Synthesis and Characterization of Isosorbide-Based Polyurethanes Exhibiting Low Cytotoxicity Towards HaCaT Human Skin Cells


 , , ,
, , ,  ,
,  and
and
Abstract
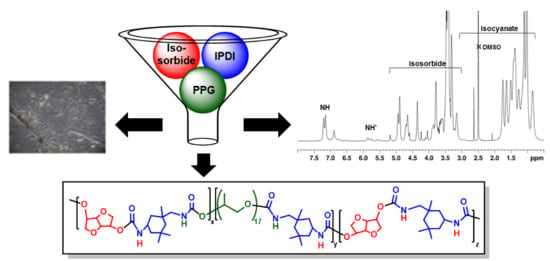
:
1. Introduction
2. Experimental Section
2.1. Materials
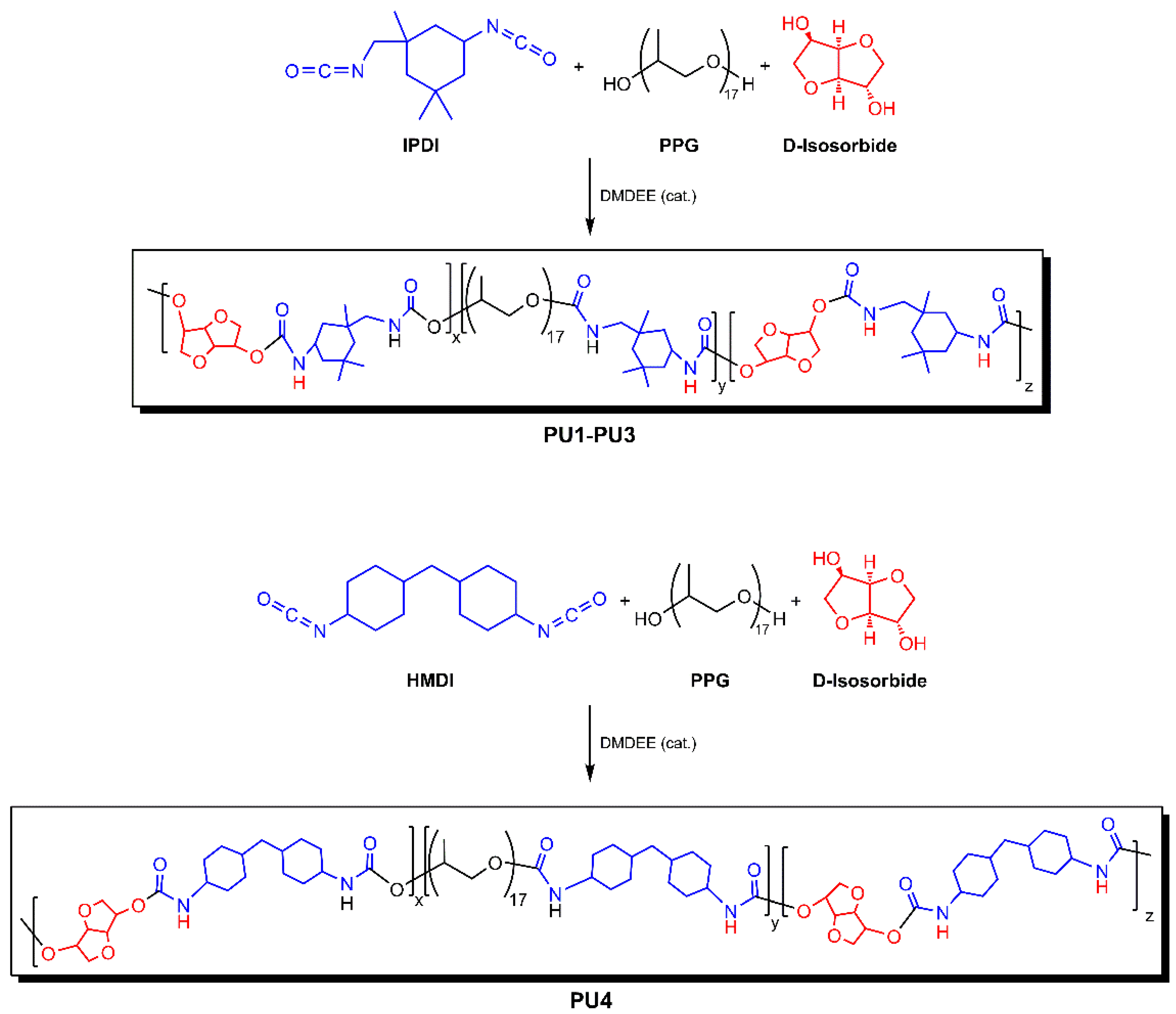
2.2. Polymer Synthesis
2.2.1. Synthesis of Quasi-Prepolymers
2.2.2. Syntheses of Polyurethanes PU1–PU4
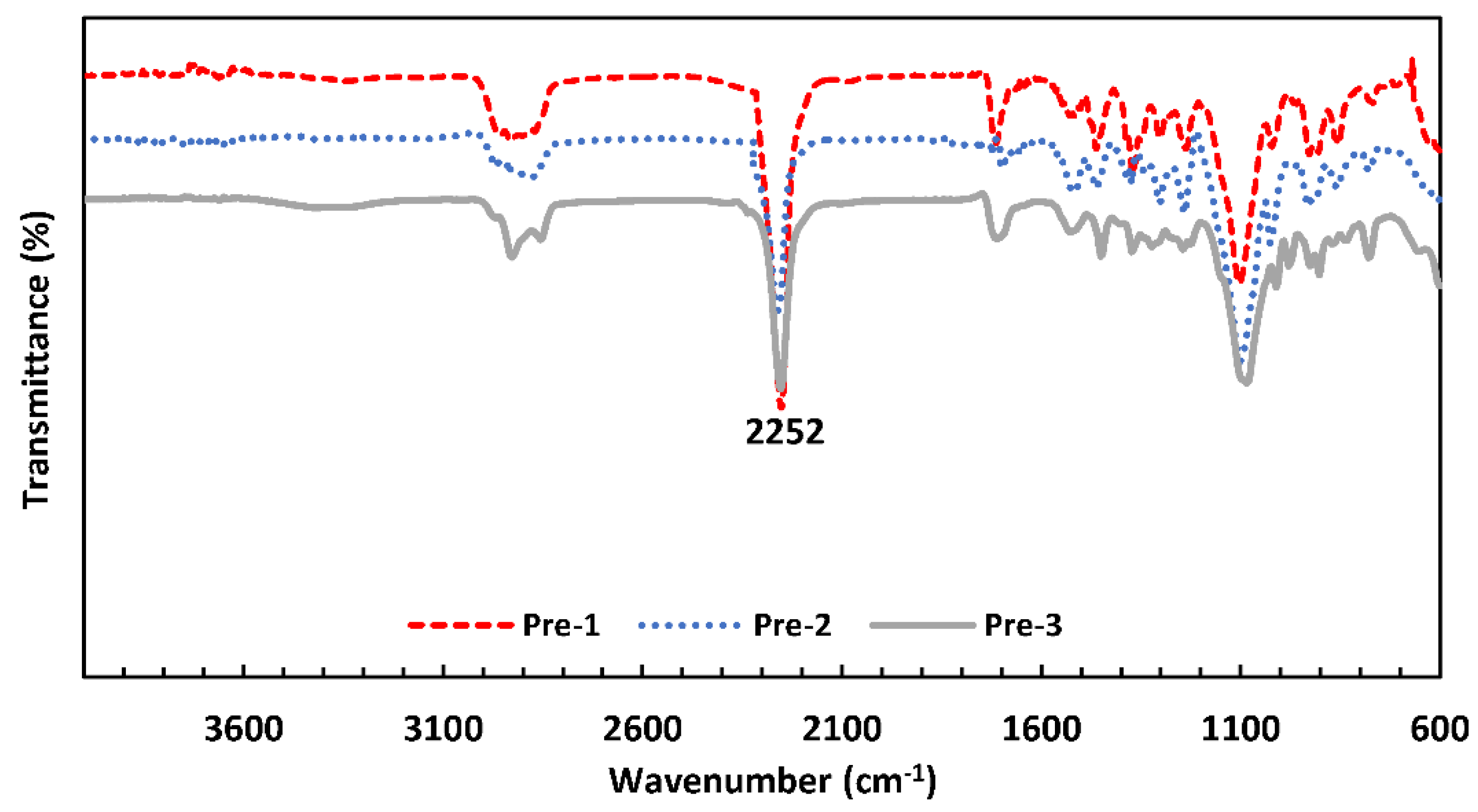
2.3. FTIR Characterization
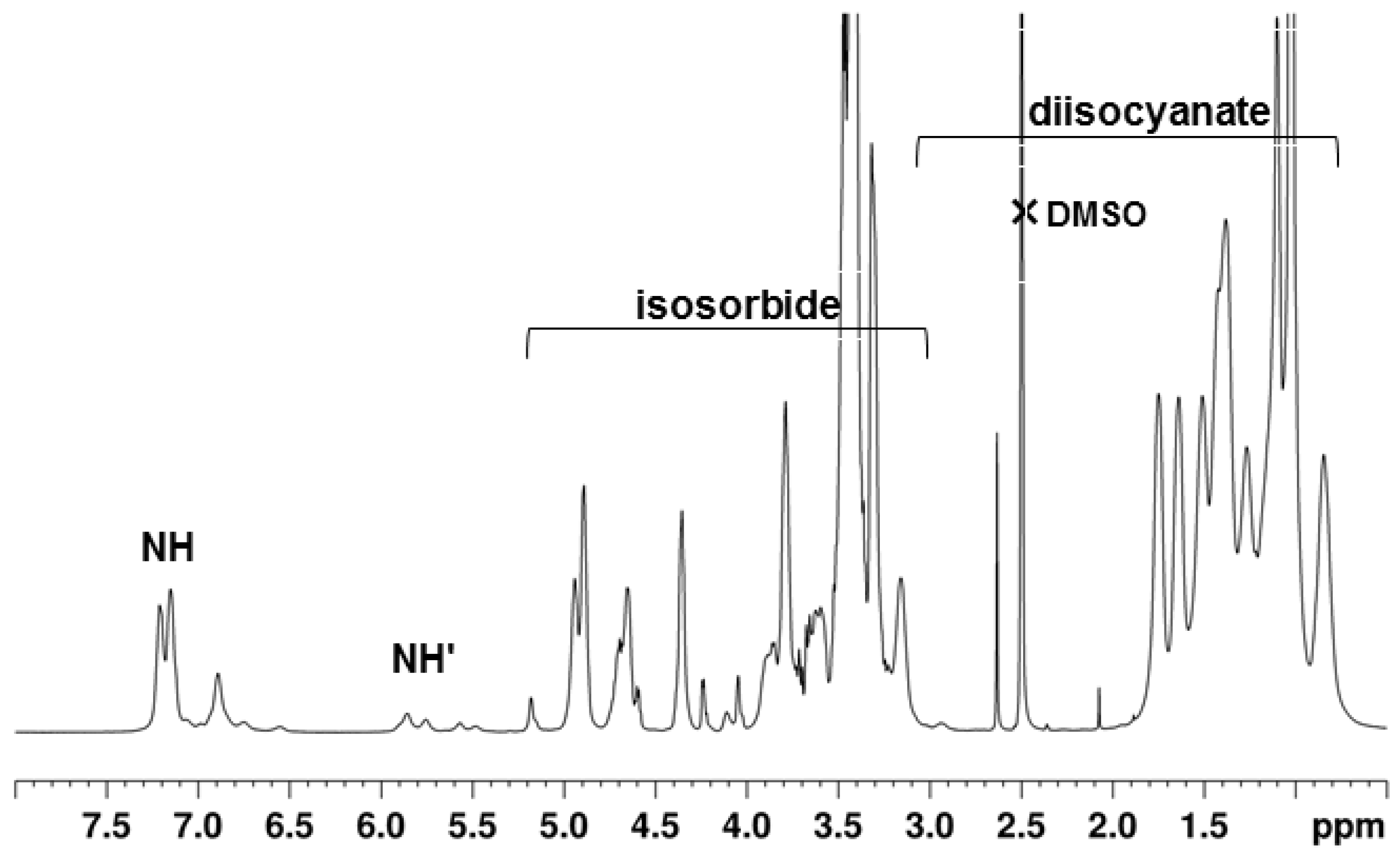
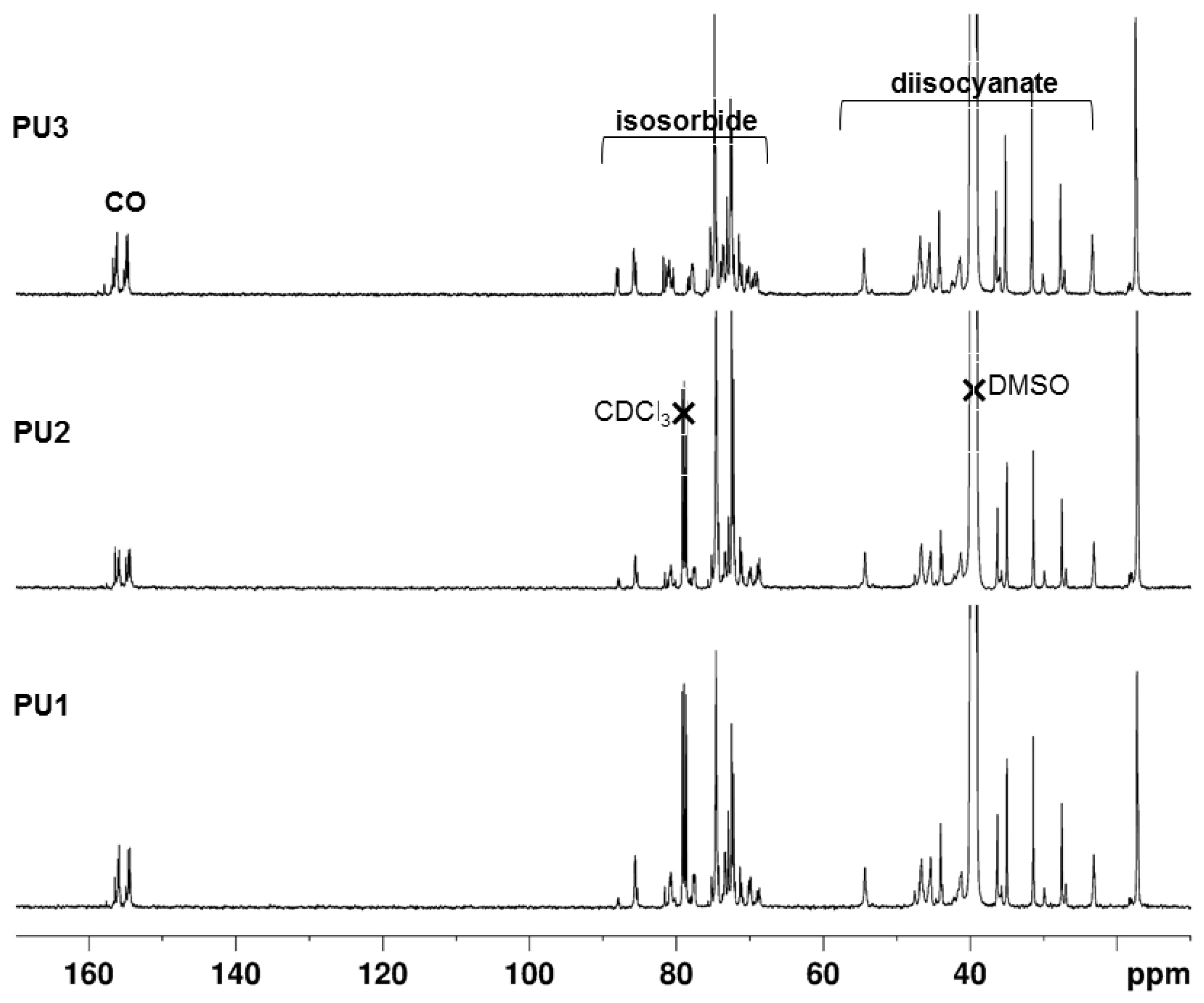
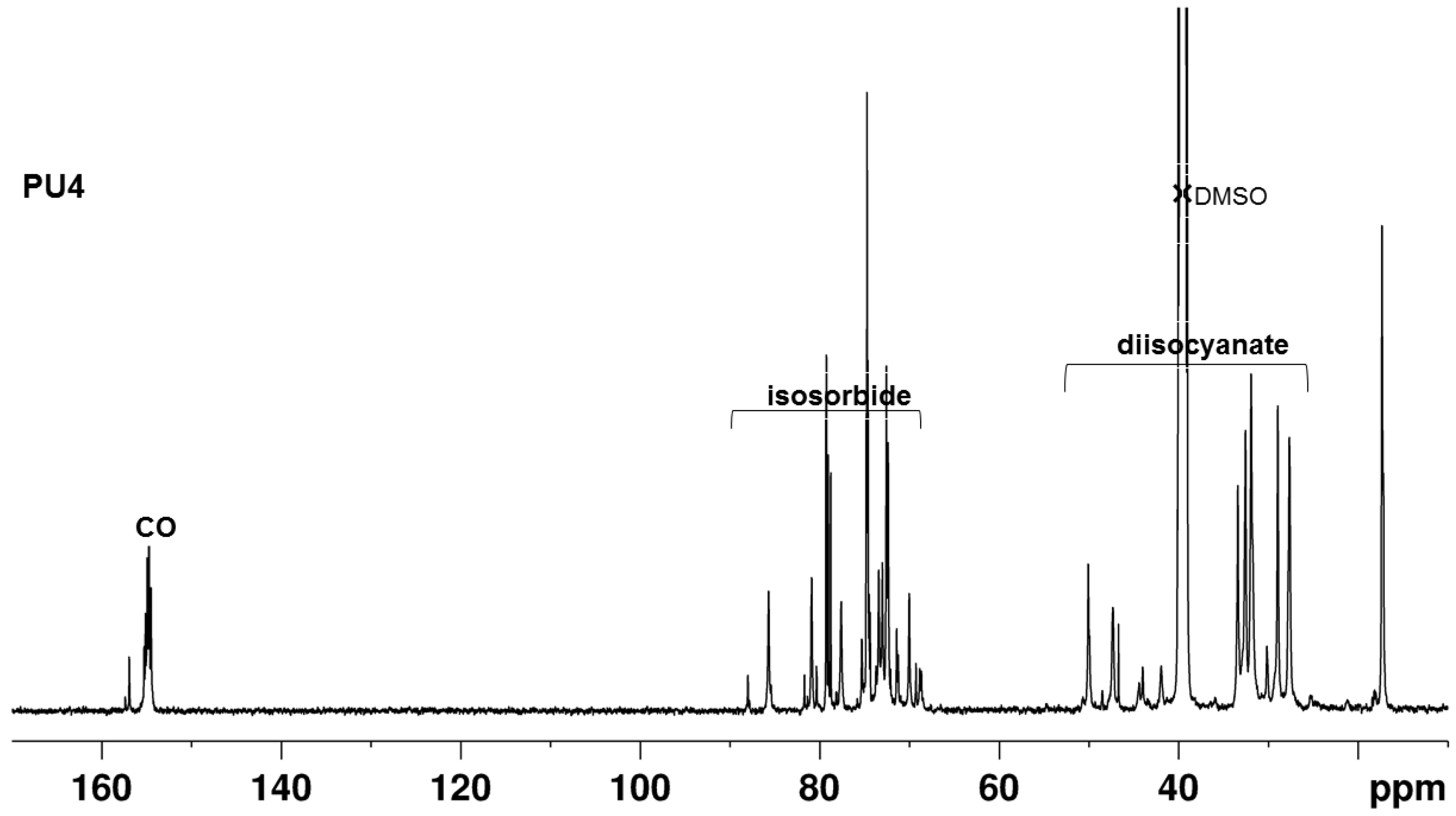
2.4. NMR Spectroscopy Characterization
2.5. Measurement of the Diffusion Coefficients by NMR Spectroscopy
2.6. Gel Permeation Chromatography/Size-Exclusion Chromatography (GPC/SEC) Characterization
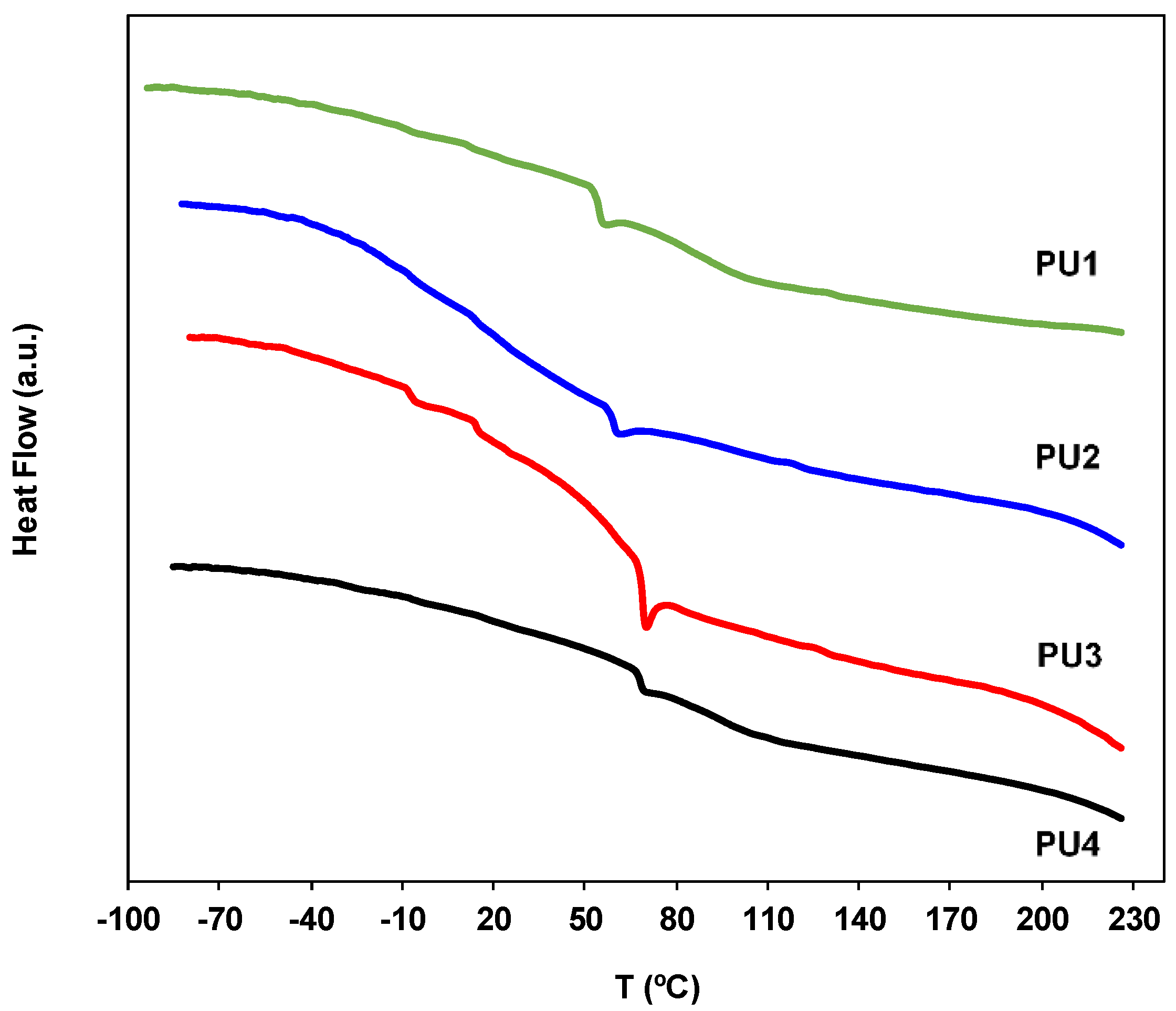
2.7. Thermal Behavior Characterization by Differential Scanning Calorimetry (DSC)
2.8. In Vitro Cytotoxicity Assay
3. Results and Discussion
3.1. Syntheses of the PUs
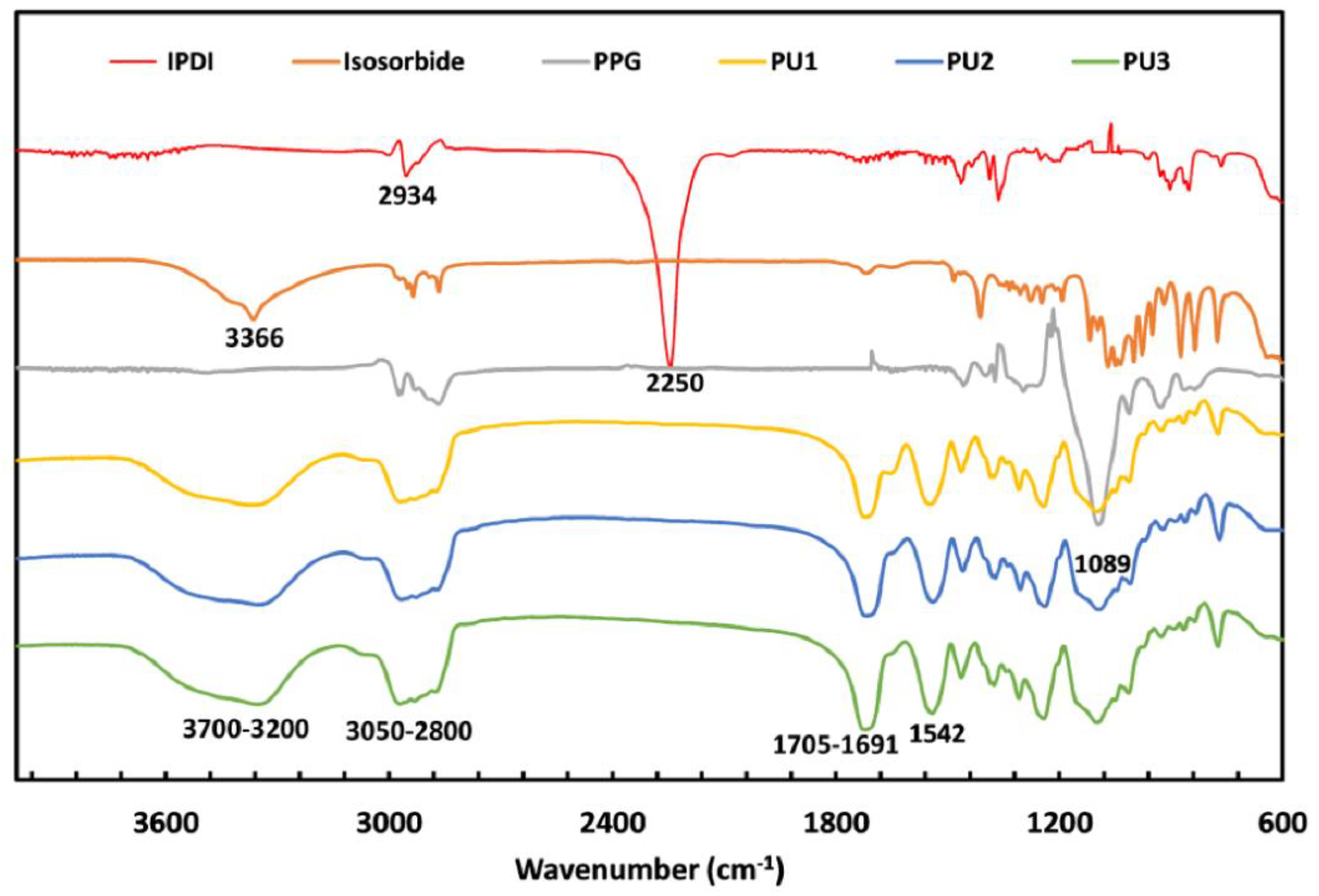
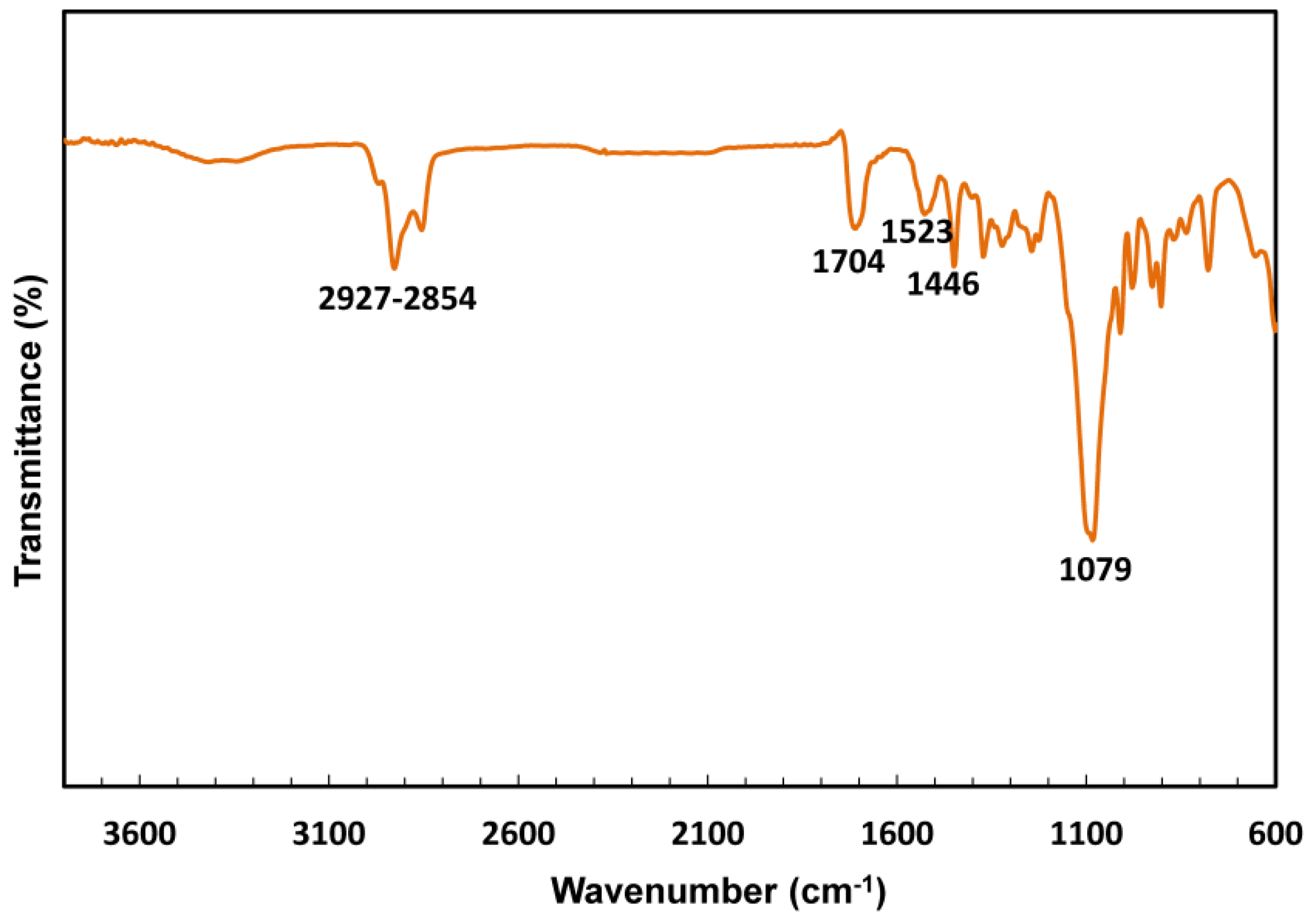
3.2. FTIR of the PUs
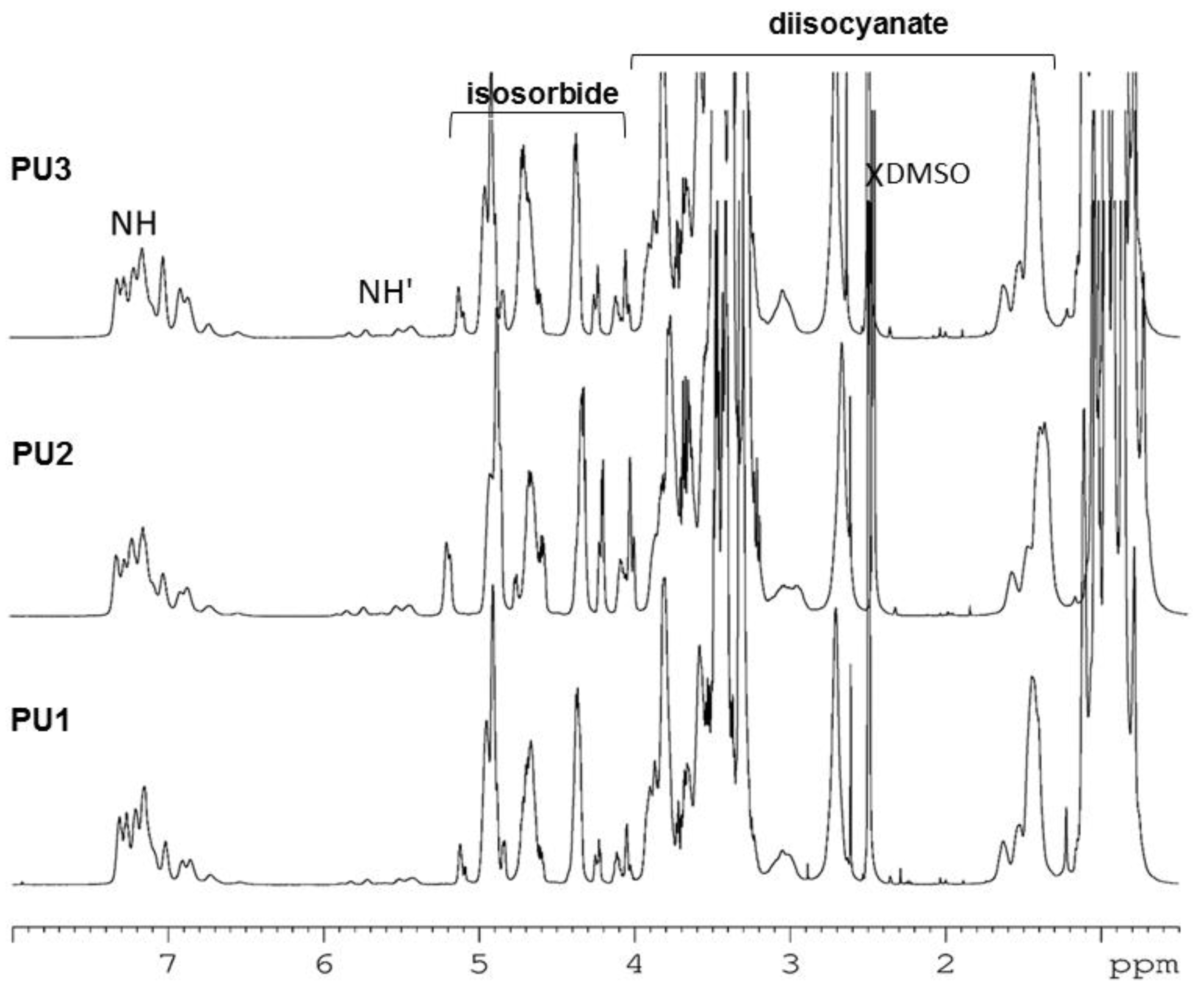
3.3. NMR of the PUs
3.4. Determination of Hydrodynamic Radii of the PUs
3.5. Determination of the Molecular Weights of the PUs
3.5.1. Determination by GPC/SEC
3.5.2. Determination by 13C NMR Spectroscopy
3.6. Thermal Properties of the PUs
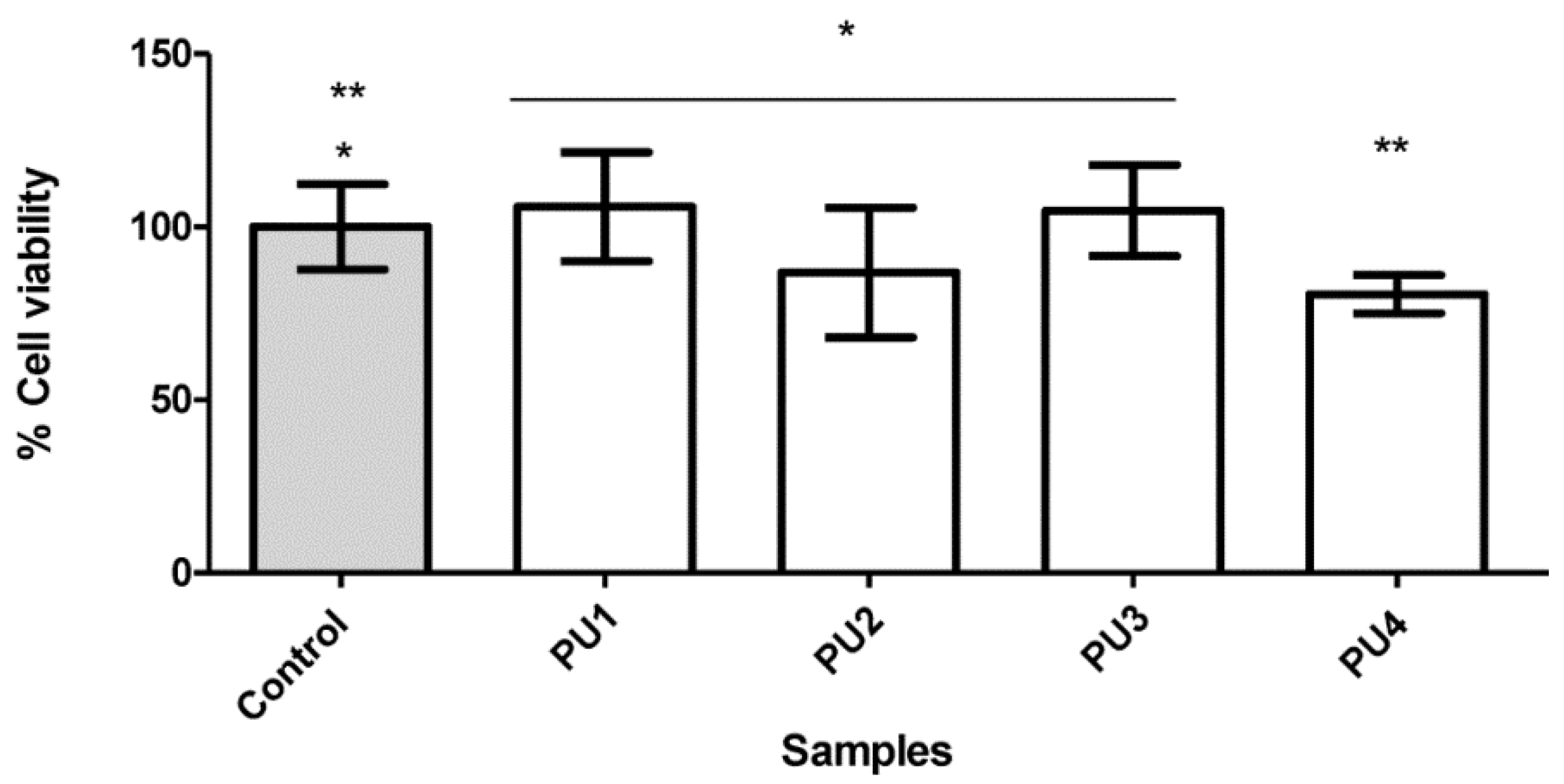
3.7. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sarkar, D.; Yang, J.C.; Sen Gupta, A.; Lopina, S.T. Synthesis and characterization of L-tyrosine based polyurethanes for biomaterial applications. J. Biomed. Mater. Res. Part A 2009, 90, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chen, B.; Ye, L.; Zhang, A.Y.; Zhang, J.; Feng, Z.G. Synthesis and characterization of biodegradable polyurethane based on poly caprolactone and L-lysine ethyl ester diisocyanate. Front. Mater. Sci. China 2009, 3, 25–32. [Google Scholar] [CrossRef]
