Comparative Nanofabrication of PLGA-Chitosan-PEG Systems Employing Microfluidics and Emulsification Solvent Evaporation Techniques


Abstract
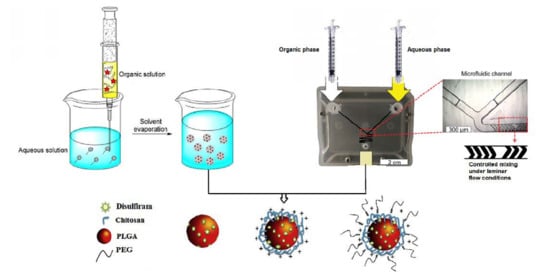
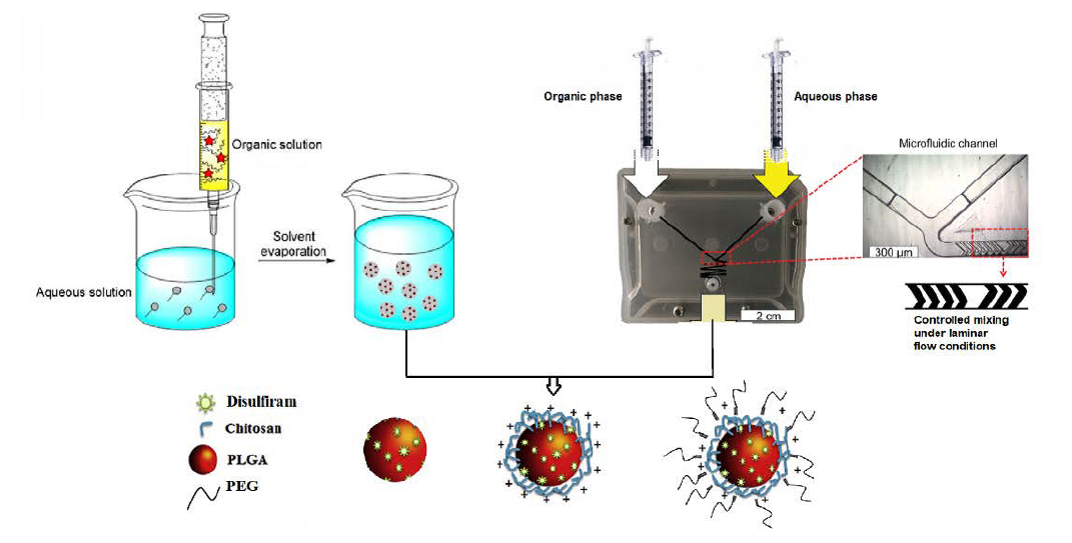
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of Nanoparticles by the Emulsification Solvent Evaporation Approach
2.3. Preparation of Nanoparticles Using the Microfluidics Approach
2.4. Determination of Particle Size and Zeta Potential
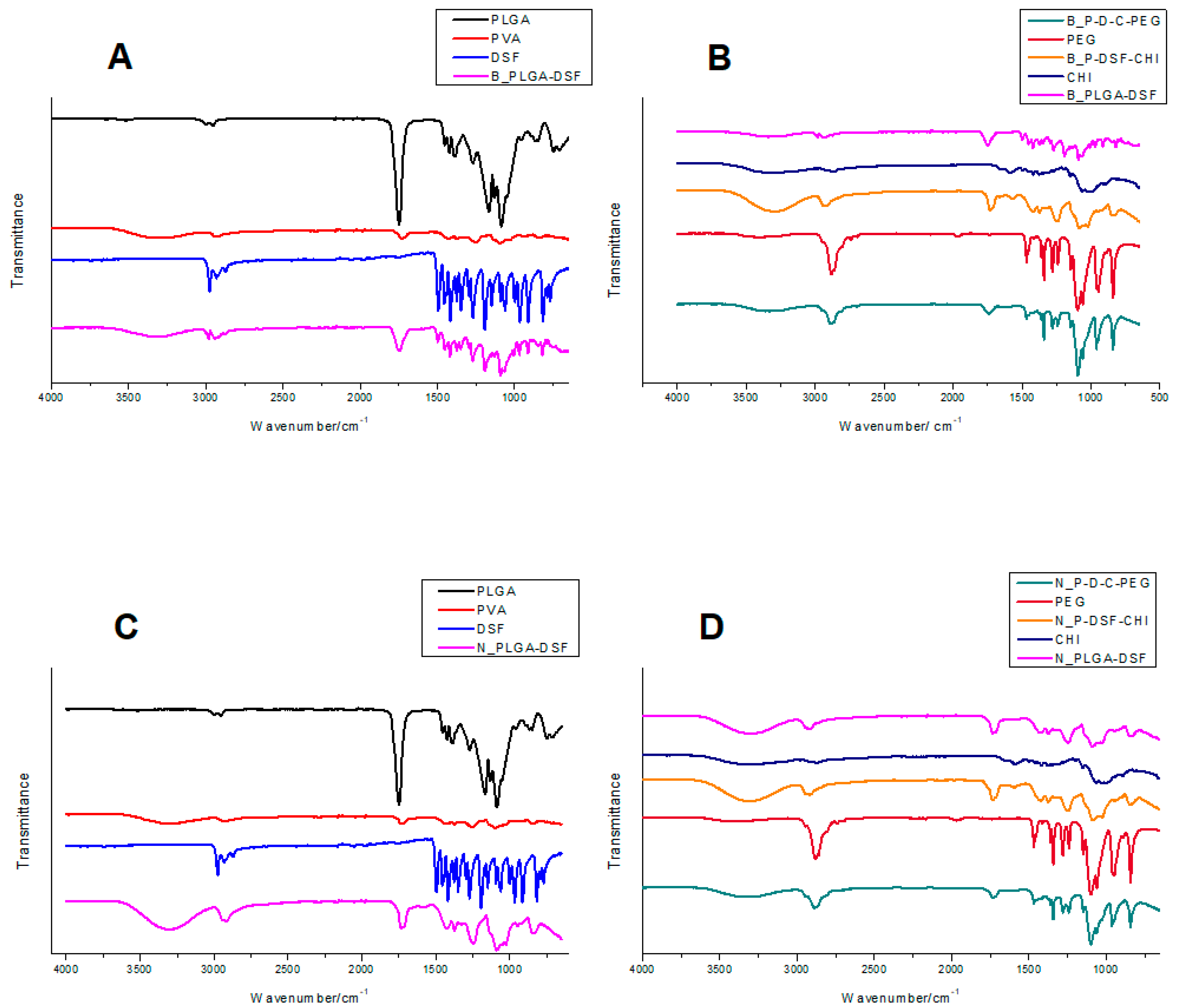
2.5. Determination of the Chemical and Structural Integrity of the Nano-Systems
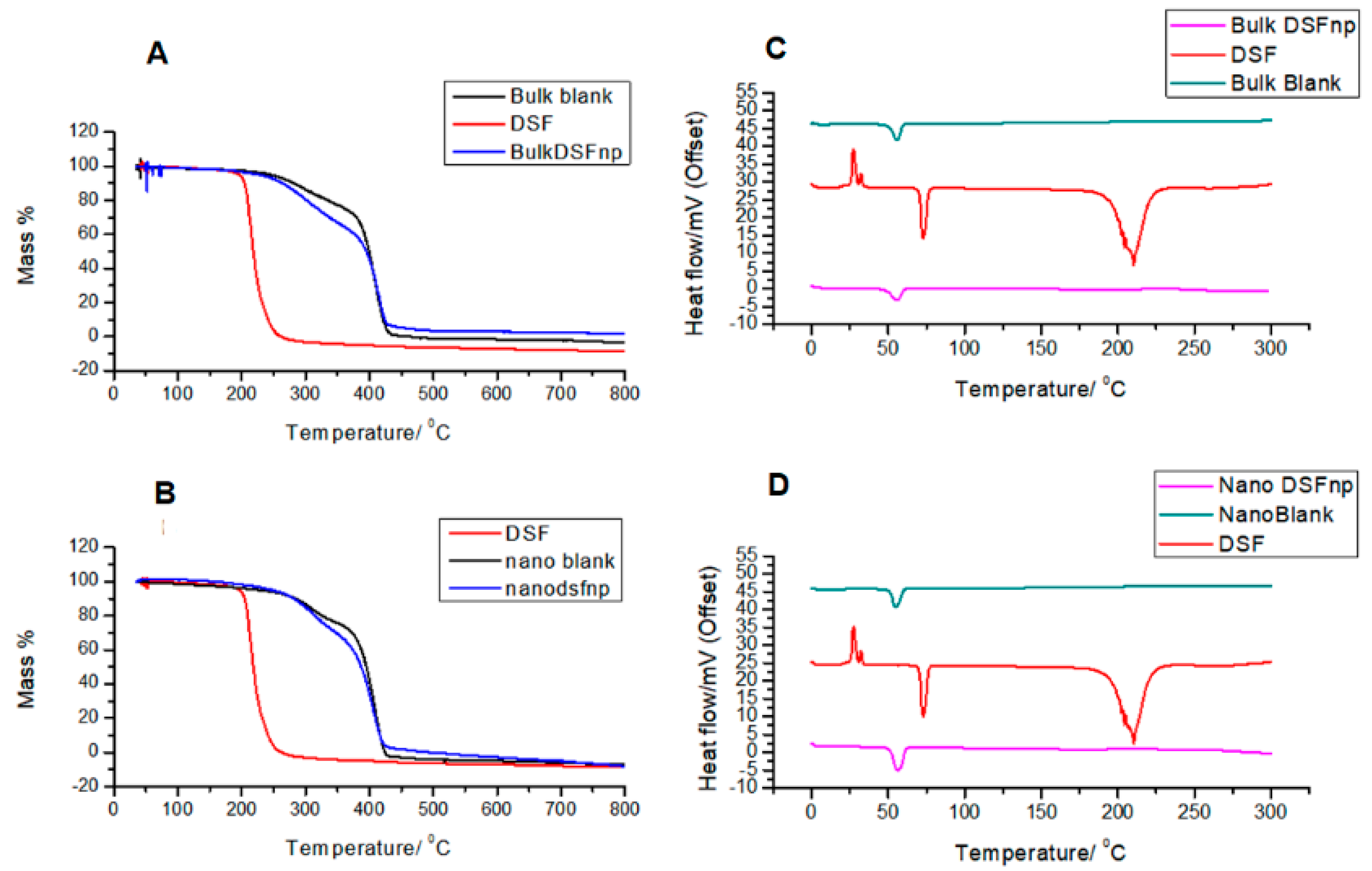
2.6. Investigation of the Thermal Properties of the Nano-Systems
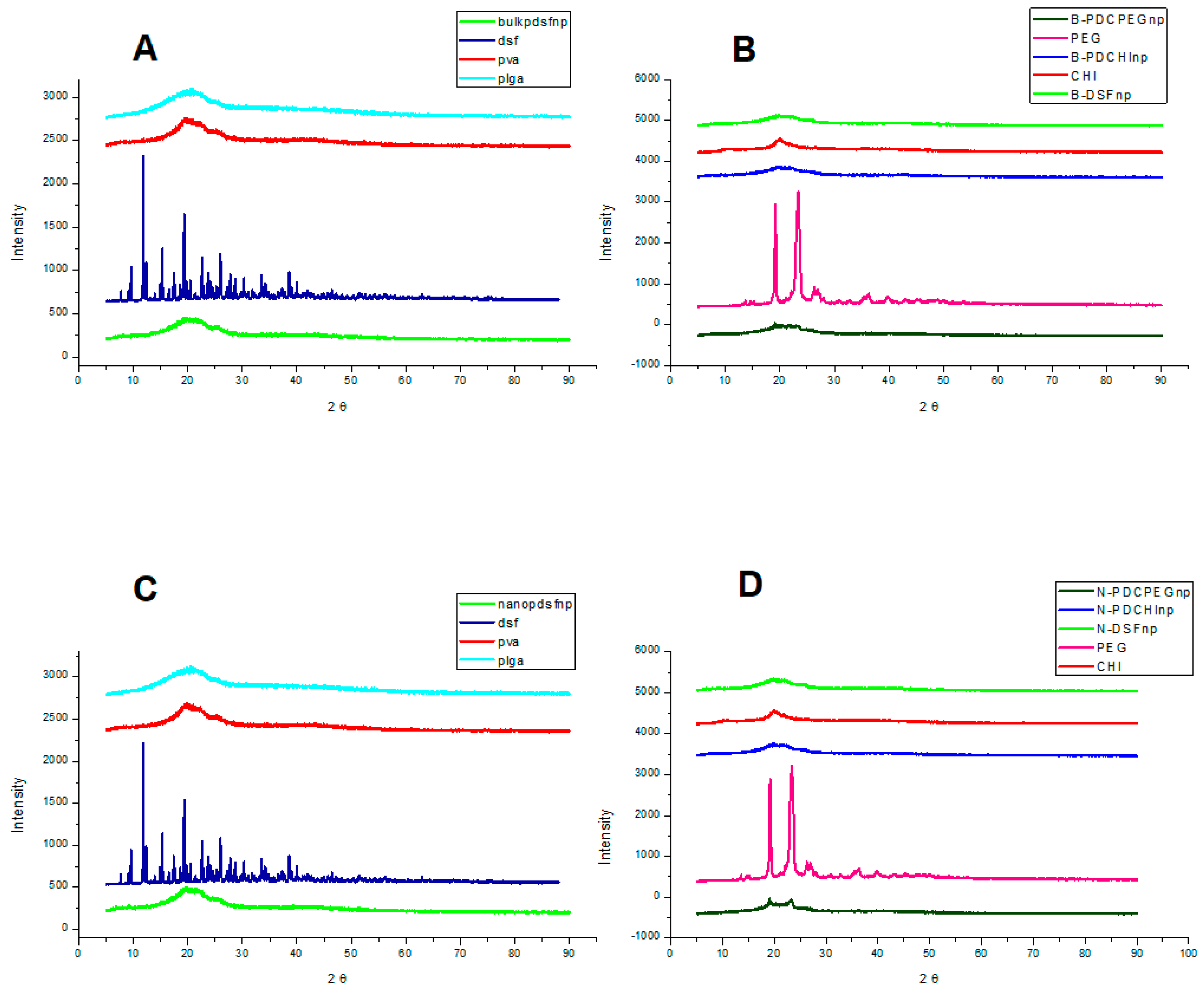
2.7. Determination of the Crystallinity of the Nano-Systems
2.8. Investigation of Surface Morphology Using Scanning Electron Microscopy (SEM)
2.9. UV Spectrophotometric Analysis to Determine Entrapment Efficiency of Disulfiram
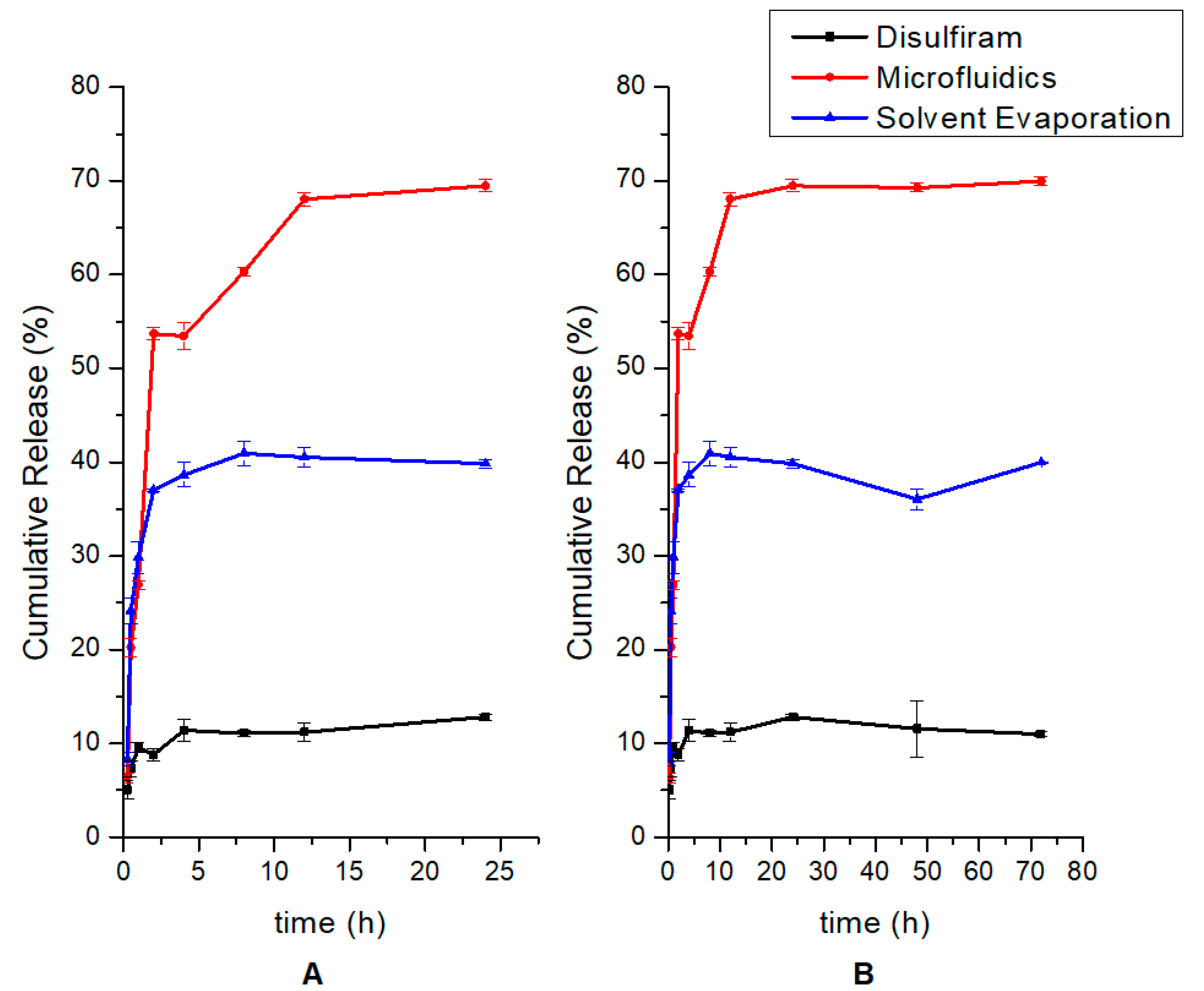
2.10. In Vitro Disulfiram Release Studies
2.11. Statistical Analysis
3. Results and Discussion
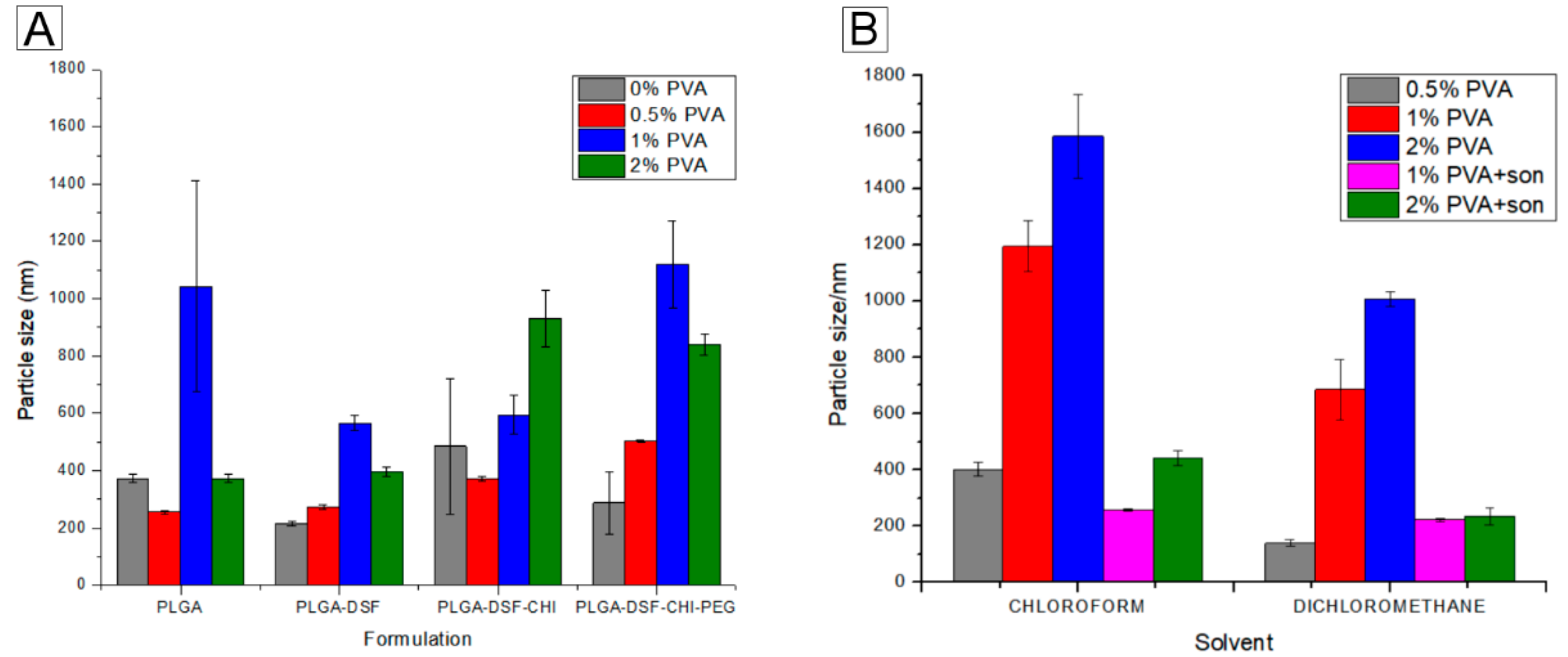
3.1. Size and Stability Analysis of Formulations Prepared by Both Methods
3.1.1. Dichloromethane as the Solvent of Choice in the Emulsification Method
3.1.2. Microfluidics-Prepared Formulations
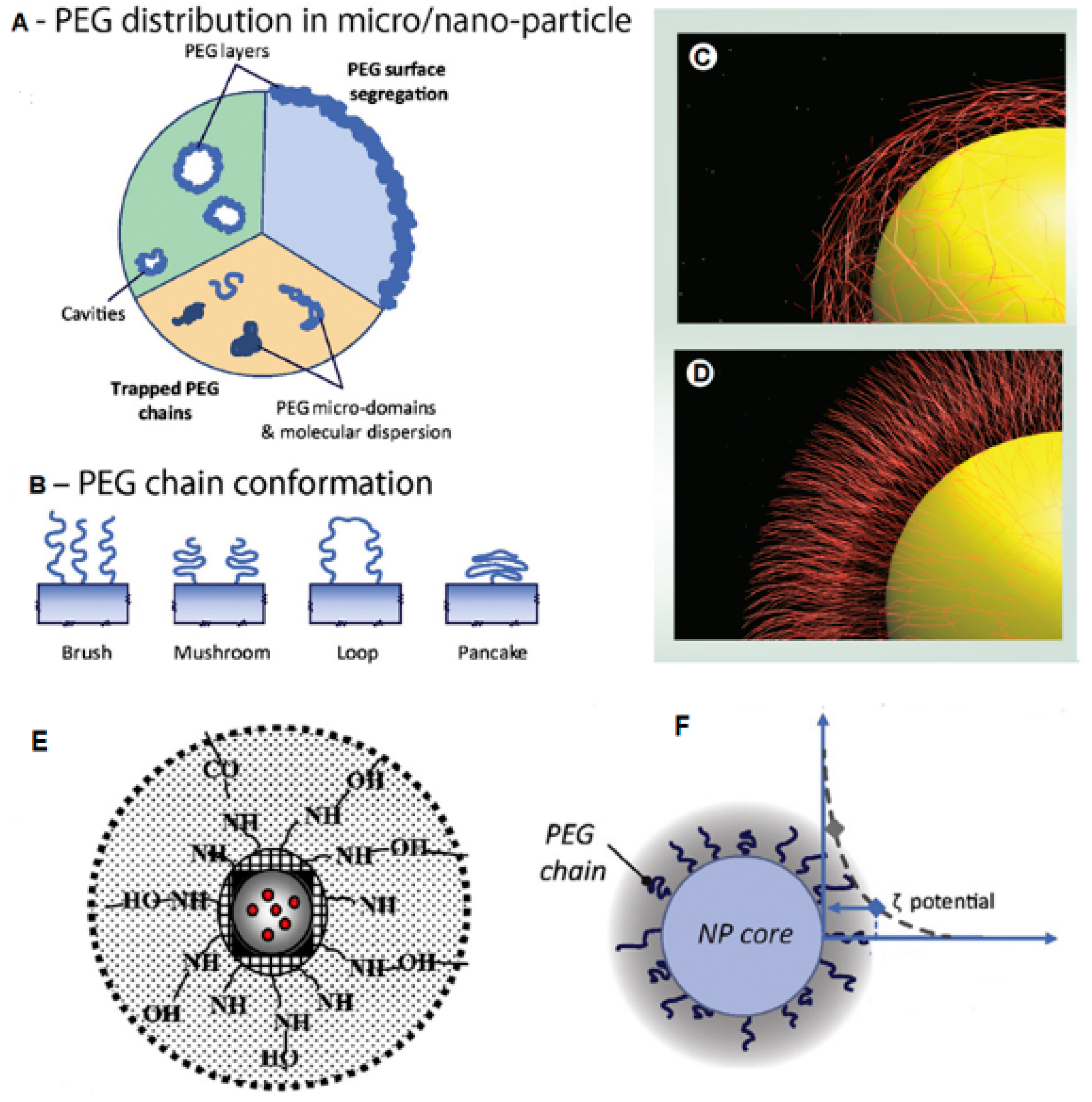
3.1.3. Analysis of Emulsification Solvent Evaporation and Microfluidics Data Showing Dependence of Properties on PEG Surface Density
