1. Introduction
Antibiotics are intensively used as human and veterinary medicines for the treatment and prevention of infectious diseases [
1]. Among them, sulfamethoxazole (SMX) is a sulfonamide bacteriostatic antibiotic that has been commonly used during the last 80 years to treat urinary tract infections due to its low cost and broad spectrum of activity to treat bacterial diseases [
2,
3]. However, the widespread and indiscriminate use of SMX, as of other antibiotics, constitutes a huge potential threat to human health and contaminates natural ecosystems by affecting aquatic and soil organisms [
4,
5]. Recently, SMX has been detected in effluents of sewage treatment plants (STP), and also in surface and groundwater [
6,
7]. Indeed, it is known that pharmaceuticals (including SMX) can reach the aquatic environment in their unchanged or transformed forms mainly through discharge of effluents from municipal STP [
7]. According to the statistics, more than 20,000 tons of SMX enter the environment worldwide every year, resulting in concentrations that range from 0.001 to 5.0 µg L
−1 in untreated or treated wastewater [
8,
9,
10]. Therefore, the problem of environmental contamination by SMX is of great concern as pathogen resistance is highly documented and has been induced even by low levels of antibiotics [
11].
To solve the above-mentioned problems, substantial research efforts have been directed worldwide to develop sustainable treatments for the removal of antibiotics, including SMX, from contaminated waters, such as membrane separation, adsorption processes, photocatalysis, and chemical oxidation [
12]. Among these treatments, adsorption-based processes have been highlighted to be efficient, easy to implement and, furthermore, avoid the generation of transformation products [
13,
14,
15]. However, the application of these processes is quite challenging due to the characteristic features of contaminated wastewaters, namely, large discharge flux, complex composition, and very low antibiotic concentrations [
16]. Increasing the adsorbent specificity has been proposed as a strategy to address these challenges and improve the efficiency of the adsorptive removal of antibiotics from such complex matrices [
17].
Molecularly imprinted technology (MIT) involves the creation of tailor-made selective binding sites in a polymeric matrix with memory of the shape, size, and functional groups of the template. Thus, molecularly imprinted polymers (MIPs) have become increasingly attractive as adsorbent materials due to their capacity to selectively bind specific targets and to their promising characteristics, such as low cost, easy synthesis, high stability to harsh chemical and physical conditions, and excellent reusability [
18,
19]. In recent years, MIPs, whose application of the extraction and analysis of organic contaminants in environmental water samples is well-established [
20], have been successfully used for the adsorptive removal of pharmaceuticals, including antibiotics, from contaminated water [
21,
22,
23,
24]. In the specific case of SMX adsorption by MIPs, few works have been published, with most of them aiming at the analytic quantification of this antibiotic. For example, Qin et al. [
5] used Fe
3-O
4-chitosan MIPs for SMX selective extraction and determination in aqueous samples, with the produced materials having attained a maximum adsorption capacity of 4.32 mg g
−1. Zhao et al. [
25] prepared core–shell MIPs on the surface of magnetic carbon nanotubes (MCNTs@MIP) for SMX, the resulting material having a maximum SMX adsorption capacity from aqueous solution of 864.9 µg g
−1. However, to the best of our knowledge, the removal of SMX from complex wastewaters using MIPs has just been assessed by Valtech et al. [
19]. Among the materials produced by these authors [
18], those having the largest maximum adsorption capacity (6.5 × 10
−5 mol g
−1 (16.5 mg g
−1)) performed similarly to a commercial activated carbon in terms of removal, but presented higher selectivity toward SMX in the presence of other pharmaceuticals and better regeneration ability.
Despite the above-mentioned advantages and applications, the preparation of MIPs by conventional MIT has two main drawbacks: (1) The imprinted polymer matrices are thick and, thus, hold a small number of recognition sites per unit volume; and (2) the template molecules are deeply embedded in the matrix, so there is a diffusion barrier for them, the mass transfer rate is low, and binding to the recognition sites is somehow hampered [
26]. Surface molecular imprinting has been proved to improve mass transfer, recognition, and binding ability relative to MIT [
27]. Among solid-support substrates used for the surface molecular imprinting process, microbial nano-magnetic materials are alternative supporters that have many advantages compared to inorganic materials [
28]: (1) They are easy to obtain and short generations can be artificially cultured [
29]; (2) there are many surface chemical functional groups and so modification steps can be avoided, reducing secondary pollution; (3) cells can guide the regulation of the growth process of inorganic materials [
30]; (4) microbial cells have a variety of structures and can provide a rich array of templates for nanomaterials by template-assisted synthesis; and (5) magnetic properties allow for a simple after-use separation of the materials.
Yeasts, which belong to the fungus kingdom, are relatively large eukaryotic and single-celled microorganisms (diameters typically measuring 2.0–4.0 µm). Their cell wall includes glucan, mannan, chitin protein, and a small amount of lipids, and it has many surface chemical groups such as carboxyl (–COOH), carbonyl (–C=O), amino (–NH
2), hydroxyl (–OH), and phosphoryl (–P=O) groups. Moreover, yeast is very cheap, easy to obtain, and environmentally friendly. These advantages make yeasts appropriate and widely used as supports for bio-nanocomposites [
31].
In the above-described context, the objectives of this study were to: (1) Prepare a bio-nanocomposite of yeast-Fe3O4 (magnetic yeast, MY) using an in situ one-step preparation of nano-Fe3O4; (2) use MY as the core to synthesize magnetic yeast-molecularly imprinted polymers (MY@MIPs) by a surface-imprinted polymerization method with MIPs as the shell and SMX as the template molecule; (3) characterize the resulting materials by Fourier-transform infrared spectroscopy (FT-IR), a vibrating sample magnetometer (VSM), X-ray diffraction (XRD), thermogravimetric analysis (TGA), specific surface area (SBET) determination, and scanning electron microscopy (SEM); (4) test the removal performance of MY@MIPs toward SMX and compare it with those of MY and MY@NIPs (magnetic molecularly imprinted polymers without template); and (5) explore the selective sorption capacity of MY@MIP in a real complex matrix (wastewater collected at a STP) and in the presence of other pharmaceuticals (diclofenac and carbamazepine).
