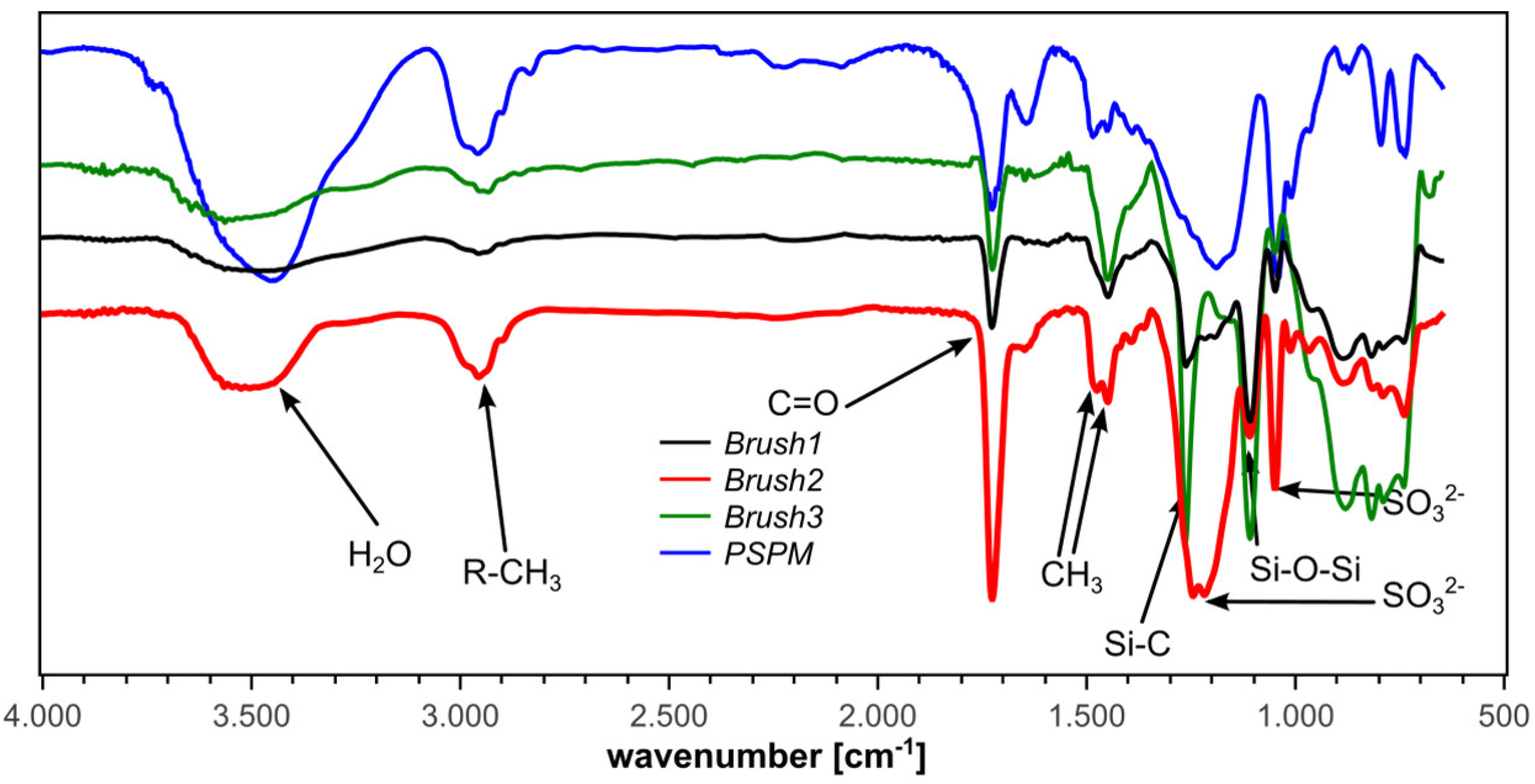
2.10. Infrared Reflection Absorption Spectroscopy (IRRAS)
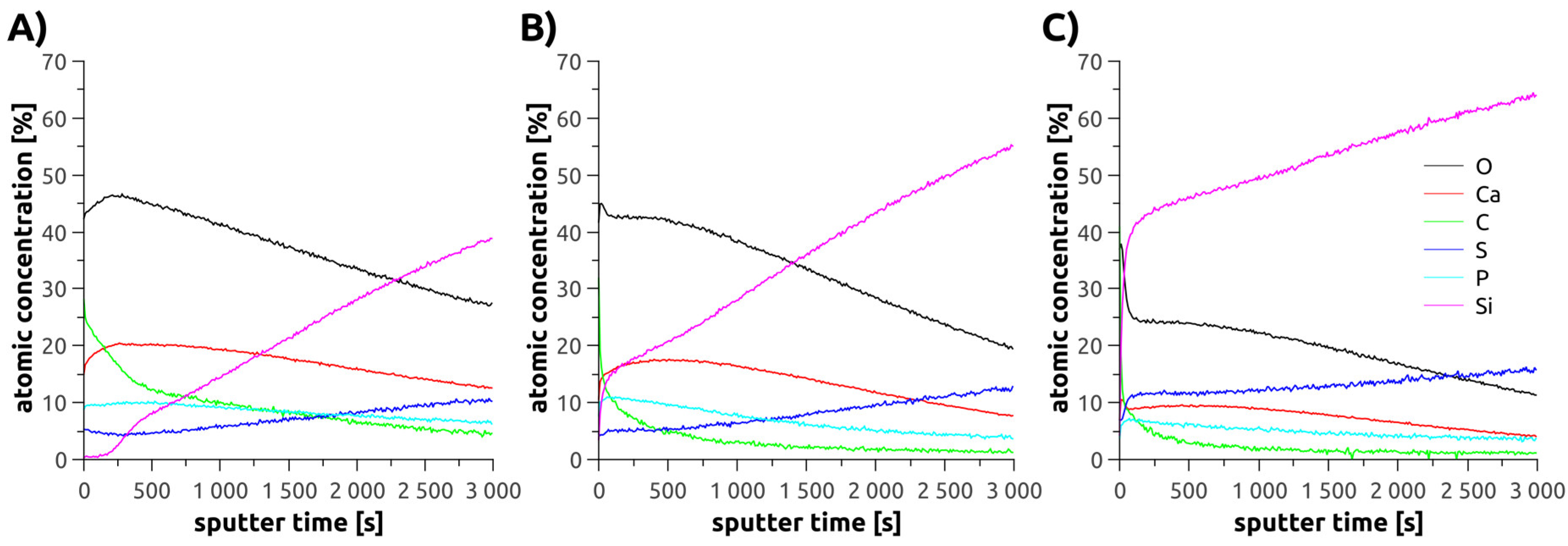
2.11. X-Ray Photoelectron Spectroscopy (XPS)
2.12. Cell Culture Experiments