- Besse, V.; Auvergne, R.; Carlotti, S.; Boutevin, G.; Otazaghine, B.; Caillol, S.; Pascault, J.P.; Boutevin, B. Synthesis of isosorbide based polyurethanes: An isocyanate free method. React. Funct. Polym. 2013, 73, 588–594. [Google Scholar] [CrossRef]
- Chen, T.K.; Tien, Y.I.; Wei, K.H. Synthesis and characterization of novel segmented polyurethane clay nanocomposite via poly(epsilon-caprolactone)/clay. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 2225–2233. [Google Scholar] [CrossRef]
- Bachmann, F.; Reimer, J.; Ruppenstein, M.; Thiem, J. Synthesis of novel polyurethanes and polyureas by polyaddition reactions of dianhydrohexitol configurated diisocyanates. Macromol. Chem. Phys. 2001, 202, 3410–3419. [Google Scholar] [CrossRef]
- Guelcher, S.A.; Gallagher, K.M.; Didier, J.E.; Klinedinst, D.B.; Doctor, J.S.; Goldstein, A.S.; Wilkes, G.L.; Beckman, E.J.; Hollinger, J.O. Synthesis of biocompatible segmented polyurethanes from aliphatic diisocyanates and diurea diol chain extenders. Acta Biomater. 2005, 1, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-J.; Kang, M.-S.; Knowles, J.C.; Gong, M.S. Synthesis of highly elastic biocompatible polyurethanes based on bio-based isosorbide and poly(tetramethylene glycol) and their properties. J. Biomater. Appl. 2014, 29, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, G.A.; Marcos-Fernández, A.; San Román, J. Bioresorbable poly(ester-ether urethane)s from L-lysine diisocyanate and triblock copolymers with different hydrophilic character. J. Biomed. Mater. Res. Part A 2006, 76, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, N.; Wen, X. Synthesis and characterization