2. Materials and Methods
2.1. Chemicals and Materials
Yeast cells (CICC 30225) were obtained from the China Center of Industrial Culture Collection (CICC). Iron salts used to produce MY were ferric chloride hexahydrate (FeCl3·6H2O) and ferrous chloride tetrahydrate (FeCl2·4H2O), purchased from Sigma-Aldrich (Stenheim, Germany). In addition, 2-vinyl pyridine (2-vpy), ethylene glycol dimethacrylate (EGDMA), acetonitrile (ACN), and azo-bis-isobutyronitrile (AIBN), which were also purchased from Sigma-Aldrich (Stenheim, Germany), were used for MIT. Other reagents used in this work included ammonium hydroxide, toluene (99.8%, Aldrich), ethanol (99.9%, Riedel-de Haën), methanol (99.99%, Fischer Chemical), and acetic acid (p.a., Merck). Ultrapure water was obtained from a Milli-Q water purification system (Millipore). SMX was purchased from TCI Europe (98%); carbamazepine (CBZ; Sigma-Aldrich, 99%); diclofenac (DCF, TCI Europe, >98%). All solutions were stored at 4 °C immediately after preparation.
2.2. Materials Preparation
2.2.1. Preparation of Magnetic Yeast (MY)
Nano-Fe
3O
4 was loaded onto the yeast cell surface by a one-step method as described by Tian et al. [
32]. Briefly, the yeast cells were cultured in ultrapure water with glucose. After reaching the exponential growth phase (6–10 h), the yeast cells were collected by centrifugation (4000 rpm). Then, collected cells (1.0 g) were suspended in 40 mL of 0.125 M FeCl
3 solution in a three-necked flask and stirred for 1 h at room temperature. After that, 0.6 g of FeCl
2·4H
2O was added under nitrogen atmosphere and stirred for another 1 h. The mixture was then heated in a water bath at 80 °C for 15 min, and the pH was adjusted to approximately 11 with 25% (
w/v) ammonium hydroxide. Stirring was kept for 30 min and then stopped to age for 1 h. The resulting magnetic yeast (MY) was then washed, separated by applying a magnetic field, and then dried in an oven (35 °C, 4 h).
2.2.2. Preparation of Magnetic Yeast-Based Molecularly Imprinted Polymer (MY@MIPs)