3.2. FTIR Spectral Analysis Showing Adsorption of Polymer Layers
3.3. Comparative Thermogravimetric Analysis and Differential Scanning Calorimetry of Disulfiram, Blank and Loaded Nanoparticles Prepared by Both Methods
3.4. XRD Spectral Analysis Using Powder Samples of Pristine Polymers and Formulations Indicating Dominance of Blank Nanoparticle and PEG Surface Properties
3.5. Scanning Electron Microscopy of Lyophilized Samples Showing Spherical Morphology and Nanoparticle Adhesion in Microfluidics Prepared Formulations
3.6. Drug Release Kinetics of Disulfiram and Emulsification Solvent Evaporation and Microfluidic Formulations Prepared Showing Higher Relative Release of Microfluidics-Based Nanoparticles
4. Conclusions and Future Work
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduct. Target. Ther. 2018, 3, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly (lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef] [PubMed]
- Dahman, Y. Poly (Lactic Acid): Green and Sustainable Plastics. Ferment. Technol. 2014, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Rezvantalab, S.; Drude, N.I.; Moraveji, M.K.; Güvener, N.; Koons, E.K.; Shi, Y. PLGA-Based Nanoparticles in Cancer Treatment. Front. Pharmacol. 2018, 9, 1260. [Google Scholar] [CrossRef] [Green Version]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [Green Version]
- Villemin, E.; Ong, Y.C.; Thomas, C.M.; Gasser, G. Polymer encapsulation of ruthenium complexes for biological and medicinal applications. Nat. Rev. Chem. 2019, 3, 261–282. [Google Scholar] [CrossRef]