of biocompatible, degradable, light-curable, polyurethane-based elastic hydrogels. J. Biomed. Mater. Res. Part A 2007, 82A, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Spiridon, I.; Popa, V.I. Hemicelluloses: Major Sources, Properties and Applications. In Monomers, Polymers and Composites from Renewable Resources; Ch. 13, Belgacem, M.N., Gandini, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 289–304. [Google Scholar]
- Gregorí Valdes, B.S.; Serro, A.P.; Gordo, P.M.; Silva, A.; Gonçalves, L.; Salgado, A.; Marto, J.; Baltazar, D.; dos Santos, R.G.; Bordado, J.M.; et al. New Polyurethane Nail Lacquers for the Delivery of Terbinafine: Formulation and Antifungal Activity Evaluation. J. Pharm. Sci. 2017, 106, 1570–1577. [Google Scholar] [CrossRef] [PubMed]
- Gogolewski, S.; Gorna, K.; Zaczynska, E.; Czarny, A. Structure-property relations and cytotoxicity of isosorbide-based biodegradable polyurethane scaffolds for tissue repair and regeneration. J. Biomed. Mater. Res. Part A 2008, 85, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Fenouillot, F.; Rousseau, A.; Colomines, G.; Saint-Loup, R.; Pascault, J.P. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): A review. Prog. Polym. Sci. 2010, 35, 578–622. [Google Scholar] [CrossRef]
- Stoss, P.; Hemmer, R. 1,4/3,6-Dianhydrohexitols. Adv. Carbohydr. Chem. Biochem. 1991, 49, 93–173. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.F.F.; Gordo, P.M.; De Lima, A.P.; Queiroz, D.P.; De Pinho, M.N.; Major, P.; Kajcsos, Z. Free-volume studies in polycaprolactone/poly(propylene oxide) urethane/urea membranes by positron lifetime spectroscopy. Acta Phys. Pol. A 2008, 113, 1359–1364. [Google Scholar] [CrossRef]