2.13. IRRAS of Brush1, Brush2, Brush3, Min1, Min2, Min3
3. Results
3.1. Polymer Brushes
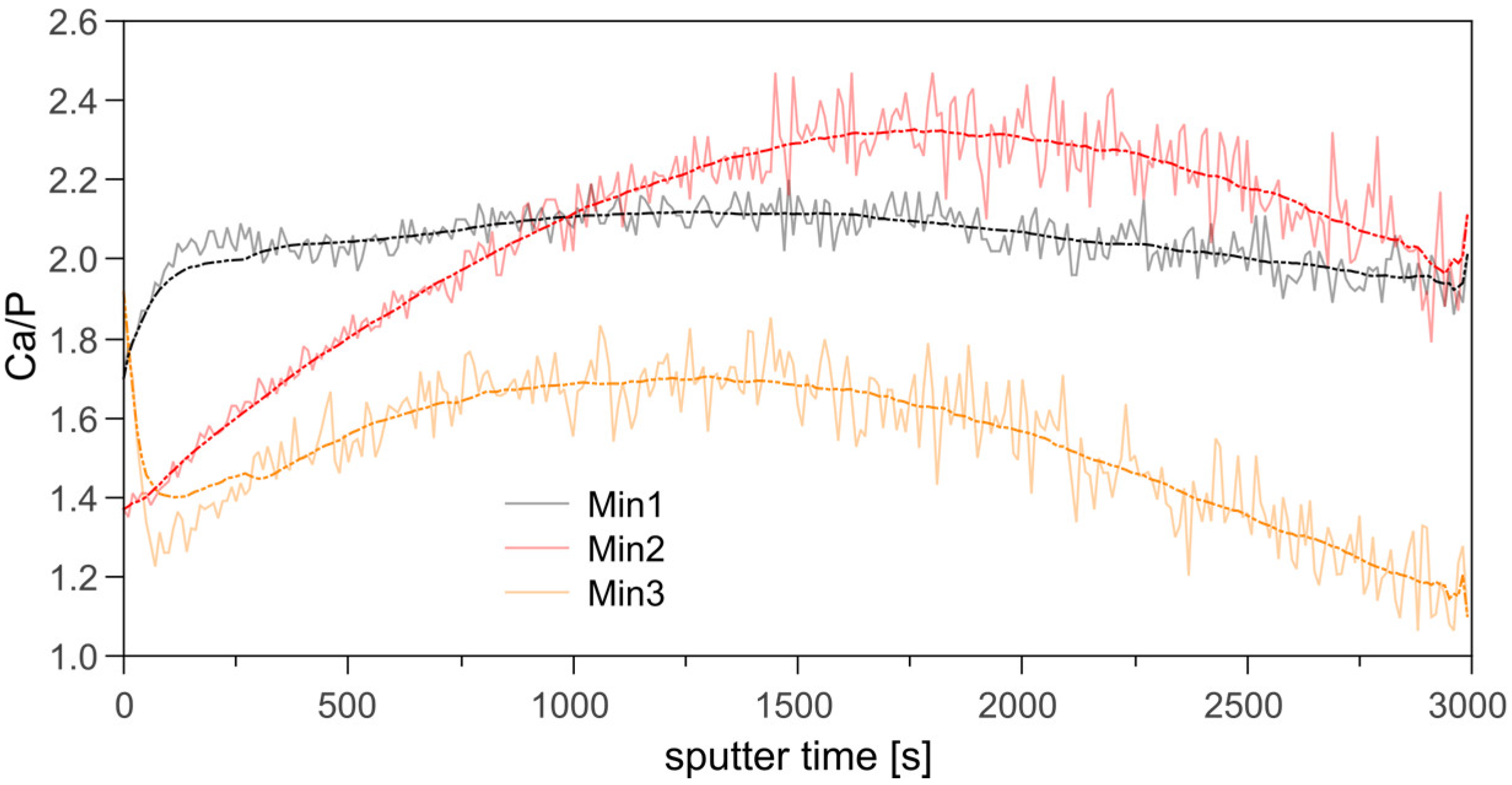
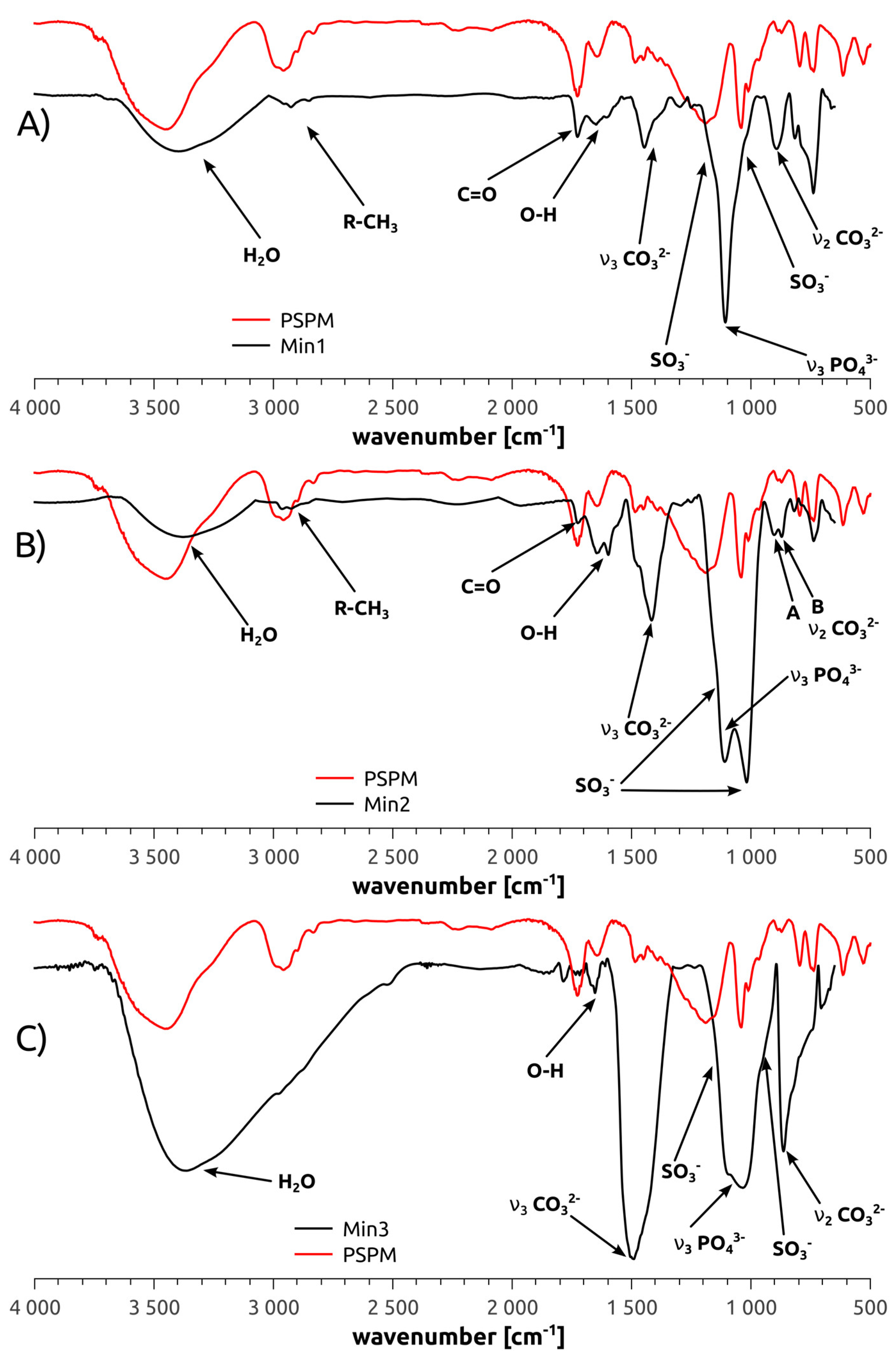
3.2. Mineralization of Polymer Brushes