- Kravanja, G.; Primožič, M.; Knez, Ž.; Leitgeb, M. Chitosan-based (Nano) materials for novel biomedical applications. Molecules 2019, 24, 1960. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Meng, Q.; Li, Q.; Liu, J.; Zhou, M.; Jin, Z. Chitosan Derivatives and Their Application in Biomedicine. Int. J. Mol. Sci. 2020, 21, 487. [Google Scholar] [CrossRef] [Green Version]
- Frank, L.; Onzi, G.; Morawski, A.; Pohlmann, A.; Guterres, S.; Contri, R.V. Chitosan as a coating material for nanoparticles intended for biomedical applications. React. Funct. Polym. 2019, 147, 104459. [Google Scholar] [CrossRef]
- Jokerst, J.V.; Lobovkina, T.; Zare, R.N.; Gambhir, S.S. Nanoparticle PEGylation for imaging and therapy. Nanomedicine (Lond) 2011, 6, 715–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabanel, J.-M.; Hildgen, P.; Banquy, X. Assessment of PEG on polymeric particles surface, a key step in drug carrier translation. J. Control Release 2014, 185, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Parveen, S.; Sahoo, S.K. Long circulating chitosan/PEG blended PLGA nanoparticle for tumor drug delivery. Eur. J. Pharmacol. 2011, 670, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, Z.; Park, J.; Lin, Q.; Yeo, Y. Low molecular-weight chitosan as a pH-sensitive stealth coating for tumor-specific drug delivery. Mol. Pharm. 2012, 9, 1262–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abouelmagd, S.A.; Ku, Y.J.; Yeo, Y. Low molecular weight chitosan-coated polymeric nanoparticles for sustained and pH-sensitive delivery of paclitaxel. Journal of drug targeting. J. Drug Target. 