- Chattopadhyay, D.K.; Raju, N.P.; Vairamani, M.; Raju, K.V.S.N. Structural investigations of polypropylene glycol (PPG) and isophorone diisocyanate (IPDI) based polyurethane prepolymer by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF)-mass spectrometry. Prog. Org. Coat. 2008, 62, 117–122. [Google Scholar] [CrossRef]
- Prabhakar, A.; Chattopadhyay, D.K.; Jagadeesh, B.; Raju, K.V.S.N. Structural investigations of polypropylene glycol (PPG) and isophorone diisocyanate (IPDI)-based polyurethane prepolymer by 1D and 2D NMR spectroscopy. J. Polym. Sci. Part A Polym. Chem. 2005, 43, 1196–1209. [Google Scholar] [CrossRef]
- Lee, C.-H.; Takagi, H.; Okamoto, H.; Kato, M.; Usuki, A. Synthesis, Characterization, and Properties of Polyurethanes Containing 1,4:3,6-Dianhydro-d-sorbitol. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 6025–6031. [Google Scholar] [CrossRef]
- Marín, R.; Alla, A.; Ilarduya, A.M.; Munõz-Guerra, S. Carbohydrate-Based Polyurethanes: A Comparative Study of Polymers Made from Isosorbide and 1,4-Butanediol. J. Appl. Polym. Sci. 2012, 123, 986–994. [Google Scholar] [CrossRef]
- Daniel-da-Silva, A.L.; Bordado, J.C.M.; Martín-Martínez, J.M. Moisture curing kinetics of isocyanate ended urethane quasi-prepolymers monitored by IR spectroscopy and DSC. J. Appl. Polym. Sci. 2008, 107, 700–709. [Google Scholar] [CrossRef]
- Gregorí Valdés, B.S.; Bordado, J.M.; Ribeiro, H.; Bello, A.; Fernández, M.; Fernandes, S.; Vázquez, NA.R. Analysis and Characterization of Quasi-Prepolymers Obtained from Polyethylene Glycol 1500 and 4,4′-Diphenylmethane-DiIsocyanate. Mater. Sci. Eng. Adv. Res. 2015, 1, 10–15. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-Field Gradient Nuclear Magnetic Resonance as a Tool for Studying Translational Diffusion. Part 1. Basic Theory. Concepts Magn. Reson. 1997, 9, 299–336. [Google Scholar] [CrossRef]