MY was treated as the core and the MIPs as the shell. The process used for the production of MY@MIPs was as follows: 1 mg of SMX (template molecule) and 4 mmol of 2-vpy (monomer) were dissolved in 60 mL of ACN/toluene (3/1; v/v). This solution was then self-polymerized for 8 h at room temperature (25 °C). Subsequently, 100 mg of MY (polymer supporter), 0.36 mmol of AIBN (initiator), and EGDMA (crosslinker) were added into the polymerized solution (template:monomer:crosslinker, 1:4:20), which was ultrasonicated for 10 min. The mixture was heated and maintained at 60 °C for 24 h under stirring with nitrogen protection. At last, the MY@MIPs were washed with methanol/formic acid (9/1; v/v) for 12 h and purified for 24 h by a Soxhlet extraction method (the extraction solution was methanol). Meanwhile, the MY@NIPs were also produced by following the above-described procedure but in the absence of the template.
2.3. Characterization of MY, MY@MIPs, and MY@NIPs
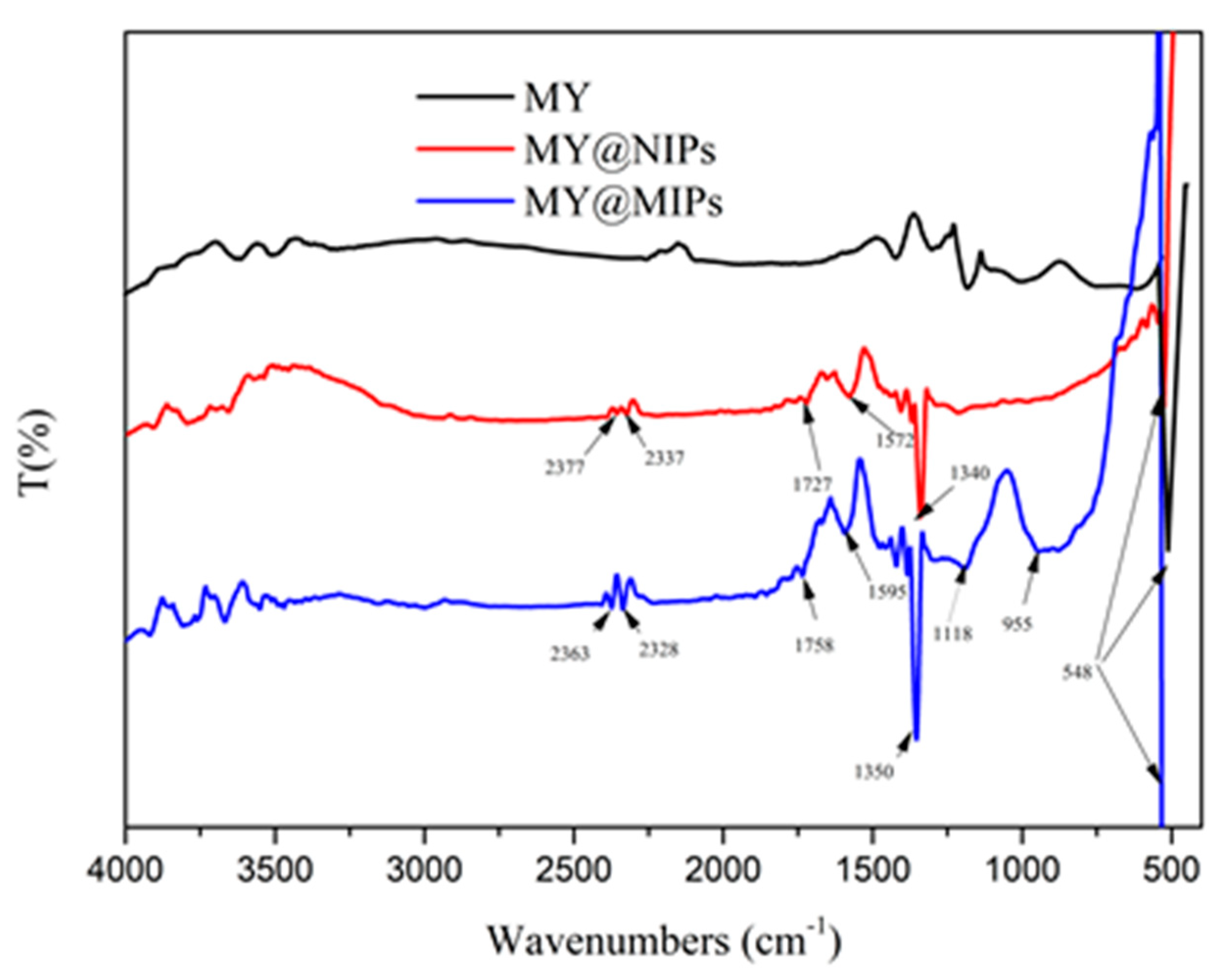
Fourier-transform infrared spectra of the produced materials were obtained in a Shimadzu-IRaffinity-1 equipment, using an ATR module (FTIR-ATR), under a nitrogen purge. The measurements were recorded in the range 500–4000 cm−1, 4.0 of resolution, 256 scans, and applying atmosphere and background correction.
A vibrating sample magnetometer (VSM EV9) with an oscillatory applied magnetic field (H) to a maximum of 22 kOe was used to determine the saturation magnetization (MS). The MS was calculated by plotting the magnetic moment versus the applied magnetic field, and it corresponded to the plateau value of the magnetic moment reached divided by the sample mass (10 mg). The sample was encapsulated in an acrylic cylindrical container (5.85 mm of diameter and 2.60 mm of height), which was coupled to the lineal motor of the VSM EV9 instrument, centered between the two polar heads of the electromagnet used to fluctuate the magnetic field. The instrument was calibrated with a disk of pure nickel (8 mm of diameter) using a procedure that establishes the determination of the magnetic field, applied at around 1 Oe, while the dispersion of the magnetic moment is inferior to 0.5%.
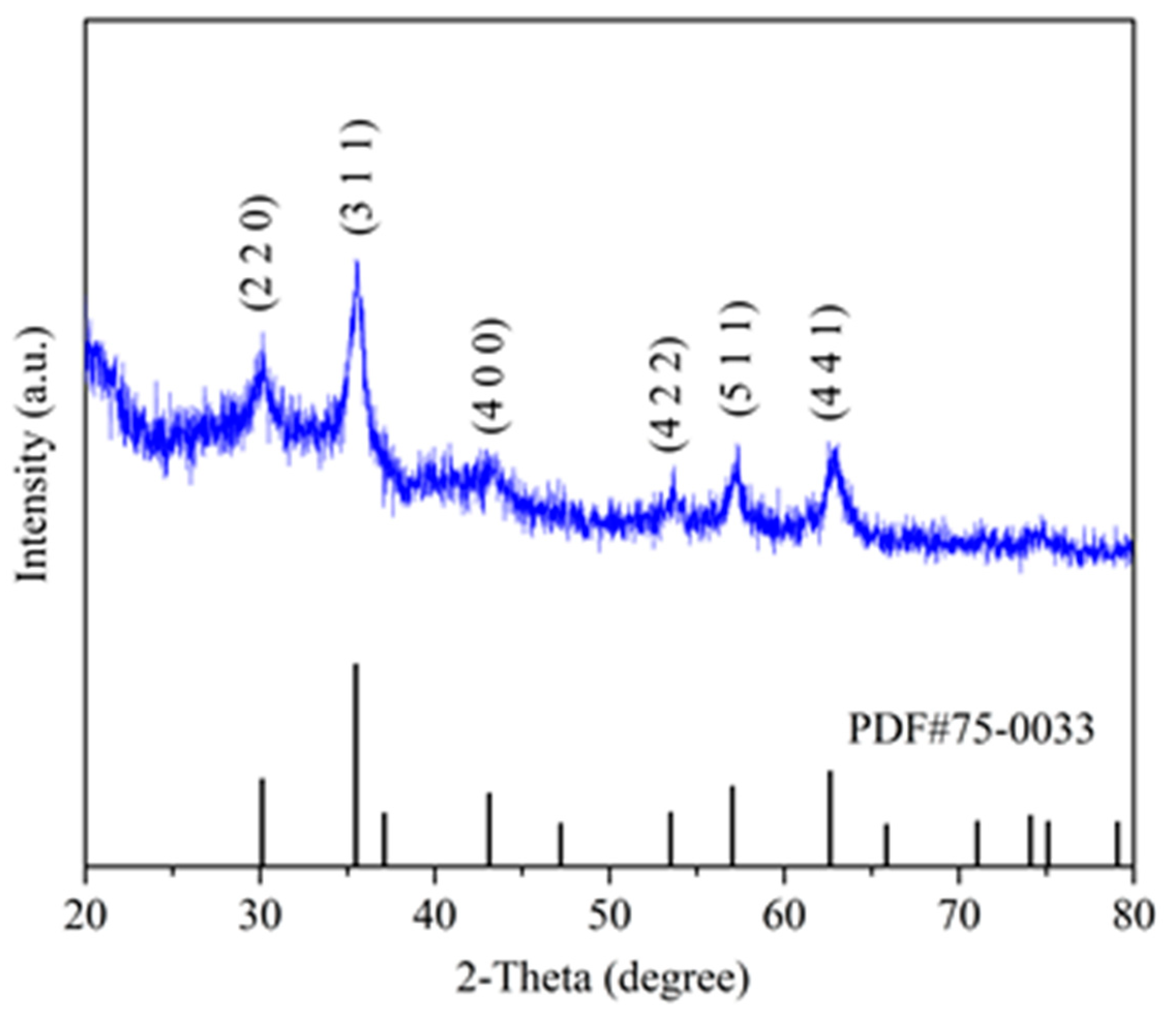
X-ray diffraction (XRD, 5–90°) was measured on a D8-Focus X-ray diffractometer (Bruker Optics) with a test rate of 10°·min-1. The results were analyzed by Jade program (9.0) and Origin (9.0).