2015, 23, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, L.; Alvarez-Puebla, R.A.; Pazos-Perez, N. Surface modifications of nanoparticles for stability in biological fluids. Materials 2018, 11, 1154. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhao, C.; Lin, W.; Hu, R.; Wang, Q.; Chen, H. Binding characteristics between polyethylene glycol (PEG) and proteins in aqueous solution. J. Mater. Chem. B 2014, 2, 2983–2992. [Google Scholar] [CrossRef]
- Zhou, H.; Fan, Z.; Li, P.Y.; Deng, J.; Arhontoulis, D.C.; Li, C.Y. Dense and dynamic polyethylene glycol shells cloak nanoparticles from uptake by liver endothelial cells for long blood circulation. ACS Nano 2018, 12, 10130–10141. [Google Scholar] [CrossRef]
- Cruje, C.; Chithrani, D.B. Polyethylene Glycol Functionalized Nanoparticles for Improved Cancer Treatment. Rev. Nanosci. Nanotechnol. 2014, 3, 20–30. [Google Scholar] [CrossRef]
- Essa, D.; Kondiah, P.P.D.; Choonara, Y.E.; Pillay, V. The Design of Poly(lactide-co-glycolide) Nanocarriers for Medical Applications. Front. Bioeng. Biotechnol. 2020, 8, 48. [Google Scholar] [CrossRef]
- Wang, Y.; Li, P.; Tran, T.T.; Zhang, J.; Kong, L. Manufacturing Techniques and Surface Engineering of Polymer Based Nanoparticles for Targeted Drug Delivery to Cancer. Nanomaterials 2016, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiesa, E.; Dorati, R.; Pisani, S.; Conti, B.; Bergamini, G.; Modena, T. The