- Geil, B. Measurement of translational molecular diffusion using ultrahigh magnetic field gradient NMR. Concepts Magn. Reson. 1998, 10, 299–321. [Google Scholar] [CrossRef]
- Moura Ramos, J.J.; Taveira-Marques, R.; Diogo, H.P. Estimation of the fragility index of indomethacin by DSC using the heating and cooling rate dependency of the glass transition. J. Pharm. Sci. 2004, 93, 1503–1507. [Google Scholar] [CrossRef] [PubMed]
- International Organization for Standardization. Biological Evaluation of Medical Devices Part 5: Tests for In Vitro Cytotoxicity; ISO 10993–5; ISO: Geneva, Switzerland, 2009; Volume 5, pp. 1–52. [Google Scholar]
- Kuan, H.-C.; Chuang, W.-P.; Ma, C.-C.M.; Chiang, C.-L.; Wu, H.-L. Synthesis and characterization of a clay/waterborne polyurethane nanocomposite. J. Mater. Sci. 2005, 40, 179–185. [Google Scholar] [CrossRef]
- Ferreira, P.; Pereira, R.; Coelho, J.F.J.; Silva, A.F.M.; Gil, M.H. Modification of the biopolymer castor oil with free isocyanate groups to be applied as bioadhesive. Int. J. Biol. Macromol. 2007, 40, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, P.; Silva, A.F.M.; Pinto, M.I.; Gil, M.H. Development of a biodegradable bioadhesive containing urethane groups. J. Mater. Sci. Mater. Med. 2008, 19, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Li, P.; Qiu, H.; Li, D.; Su, M.; Xu, K. Synthesis, characterizations and biocompatibility of alternating block polyurethanes based on P3/4HB and PPG-PEG-PPG. J. Biomed. Mater. Res. Part A. 2011, 98A, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Alves, P.; Coelho, J.F.J.; Haack, J.; Rota, A.; Bruinink, A.; Gil, M.H. Surface modification and characterization of thermoplastic polyurethane. Eur. Polym. J. 2009, 45, 1412–1419. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Hasneen, A.; Chung, I.; Kim, H.; Lee, W.-K.; Chun, J.H. Synthesis and properties of polyurethane coatings: The effect of different types of soft segments and their ratios. Compos. Interfaces 2013, 20, 15–26. [Google Scholar] [CrossRef]