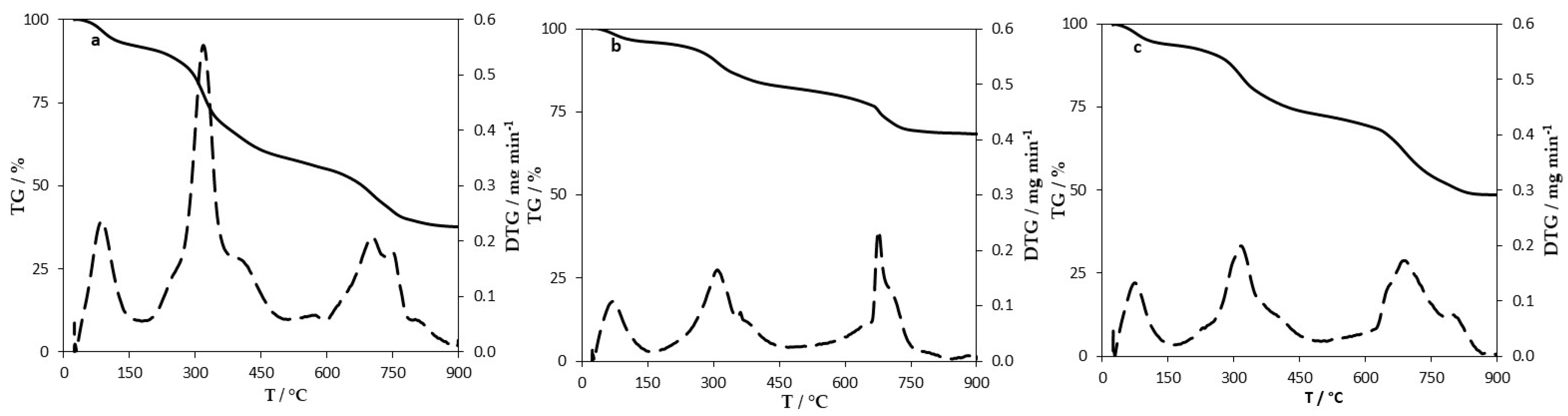
Thermogravimetric analysis (TGA) was performed in a thermogravimetric balance Setsys Evolution 1750, Setaram, TGA mode (S type sensor). The samples were heated at a heating rate of 10 °C min−1, under nitrogen atmosphere, from room temperature to 105 °C and from 105 °C to 900 °C, maintaining constant temperature until total stabilization of the sample mass at the end of both stages (approximately 30 min).
The SBET and micropore volume (W0) were determined by nitrogen adsorption isotherms, acquired at 77 K using a Micromeritics Instrument, Gemini VII 2380, after outgassing the materials overnight at 120 °C. SBET was calculated from the Brunauer–Emmett–Teller equation in the relative pressure range 0.01–0.1. Pore volume (Vp) was estimated from the amount of nitrogen adsorbed at a relative pressure of 0.99.
The surface morphology of the materials was analyzed by scanning electron microscopy (SEM) using a Hitachi S4100. The images were obtained at magnifications of 500, 3000, and 10,000×.
2.4. Adsorptive Removal of SMX by the Produced Materials
The produced materials (MY, MY@MIPs, MY@NIPs) were used as adsorbents for the removal of SMX under batch operation conditions. Summarizing, the materials were put in contact with a 5 mg L
−1 SMX solution in polypropylene tubes, which were shaken in a head-over-head shaker (80 rpm) for a predetermined period of time at controlled temperature (32 °C). The corresponding adsorbent material was separated from the suspension liquid by an external magnetic field. At last, the concentration of SMX in the liquid phase was measured by micellar electrokinetic chromatography (MEKC), using a methodology adapted from Silva et al. (2019) [
33]. The experiments were conducted in triplicate, and control experiments without adsorbent were run in parallel. The performance of the materials was evaluated by carrying out kinetic, equilibrium, pH, selectivity, and regeneration/reutilization studies, described in detail in the next subsections.