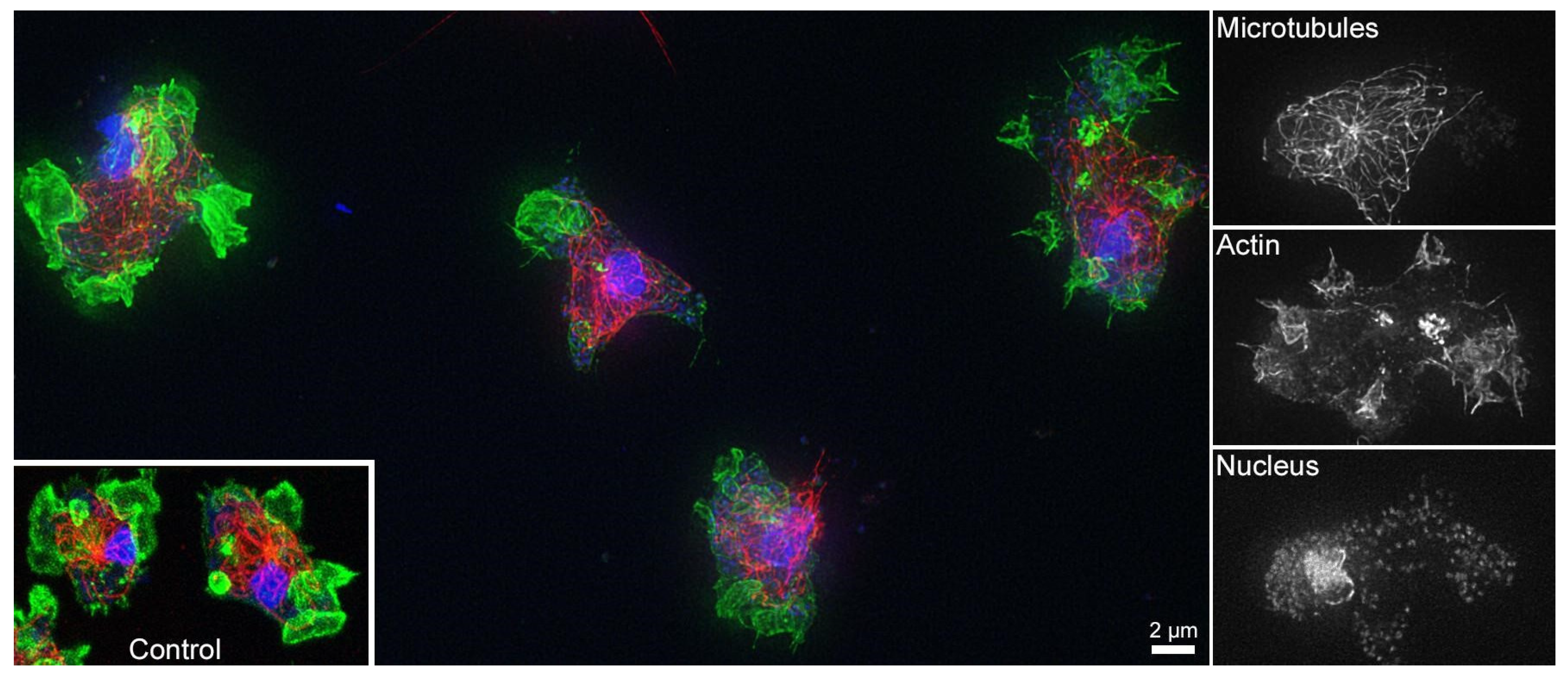
3.3. Cell Compatibility of Non-Mineralized and Mineralized Polymer Brushes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- DESTATIS. Durchschnittliche Weitere Lebenserwartung Nach Altersstufen ab 1871-81 bis 2010-12. 22.04.2015. 2015. Available online: https://www.destatis.de/DE/ZahlenFakten/GesellschaftStaat/Bevoelkerung/Sterbefaelle/Sterbefaelle.html#tab194502No2 (accessed on 28 May 2015).