- Caracciolo, P.C.; Buffa, F.; Abraham, G.A. Effect of the hard segment chemistry and structure on the thermal and mechanical properties of novel biomedical segmented poly(esterurethanes). J. Mater. Sci. Mater. Med. 2009, 20, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Kricheldorf, H.R.; Chatti, S.; Schwarz, G.; Krüger, R.P. Macrocycles 27: Cyclic Aliphatic Polyesters of Isosorbide. J. Polym. Sci. Part A Polym. Chem. 2003, 41, 3414–3424. [Google Scholar] [CrossRef]
- Ferreira, P.; Coelho, J.F.J.; Pereira, R.; Silva, A.F.M.; Gil, M.H. Synthesis and characterization of a poly(ethylene glycol) prepolymer to be applied as a bioadhesive. J. Appl. Polym. Sci. 2007, 105, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Vieira, A.P.; Ferreira, P.; Coelho, J.F.; Gil, M.H. Photocrosslinkable Polymers for Biomedical Applications. Int. J. Biol. Macromol. 2008, 43, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.; Coelho, J.F.; Gil, M.H. Developmet of a new photocrosslinkable biodegradable bioadhesive. Int. J. Pharm. 2008, 352, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Daniel da Silva, A.L.; Martín-Martinez, J.M.; Bordado, J.C.M. Influence of the storage of reactive urethane quasi-prepolymers in their compositions and adhesion properties. Int. J. Adhes. Adhes. 2007, 28, 29–37. [Google Scholar] [CrossRef]
- Zhu, R.; Wang, Y.; Zhang, Z.; Ma, D.; Wang, X. Synthesis of polycarbonate urethane elastomers and effects of the chemical structures on their thermal, mechanical and biocompatibility properties. Heliyon 2016, 2, e00125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cheng, L.; Hu, J. NMR studies of water-borne polyurethanes. J. Appl. Polym. Sci. 2003, 90, 257–260. [Google Scholar] [CrossRef]