Microfluidic Technique and the Manufacturing of Polysaccharide Nanoparticles. Pharmaceutics 2018, 10, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.T.; Lee, J.Y.; Kim, D.D.; Yoon, I.S.; Cho, H.J. Recent Progress in the Development of Poly(lactic-co-glycolic acid)-Based Nanostructures for Cancer Imaging and Therapy. Pharmaceutics 2019, 11, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauna, Z.E.; Shukla, S.; Ambudkar, S.V. Disulfiram, an old drug with new potential therapeutic uses for human cancers and fungal infections. Mol. Biosyst. 2005, 1, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Elmira, E.; Sagar, R.; Raheel, K.; Qingping, P.D. Repurposing Disulfiram as An Anti-Cancer Agent: Updated Review on Literature and Patents. Recent Pat. Anti Cancer Drug Discov. 2019, 14, 113–132. [Google Scholar]
- He, H.; Markoutsa, E.; Li, J.; Xu, P. Repurposing disulfiram for cancer therapy via targeted nanotechnology through enhanced tumor mass penetration and disassembly. Acta Biomater. 2018, 68, 113–124. [Google Scholar] [CrossRef]
- Ding, D.; Zhu, Q. Recent advances of PLGA micro/nanoparticles for the delivery of biomacromolecular therapeutics. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 1041–1060. [Google Scholar] [CrossRef]
- Donno, R.; Gennari, A.; Lallana, E.; De La Rosa, J.M.R.; d’Arcy, R.; Treacher, K. Nanomanufacturing through microfluidic-assisted nanoprecipitation: Advanced analytics and structure-activity relationships. Int. J. Pharm. 2017, 534, 97–107. [Google Scholar] [CrossRef]