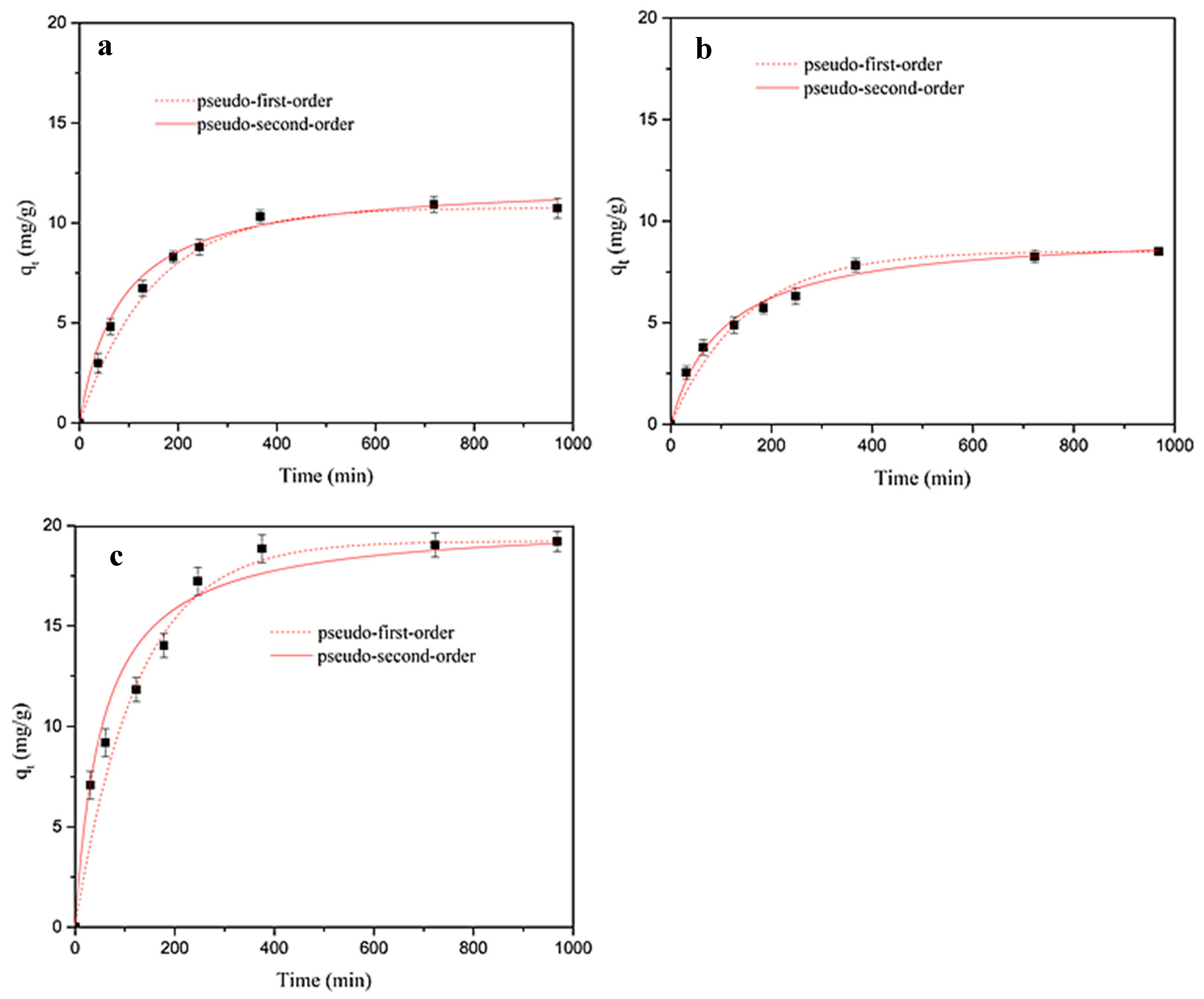
2.4.1. Kinetic Adsorption Studies in Ultrapure Water
In the kinetic study, tubes containing 250 mg of adsorbent material (MY, MY@NIPs, or MY@MIPs), together with 10 mL of a 5 mg L
−1 SMX solution in ultrapure water, were incubated and shaken as described above. After shaking during defined periods of time (
t, min), at intervals from 0 to 24 h, the materials were separated from the aqueous phase and the remaining SMX concentration in solution was measured by MEKC. At each time, the corresponding value of the adsorbed concentration (
qt, mg·g
−1) was determined as follows:
where
Ct (mg L
−1) is the residual SMX concentration at time
t,
C0 is the initial SMX concentration (mg L
−1), and
Cm is the adsorbent dosage (mg·L
−1).
When adsorption equilibrium was attained, the percentage of adsorption R (%) was determined as:
where
Ce (mg·L
−1) is the residual SMX concentration at equilibrium.
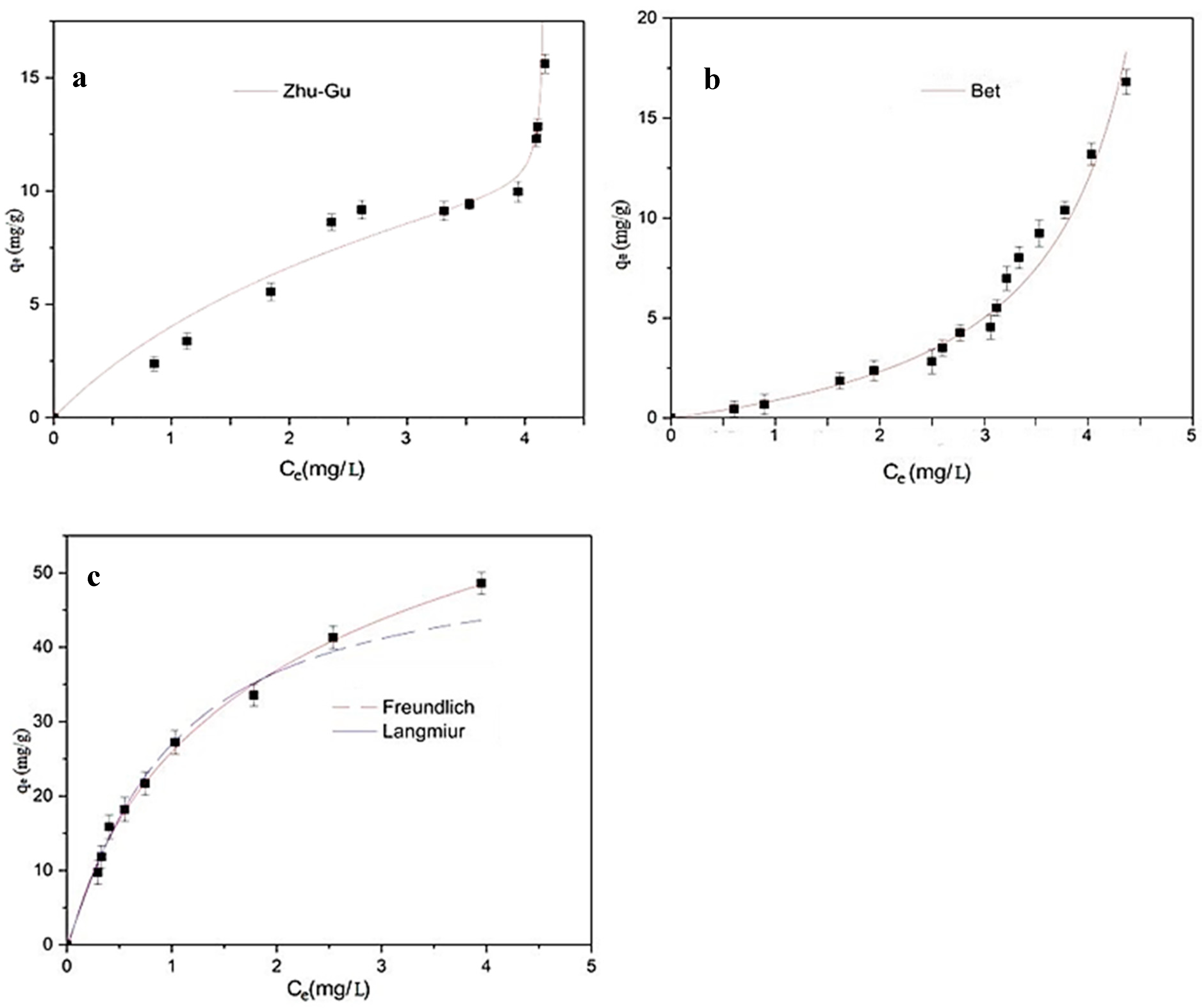
2.4.2. Equilibrium Adsorption Studies in Ultrapure Water