- Rahman, M.M.; Hasneen, A.; Jo, N.J.; Kim, H.I.; Lee, W.K. Properties of Waterborne Polyurethane Adhesives with Aliphatic and Aromatic Diisocyanates. J. Adhes. Sci. Technol. 2011, 25, 2051–2062. [Google Scholar] [CrossRef]
- Götz, H.; Beginn, U.; Bartelink, C.F.; Grünbauer, H.J.M.; Möller, M. Preparation of isophorone diisocyanate terminated star polyethers. Macromol. Mater. Eng. 2002, 287, 223–230. [Google Scholar] [CrossRef]
- Wang, H.; Kao, H.; Digar, M.; Wen, T. FTIR and Solid State 13 C NMR Studies on the Interaction of Lithium Cations with Polyether Poly(urethane urea). Macromolecules 2011, 34, 529–537. [Google Scholar] [CrossRef]
- Mondiot, F.; Loudet, J.C.; Mondain-Monval, O.; Snabre, P.; Vilquin, A.; Würger, A. Stokes-Einstein diffusion of colloids in nematics. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2012, 86, 010401. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.; Mao, X.A.; Seiferling, D.; Sacco, A. Experimental study of dynamic isotope effects in molecular liquids: Detection of translation-rotation coupling. J. Chem. Phys. 1996, 104, 669–679. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomers | Pre-1 | Pre-2 | Pre-3 |
|---|---|---|---|
| IPDI | 6 | 6 | - |
| HMDI | - | - | 6 |
| PPG | 1 | 2 | 1 |
| Monomers | Polymers | |||
|---|---|---|---|---|
| PU1 n (mol)/m (g) | PU2 n (mol)/m (g) | PU3 n (mol)/m (g) | PU4 n (mol)/m (g) | |
| IPDI | 0.06/13.33 | 0.06/13.33 | 0.06/13.33 | - |
| HMDI | - | - | - | 0.06/15.74 |
| PPG | 0.01/10.00 | 0.02/20.00 | 0.01/10.00 | 0.01/10.00 |
| D-isosorbide | 0.05/7.30 | 0.05/7.30 | 0.06/8.76 | 0.05/7.30 |
| Hard segment content (wt %) a | 67.4 | 50.8 | 68.8 | 69.7 |
| Polyurethane | D0 × 1011 (m2/s−1) | rh (nm) |