- Streck, S.; Clulow, A.J.; Nielsen, H.M.; Rades, T.; Boyd, B.J.; McDowell, A. The distribution of cell-penetrating peptides on polymeric nanoparticles prepared using microfluidics and elucidated with small angle X-ray scattering. J. Colloid Interface Sci. 2019, 555, 438–448. [Google Scholar] [CrossRef]
- Madala, H.R.; Punganuru, S.R.; Ali-Osman, F.; Zhang, R.; Srivenugopal, K.S. Brain- and brain tumor-penetrating disulfiram nanoparticles: Sequence of cytotoxic events and efficacy in human glioma cell lines and intracranial xenografts. Oncotarget 2018, 9, 3459–3482. [Google Scholar] [CrossRef]
- Garg, S.; Heuck, G.; Ip, S.; Ramsay, E. Microfluidics: A transformational tool for nanomedicine development and production. J. Drug Target. 2016, 24, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Kothawade, N.P.; Katti, S.A. UV method development and validation for the estimation of disulfiram in marketed tablet preparation. Int. J. Res. Pharm. Chem. 2016, 6, 528–533. [Google Scholar]
- Bamrungsap, S.; Zhao, Z.; Chen, T.; Wang, L.; Li, C.; Fu, T.; Tan, W. Nanotechnology in therapeutics: A focus on nanoparticles as a drug delivery system. Nanomedicine (Lond) 2012, 7, 1253–1271. [Google Scholar] [CrossRef] [PubMed]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Mohammadi-Samani, S.; Taghipour, B. technology. PLGA micro and nanoparticles in delivery of peptides and proteins; problems and approaches. Pharm. Dev. Technol. 2015, 20, 385–393. [Google Scholar] [CrossRef]