For the equilibrium studies, the corresponding adsorbent material (MY, MY@NIPs, or MY@MIPs), with doses ranging from 50 to 2000 mg L
−1, was added to 10 mL of a 5 mg L
−1 solution of SMX in ultrapure water. Tubes with the mixtures were shaken for 16 h, which allowed equilibrium to be reached. The materials were recovered from the suspension by the application of a magnetic field and the residual concentration of SMX was determined by MEKC. Then, for the different doses of material, the adsorbed concentration at the equilibrium (
qe, mg·g
−1) was determined as follows:
where
Ce (mg L
−1) is the SMX concentration in the liquid phase at equilibrium.
2.5. Adsorptive Performance of MY@MIPs
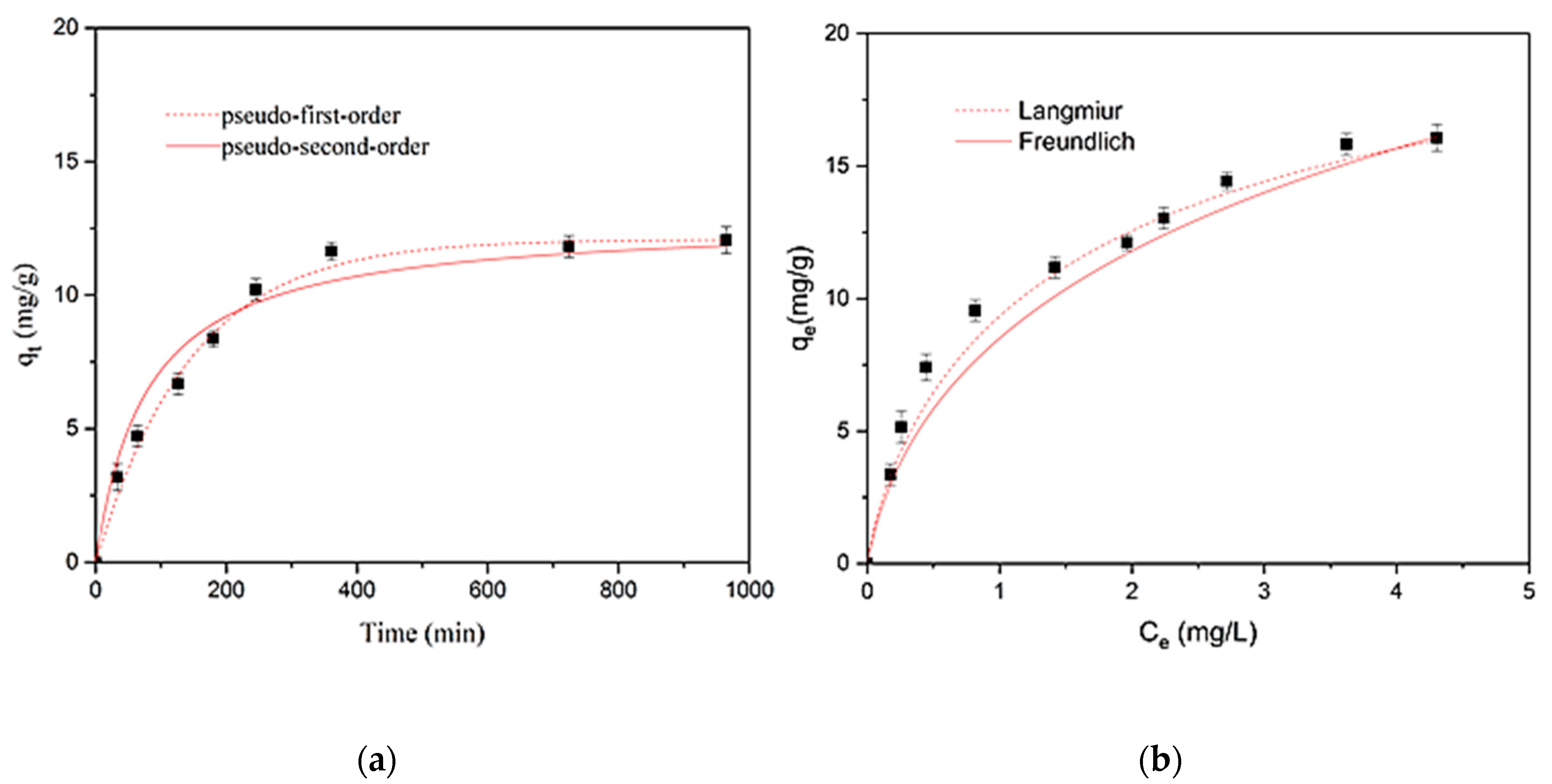
From the results of the above-mentioned kinetic and equilibrium studies in ultra-pure water, the most efficient material for removal of SMX was MY@MIPs. Thus, in order to assess the practical application of this material, further studies were carried out on the adsorptive performance of MY@MIPs under different experimental conditions.