|---|---|---|
| PU1 | 7.04 | 1.62 ± 0.04 |
| PU2 | 4.49 | 2.65 ± 0.15 |
| PU3 | 6.74 | 1.69 ± 0.04 |
| PU4 | 4.51 | 2.52 ± 0.19 |
| Polyurethane No. | Mwa (Dalton) | Mna (Dalton) | Ð (Mw/Mn) |
|---|---|---|---|
| PU1 | 15,600 | 10,300 | 1.50 |
| PU2 | 19,200 | 12,800 | 1.48 |
| PU3 | 9200 | 7200 | 1.27 |
| PU4 | 23,500 | 15,100 | 1.55 |
| Polyurethane No. | Number of Isosorbide Units a | Number of IPDI Units a | Number of HDMI Units a | Number of PPG Segments | Mn b (Dalton) |
|---|---|---|---|---|---|
| PU1 | 5 (t) | 6 (t) | - | 1 | 3040 |
| PU2 | 5 (t) | 7 (t,b) | - | 2 | 4270 |
| PU3 | 6 (t) | 6 | - | 1 | 3210 |
| PU4 | 5 (t) | - | 6 (t) | 1 | 3380 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gregorí Valdés, B.S.; Gomes, C.S.B.; Gomes, P.T.; Ascenso, J.R.; Diogo, H.P.; Gonçalves, L.M.; Galhano dos Santos, R.; Ribeiro, H.M.; Bordado, J.C. Synthesis and Characterization of Isosorbide-Based Polyurethanes Exhibiting Low Cytotoxicity Towards HaCaT Human Skin Cells. Polymers 2018, 10, 1170. https://0-doi-org.brum.beds.ac.uk/10.3390/polym10101170
Gregorí Valdés BS, Gomes CSB, Gomes PT, Ascenso JR, Diogo HP, Gonçalves LM, Galhano dos Santos R, Ribeiro HM, Bordado JC. Synthesis and Characterization of Isosorbide-Based Polyurethanes Exhibiting Low Cytotoxicity Towards HaCaT Human Skin Cells. Polymers. 2018; 10(10):1170. https://0-doi-org.brum.beds.ac.uk/10.3390/polym10101170
Chicago/Turabian StyleGregorí Valdés, Barbara S., Clara S. B. Gomes, Pedro T. Gomes, José R. Ascenso, Hermínio P. Diogo, Lídia M. Gonçalves, Rui Galhano dos Santos, Helena M. Ribeiro, and João C. Bordado. 2018. "Synthesis and Characterization of Isosorbide-Based Polyurethanes Exhibiting Low Cytotoxicity Towards HaCaT Human Skin Cells" Polymers 10, no. 10: 1170. https://0-doi-org.brum.beds.ac.uk/10.3390/polym10101170