- Abd, E.l.; Hady, W.E.; Mohamed, E.A.; Soliman, O.A.E.; El-Sabbagh, H.M. In vitro-in vivo evaluation of chitosan-PLGA nanoparticles for potentiated gastric retention and anti-ulcer activity of diosmin. Int. J. Nanomed. 2019, 14, 7191–7213. [Google Scholar]
- Al-Nemrawi, N.K.; Alshraiedeh, N.A.H.; Zayed, A.L.; Altaani, B.M. Low molecular weight chitosan-coated PLGA nanoparticles for pulmonary delivery of tobramycin for cystic fibrosis. Pharmaceuticals 2018, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Fasehee, H.; Ghavamzadeh, A.; Alimoghaddam, K.; Ghaffari, S.-H.; Faghihi, S. A Comparative Cytotoxic Evaluation of Disulfiram Encapsulated PLGA Nanoparticles on MCF-7 Cells. Int. J. Hematol. Oncol. Stem. Cell Res. 2017, 11, 102–107. [Google Scholar]
- Wilkosz, N.; Łazarski, G.; Kovacik, L.; Gargas, P.; Nowakowska, M.; Jamróz, D. Molecular insight into drug-loading capacity of PEG–PLGA nanoparticles for itraconazole. J. Phys. Chem. B 2018, 122, 7080–7090. [Google Scholar] [CrossRef]
- Wu, C. How does a polymer brush repel proteins? Chin. J. Polym. Sci. 2014, 32, 1575–1580. [Google Scholar] [CrossRef]
- De Gennes, P.G. Polymers at an interface; a simplified view. Adv. Colloid Interface Sci. 1987, 27, 189–209. [Google Scholar] [CrossRef]
- Jiang, W.; Han, S.J. Study of interaction between polyethylene glycol and chitosan by viscosity method. Int. J. Polym. Sci. Part B Polym. Phys. 1998, 36, 1275–1281. [Google Scholar] [CrossRef]