- Dey, A.; Bomans, P.H.H.; Müller, F.A.; Will, J.; Frederik, P.M.; de With, G.; Sommerdijk, N.A.J.M. The role of prenucleation clusters in surface-induced calcium phosphate crystallization. Nat. Mater. 2010, 9, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; de With, G.; Sommerdijk, N.J.M. In situ techniques in biomimetic mineralization studies of calcium carbonate. Chem. Soc. Rev. 2010, 39, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Hentrich, D.; Junginger, M.; Bruns, M.; Börner, H.G.; Brandt, J.; Brezesinski, G.; Taubert, A. Interface-controlled calcium phosphate mineralization: effect of oligo(aspartic acid)-rich interfaces. CrystEngComm 2015, 17, 6901–6913. [Google Scholar] [CrossRef] [Green Version]
- Löbbicke, R.; Chanana, M.; Schlaad, H.; Pilz-Allen, C.; Günter, C.; Möhwald, H.; Taubert, A. Polymer brush controlled bioinspired calcium phosphate mineralization and bone cell growth. Biomacromolecules 2011, 12, 3753–3760. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-J.; Liu, H.-G.; Feng, X.-S.; Zhang, R.-J.; Zhang, L.; Mu, Y.-D.; Hao, J.-C.; Qian, D.-J.; Lou, Y.-F. Mineralization mechanism of calcium phosphates under three kinds of Langmuir monolayers. Langmuir 2004, 20, 2243–2249. [Google Scholar] [CrossRef] [PubMed]
- Hentrich, D.; Brezesinski, G.; Kübel, C.; Bruns, M.; Taubert, A. Cholesteryl Hemisuccinate Monolayers Efficiently Control Calcium Phosphate Nucleation and Growth. Cryst. Growth Des. 2017, 17, 5764–5774. [Google Scholar] [CrossRef]
- Hentrich, D.; Taabache, S.; Brezesinski, G.; Lange, N.; Unger, W.; Kübel, C.; Bertin, A.; Taubert, A. A Dendritic Amphiphile for Efficient Control of Biomimetic Calcium Phosphate Mineralization. Macromol. Biosci. 2017, 17, 1600524. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, M.; Voccia, S.; Gabriel, S.; Gilbert, B.; Cossement, D.; Jérôme, R.; Jérôme, C. Stainless steel grafting of hyperbranched polymer brushes with an antibacterial activity: Synthesis, characterization, and properties. Langmuir 2009, 25, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Letsche, S.A.; Steinbach, A.M.; Pluntke, M.; Marti, O.; Ignatius, A.; Volkmer, D. Usage of polymer brushes as substrates of bone cells. Front. Mater. Sci. China 2009, 3, 132–144. [Google Scholar] [CrossRef]
- Liu, W.; Lipner, J.; Xie, J.; Manning, C.N.; Thomopoulos, S.; Xia, Y. Nanofiber Scaffolds with Gradients in Mineral Content for Spatial Control of Osteogenesis. ACS Appl. Mater. Interfaces 2014, 6, 2842–2849. [Google Scholar] [CrossRef] [PubMed]
- Ramstedt, M.; Cheng, N.; Azzaroni, O.; Mossialos, D.; Mathieu, H.J.; Huck, W.T.S. Synthesis and characterization of poly(3-sulfopropylmethacrylate) brushes for potential antibacterial applications. Langmuir 2007, 23, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Sin, M.C.; Sun, Y.M.; Chang, Y. Zwitterionic-Based Stainless Steel with Well-Defined Polysulfobetaine Brushes for General Bioadhesive Control. ACS Appl. Mater. Interfaces 2014, 6, 861–873. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, A.; Tautzenberger, A.; Ignatius, A.; Pluntke, M.; Marti, O.; Volkmer, D. Coatings from micropatterned sulfobetaine polymer brushes as substrates for MC3T3-E1 cells. J. Mater. Sci. Mater. Med. 2012, 23, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Mai, T.; Rakhmatullina, E.; Bleek, K.; Boye, S.; Yuan, J.; Völkel, A.; Gräwert, M.; Cheaib, Z.; Eick, S.; Günter, C.; et al. Poly(ethylene oxide)-b-poly(3-sulfopropyl methacrylate) Block Copolymers for Calcium Phosphate Mineralization and Biofilm Inhibition. Biomacromolecules 2014, 15, 3901–3914. [Google Scholar] [CrossRef] [PubMed]
- Aizenberg, J.; Black, A.J.; Whitesides, G.M. Oriented Growth of Calcite Controlled by Self-Assembled Monolayers of Functionalized Alkanethiols Supported on Gold and Silver. J. Am. Chem. Soc. 1999, 121, 4500–4509. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C. Purification of Laboratory Chemicals, 6th ed.; Elsevier Science: Amsterdam, The Netherlands, 2009; ISBN 978-1-85617-567-8. [Google Scholar]
- Mai, T.; Boye, S.; Yuan, J.; Völkel, A.; Gräwert, M.; Günter, C.; Lederer, A.; Taubert, A. Poly(ethylene oxide)-based block copolymers with very high molecular weights for biomimetic calcium phosphate mineralization. RSC Adv. 2015, 5, 103494–103505. [Google Scholar] [CrossRef] [Green Version]