2.5.1. Kinetic and Equilibrium Adsorption Studies in STP Effluent
The kinetic and equilibrium procedures described in
Section 2.4. were carried out using MY@MIPs for the adsorptive removal of SMX from a real matrix, namely the effluent from a STP. In this case, 5 mg L
−1 solutions of SMX were prepared using a STP effluent instead of ultrapure water. The effluent was collected from an urban STP in Aveiro (Portugal) that is designed to serve 159,700 population equivalents. This STP consists of primary and biological treatment stages. For this work, water was collected at the outlet of the biological decanter, as this is the final treated effluent that is discharged from the STP into the aquatic environment. Immediately after collection, the effluent was filtered through 0.45 μm, 293 mm Supor
® membrane disk filters (Gelman Sciences) and stored at 4 °C until use, which occurred within a maximum of 15 days. The collected effluent had a pH of 7.99, conductivity of 3.03 mS cm
−1, and total organic carbon content of 21.5 mg L
−1.
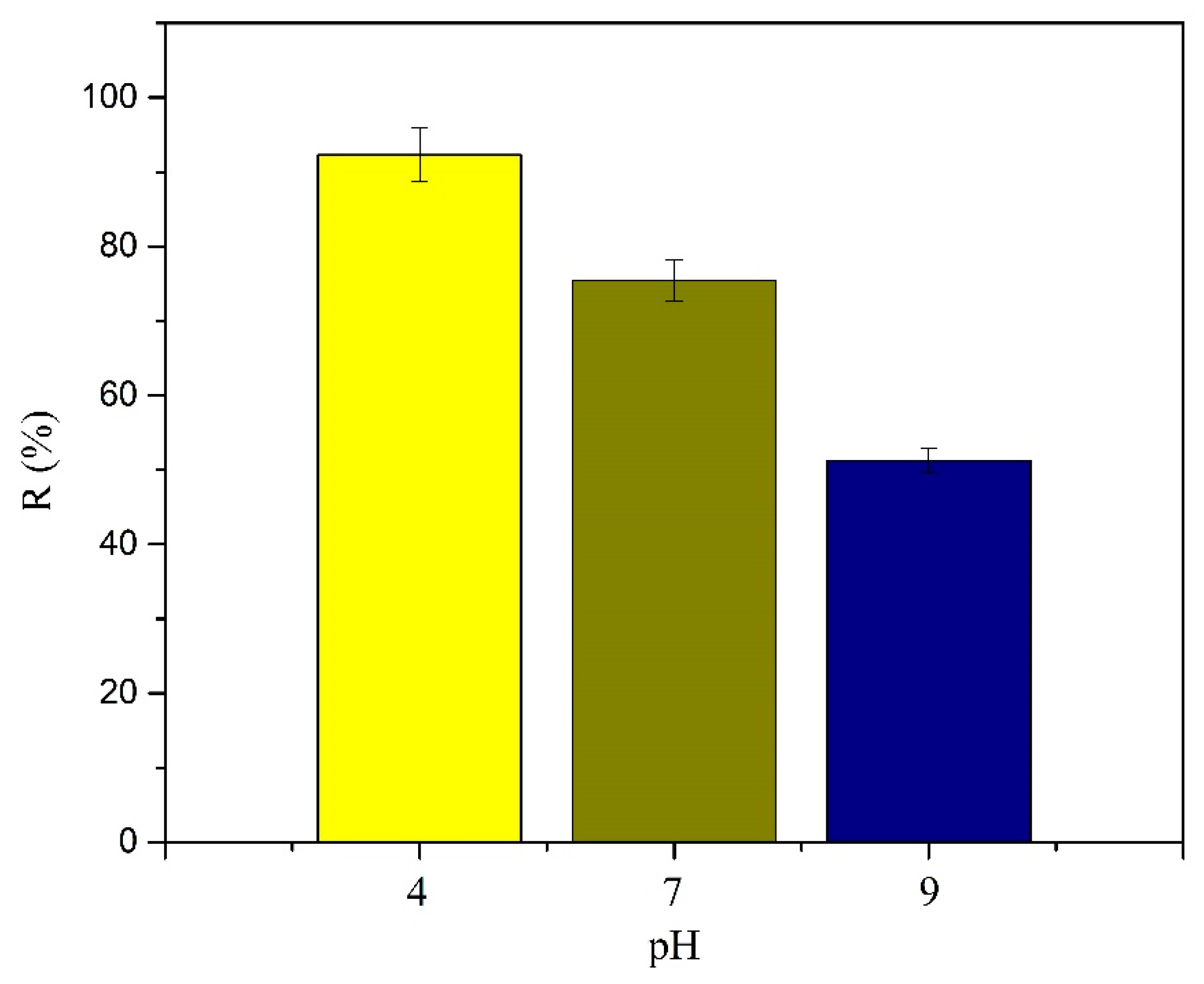
2.5.2. pH Study
Adsorption studies on the effect of pH were carried out at 32 °C with the initial conditions of C0 = 5 mg L−1 in ultrapure water and Cm = 300 mg L−1. Experiments were carried out at three different pHs, namely 4, 7, and 8 (pH was adjusted by adding HCl or NaOH, 1 M). After shaking during 16 h, MY@MIPs were separated from the liquid suspensions, the residual concentration of SMX was analyzed by MEKC, and the corresponding qe (mg g−1) at each pH was determined using Equation (3).