- Nečas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Cent. Eur. J. Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Wiemann, M.; Bingmann, D.; Franzka, S.; Hartmann, N.; Urch, H.; Epple, M. Oriented Growth of Osteoblast-like Cells on Two- Dimensionally Structured Films of Functionalized Calcium Phosphate Nanoparticles on a Silicon Substrate. Adv. Eng. Mater. 2007, 9, 1077–1081. [Google Scholar] [CrossRef]
- Wolski, K.; Szuwarzyński, M.; Zapotoczny, S. A facile route to electronically conductive polyelectrolyte brushes as platforms of molecular wires. Chem. Sci. 2015, 6, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parry, K.L.; Shard, A.G.; Short, R.D.; White, R.G.; Whittle, J.D.; Wright, A. ARXPS characterisation of plasma polymerised surface chemical gradients. Surf. Interface Anal. 2006, 38, 1497–1504. [Google Scholar] [CrossRef]
- Scofield, J.H. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron Spectros. Relat. Phenomena 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Tanuma, S.; Powell, C.J.; Penn, D.R. Calculations of electron inelastic mean free paths. V. Data for 14 organic compounds over the 50–2000 eV range. Surf. Interface Anal. 1994, 21, 165–176. [Google Scholar] [CrossRef]
- Bretschneider, T.; Diez, S.; Anderson, K.; Heuser, J.; Clarke, M.; Müller-Taubenberger, A.; Köhler, J.; Gerisch, G. Dynamic Actin Patterns and Arp2/3 Assembly at the Substrate-Attached Surface of Motile Cells. Curr. Biol. 2004, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batsios, P.; Baumann, O.; Gräf, R.; Meyer, I. Isolation of Dictyostelium Nuclei for Light and Electron Microscopy. In Dictyostelium Discoideum Protocols; Eichinger, L., Rivero, F., Eds.; Humana Press: New York, NY, USA, 2013; pp. 283–294. ISBN 978-1-62703-302-2. [Google Scholar]
- Karakassides, M.A. An Infrared Reflectance Study of Si-O Vibrations in Thermally Treated Alkali-Saturated Montmorillonites. Clay Miner. 1999, 34, 429–438. [Google Scholar] [CrossRef]
- Luna-López, J.A.; Carrillo-López, J.; Aceves-Mijares, M.; Morales-Sánchez, A.; Falcony, C.; Luna-Lope, J.A.; Carrillo-Lopez, J.; Aceves-Mijares, M.; Morales-Sanchez, A.; Falcony, C. FTIR and photoluminescence of annealed silicon rich oxide films. Superf. Vacio 2009, 22, 11–14. [Google Scholar]
- Thomas, S. Online Programme zur Spektroskopie. Available online: http://www.spec-online.de/ (accessed on 10 May 2018).
- Koutsopoulos, S. Synthesis and characterization of hydroxyapatite crystals: A review study on the analytical methods. J. Biomed. Mater. Res. 2002, 62, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Ren, F.; Ding, Y.; Leng, Y. Infrared spectroscopic characterization of carbonated apatite: A combined experimental and computational study. J. Biomed. Mater. Res. Part A 2014, 102, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Shimizu, M.; Collins, B.; Glimcher, M.J. Resolution-enhanced fourier transform infrared spectroscopy study of the environment of phosphate ions in the early deposits of a solid phase of calcium-phosphate in bone and enamel, and their evolution with age. I: Investigations in thev 4 PO4 domain. Calcif. Tissue Int. 1990, 46, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Boyd, I.W.; Wilson, J.I.B. A study of thin silicon dioxide films using infrared absorption techniques. J. Appl. Phys. 1982, 53, 4166–4172. [Google Scholar] [CrossRef]
- Mansour, N.; Momeni, A.; Karimzadeh, R.; Amini, M. Surface effects on the luminescence properties of colloidal silicon nanocrystals in water. Phys. Scr. 2013, 87, 035701. [Google Scholar] [CrossRef]
- Pluchery, O.; Costantini, J.-M. Infrared spectroscopy characterization of 3C–SiC epitaxial layers on silicon. J. Phys. D Appl. Phys. 2012, 45, 495101. [Google Scholar] [CrossRef]
- Bohner, M. Calcium orthophosphates in medicine: from ceramics to calcium phosphate cements. Injury 2000, 31 (Suppl. 4), 37–47. [Google Scholar] [CrossRef]
- Schweizer, S.; Taubert, A. Polymer-controlled, bio-inspired calcium phosphate mineralization from aqueous solution. Macromol. Biosci. 2007, 7, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Wopenka, B.; Pasteris, J.D. A mineralogical perspective on the apatite in bone. Mater. Sci. Eng. C 2005, 25, 131–143. [Google Scholar] [CrossRef]
- Goldmann, A.S.; Tischer, T.; Barner, L.; Bruns, M.; Barner-Kowollik, C. Mild and Modular Surface Modification of Cellulose via Hetero Diels−Alder (HDA) Cycloaddition. Biomacromolecules 2011, 12, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Zydziak, N.; Hübner, C.; Bruns, M.; Barner-Kowollik, C. One-Step Functionalization of Single-Walled Carbon Nanotubes (SWCNTs) with Cyclopentadienyl-Capped Macromolecules via Diels−Alder Chemistry. Macromolecules 2011, 44, 3374–3380. [Google Scholar] [CrossRef]
- Chusuei, C.C.; Goodman, D.W.; van Stipdonk, M.J.; Justes, D.R.; Schweikert, E.A. Calcium Phosphate Phase Identification Using XPS and Time-of-Flight Cluster SIMS. Anal. Chem. 1999, 71, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Fleet, M.E.; Liu, X. Coupled substitution of type A and B carbonate in sodium-bearing apatite. Biomaterials 2007, 28, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Fleet, M.E.; Liu, X.; King, P.L. Accommodation of the carbonate ion in apatite: An FTIR and X-ray structure study of crystals synthesized at 2-4 GPa. Am. Mineral. 2004, 89, 1422–1432. [Google Scholar] [CrossRef]
- Vignoles, M.; Bonel, G.; Holcomb, D.W.; Young, R.A. Influence of preparation conditions on the composition of type B carbonated hydroxyapatite and on the localization of the carbonate ions. Calcif. Tissue Int. 1988, 43, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Pelorgeas, S.; Martin, J.B.; Satre, M. Cytotoxicity of dichloromethane diphosphonate and of 1-hydroxyethane-1,1-diphosphonate in the amoebae of the slime mould Dictyostelium discoideum. A 31P NMR study. Biochem. Pharmacol. 1992, 44, 2157–2163. [Google Scholar] [CrossRef]
- Pozzolini, M.; Sturla, L.; Cerrano, C.; Bavestrello, G.; Camardella, L.; Parodi, A.M.; Raheli, F.; Benatti, U.; Müller, W.E.G.; Giovine, M. Molecular cloning of silicatein gene from marine sponge Petrosia ficiformis (Porifera, Demospongiae) and development of primmorphs as a model for biosilicification studies. Mar. Biotechnol. (N. Y.) 2005, 6, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Shkilnyy, A.; Gräf, R.; Hiebl, B.; Neffe, A.T.; Friedrich, A.; Hartmann, J.; Taubert, A. Unprecedented, low cytotoxicity of spongelike calcium phosphate/poly(ethylene imine) hydrogel composites. Macromol. Biosci. 2009, 9, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Barbey, R.R.; Lavanant, L.; Paripovic, D.; Schüwer, N.; Sugnaux, C.; Tugulu, S.; Klok, H.-A.; Schuwer, N.; Sugnaux, C.; Tugulu, S.; et al. Polymer brushes via surface-initiated controlled radical polymerization: synthesis, characterization, properties, and applications. Chem. Rev. 2009, 109, 5437–5527. [Google Scholar] [CrossRef] [PubMed]
- Brinks, M.K.; Studer, A. Polymer Brushes by Nitroxide-Mediated Polymerization. Macromol. Rapid Commun. 2009, 30, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Howarter, J.A.; Youngblood, J.P. Self-Cleaning and Anti-Fog Surfaces via Stimuli-Responsive Polymer Brushes. Adv. Mater. 2007, 19, 3838–3843. [Google Scholar] [CrossRef]
- Kobayashi, M.; Terayama, Y.; Yamaguchi, H.; Terada, M.; Murakami, D.; Ishihara, K.; Takahara, A. Wettability and antifouling behavior on the surfaces of superhydrophilic polymer brushes. Langmuir 2012, 28, 7212–7222. [Google Scholar] [CrossRef] [PubMed]
- Mittal, V. Polymer Brushes: Substrates, Technologies, and Properties; CRC Press: Boca Raton, FL, USA, 2012. [Google Scholar]
- Zhou, F.; Huck, W.T.S. Surface grafted polymer brushes as ideal building blocks for “smart“ surfaces. Phys. Chem. Chem. Phys. 2006, 8, 3815–3818. [Google Scholar] [CrossRef] [PubMed]
- Minko, S. Grafting on Solid Surfaces: “Grafting to” and “Grafting from” Methods. In Polymer Surfaces and Interfaces; Springer: Berlin/Heidelberg, Germany, 2008; pp. 215–234. ISBN 9783540738640. [Google Scholar]
- Matyjaszewski, K. Atom Transfer Radical Polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Szwarc, M. ‘Living’ Polymers. Nature 1956, 178, 1168–1169. [Google Scholar] [CrossRef]