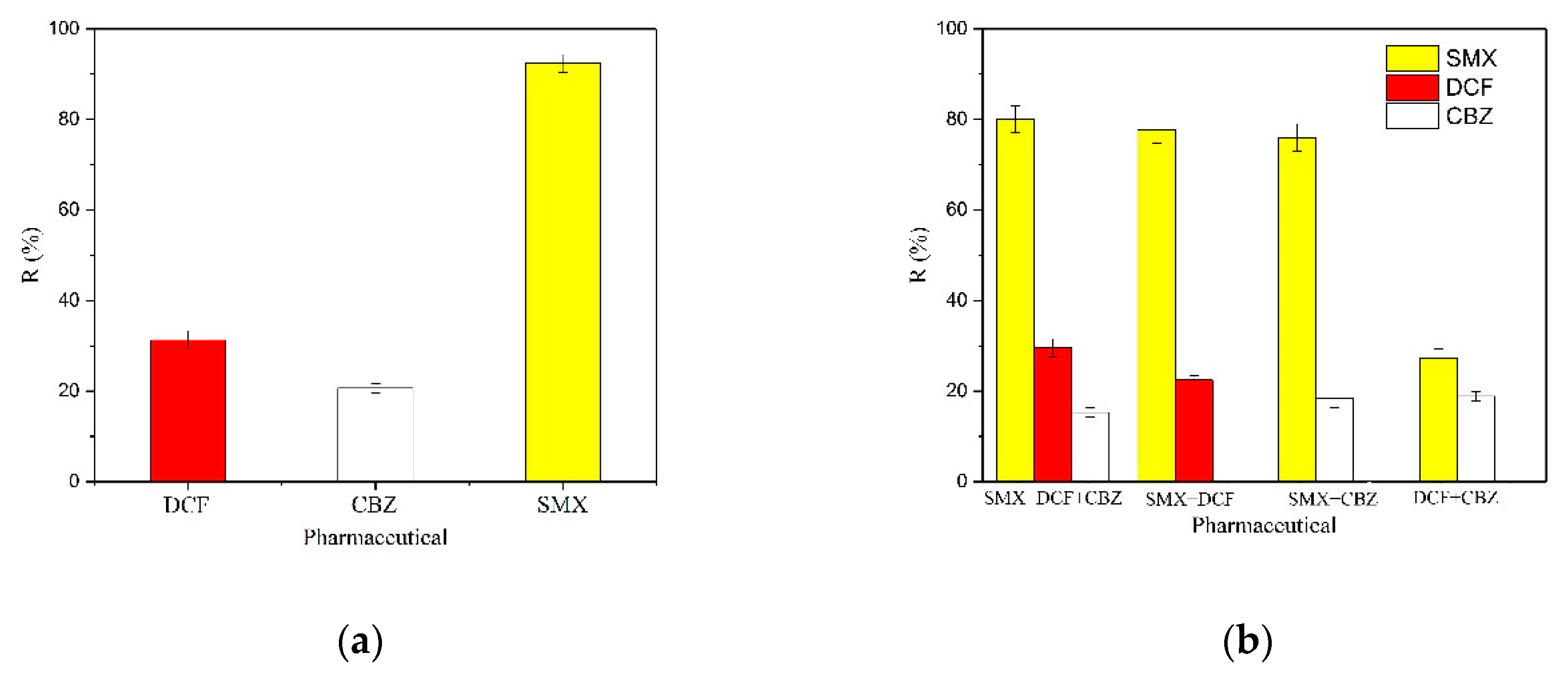
2.5.3. Selective Adsorption
To study the selective capacity of MY@MIPs toward SMX, diclofenac (DCF) and carbamazepine (CBZ) were used as competing species in the adsorption experiments. These pharmaceuticals were selected due to their high global frequency of occurrence in wastewater, surface water, and groundwater and their recalcitrant properties, with low removal rates after conventional STP treatments [
34]. The concentration of DCF and CBZ in ultrapure water solution was the same as that of SMX (5 mg L
−1), the
Cm was 300 mg L
−1, the incubation temperature was 32 °C, the pH was 4, and shaking was maintained during 16 h. Then, the residual concentration of SMX at equilibrium was analyzed and the corresponding
qe (mg g
−1) was determined with Equation (3).
2.5.4. Regeneration and Reutilization
In order to evaluate the adsorptive performance after regeneration, after SMX saturation in ultrapure water, MY@MIPs were regenerated and then tested for the adsorption of SMX in four subsequent cycles. For the regeneration, saturated MY@MIPs were washed by methanol/acetic acid (9/1, v/v) through Soxhlet extraction during 72 h. Then, the regenerated material was used in adsorption experiments as described in previous sections (shaking during 16 h at 32 °C with the initial conditions of C0 = 5 mg L−1 in ultrapure water and Cm = 300 mg L−1). The residual concentration of SMX at the equilibrium was analyzed and the corresponding R (%) was determined as for Equation (2).


 and
and 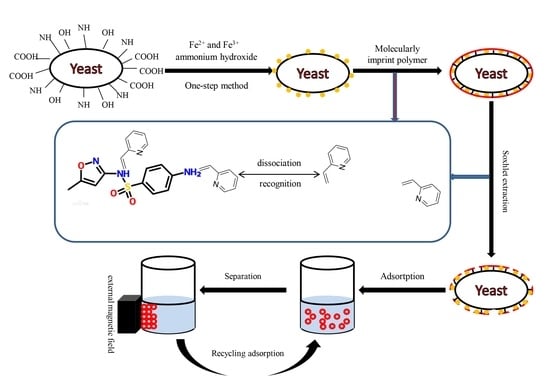











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}