- Szwarc, M. Living polymers. Their discovery, characterization, and properties. J. Polym. Sci. Part A Polym. Chem. 1998, 36, ix–xv. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Amorphous Calcium Orthophosphates: Nature, Chemistry and Biomedical Applications. Int. J. Mater. Chem. 2012, 2, 19–46. [Google Scholar] [CrossRef]
- Elyada, A.; Garti, N.; Füredi-Milhofer, H. Polyelectrolyte multilayer-calcium phosphate composite coatings for metal implants. Biomacromolecules 2014, 15, 3511–3521. [Google Scholar] [CrossRef] [PubMed]
- LeGeros, R.Z. Properties of osteoconductive biomaterials: Calcium phosphates. Clin. Orthop. Relat. Res. 2002, 81–98. [Google Scholar] [CrossRef]
- Eanes, E.D. Amorphous Calcium Phosphate. In Octacalcium Phosphate; KARGER: Basel, Switzerland, 2001; pp. 130–147. [Google Scholar]
- Gibson, I.R.; Bonfield, W. Novel synthesis and characterization of an AB-type carbonate-substituted hydroxyapatite. J. Biomed. Mater. Res. 2002, 59, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Boonen, S.; Vanderschueren, D.; Callewaert, F.; Haentjens, P. Aging and Bone Loss. Osteoporos. Men 2010, 89, 207–219. [Google Scholar] [CrossRef]
- Li, Z.; Pasteris, J.D. Tracing the pathway of compositional changes in bone mineral with age: Preliminary study of bioapatite aging in hypermineralized dolphin’s bulla. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Yerramshetty, J.S.; Lind, C.; Akkus, O. The compositional and physicochemical homogeneity of male femoral cortex increases after the sixth decade. Bone 2006, 39, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Rey, C.; Shimizu, M.; Collins, B.; Glimcher, M.J. Resolution-enhanced fourier transform infrared spectroscopy study of the environment of phosphate ion in the early deposits of a solid phase of calcium phosphate in bone and enamel and their evolution with age: 2. Investigations in the v3 PO4 domain. Calcif. Tissue Int. 1991, 49, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Legros, R.; Balmain, N.; Bonel, G. Age-related changes in mineral of rat and bovine cortical bone. Calcif. Tissue Int. 1987, 41, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Tugulu, S.; Harms, M.; Fricke, M.; Volkmer, D.; Klok, H.-A. Polymerbürsten als ionotrope Matrices für die gezielte Herstellung mikrostrukturierter dünner Calcit-Filme. Angew. Chem. 2006, 118, 7619–7623. [Google Scholar] [CrossRef]
- Tugulu, S.; Barbey, R.; Harms, M.; Fricke, M.; Volkmer, D.; Rossi, A.; Klok, H.-A. Synthesis of Poly(methacrylic acid) Brushes via Surface-Initiated Atom Transfer Radical Polymerization of Sodium Methacrylate and Their Use as Substrates for the Mineralization of Calcium Carbonate. Macromolecules 2007, 40, 168–177. [Google Scholar] [CrossRef]
- Van den Beucken, J.J.J.P.; Walboomers, X.F.; Leeuwenburgh, S.; Vos, M.F.J.; Sommerdijk, N.A.J.M.; Nolte, R.J.M.; Jansen, J.A. DNA-Coatings: Bioactive Properties and Effects on Osteoblast-Like Cells. Key Eng. Mater. 2008, 361–363, 605–608. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identifier | Prec1 | Ini1 | Brush1 | Prec2 | Ini2 | Brush2 | Prec3 | Ini3 | Brush3 |
|---|---|---|---|---|---|---|---|---|---|
| Contact angle (°) | 20 ± 10 | 75 ± 5 | 7 ± 5 | 20 ± 10 | 7 5± 5 | 7 ± 5 | 15 ± 5 | 50 ± 5 | 7 ± 5 |
| Thickness by AFM (nm) | 78 ± 6 | 92 ± 10 | 30–300 1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mai, T.; Wolski, K.; Puciul-Malinowska, A.; Kopyshev, A.; Gräf, R.; Bruns, M.; Zapotoczny, S.; Taubert, A. Anionic Polymer Brushes for Biomimetic Calcium Phosphate Mineralization—A Surface with Application Potential in Biomaterials. Polymers 2018, 10, 1165. https://0-doi-org.brum.beds.ac.uk/10.3390/polym10101165
Mai T, Wolski K, Puciul-Malinowska A, Kopyshev A, Gräf R, Bruns M, Zapotoczny S, Taubert A. Anionic Polymer Brushes for Biomimetic Calcium Phosphate Mineralization—A Surface with Application Potential in Biomaterials. Polymers. 2018; 10(10):1165. https://0-doi-org.brum.beds.ac.uk/10.3390/polym10101165
Chicago/Turabian StyleMai, Tobias, Karol Wolski, Agnieszka Puciul-Malinowska, Alexey Kopyshev, Ralph Gräf, Michael Bruns, Szczepan Zapotoczny, and Andreas Taubert. 2018. "Anionic Polymer Brushes for Biomimetic Calcium Phosphate Mineralization—A Surface with Application Potential in Biomaterials" Polymers 10, no. 10: 1165. https://0-doi-org.brum.beds.ac.uk/10.3390/polym10101165