- Trivedi, M.K.; Branton, A.; Trivedi, D.; Nayak, G.; Bairwa, K.; Jana, S. Spectroscopic characterization of disulfiram and nicotinic acid after biofield treatment. J. Anal. Bioanal. Tech. 2015, 6, 1000265. [Google Scholar]
- Lu, B.; Lv, X.; Le, Y. Chitosan-Modified PLGA Nanoparticles for Control-Released Drug Delivery. Polymers 2019, 11, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.; Tang, S.; Tong, Q. Oleanolic acid liposomes with polyethylene glycol modification: Promising antitumor drug delivery. Int. J. Nanomed. 2012, 7, 3517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyukova, V.; Kedik, S.; Panov, A.; Zhavoronok, E.; Mendeleev, D.; Senchikhin, I. Synthesis of a Disulfuram Inclusion Complex with Hydroxypropyl-β-Cyclodextrin and Its Effect on Cataract Development in Rats. Pharm. Chem. J. 2020, 53, 1158–1163. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, F.; Gao, Q.; Liu, Y. Physicochemical Properties of Poly (lactic acid-co-glycolic acid) Film Modified via Blending with Poly (butyl acrylate-co-methyl methacrylate). Polímeros 2013, 23, 619–623. [Google Scholar] [CrossRef]
- Arya, G.; Das, M.; Sahoo, S.K.S. Evaluation of curcumin loaded chitosan/PEG blended PLGA nanoparticles for effective treatment of pancreatic cancer. Biomed. Pharm. 2018, 102, 555–566. [Google Scholar] [CrossRef]
- Brzeziński, M.; Socka, M.; Kost, B. Microfluidics for producing polylactide nanoparticles and microparticles and their drug delivery application. Polym. Int. 2019, 68, 997–1014. [Google Scholar] [CrossRef]
- Shergill, M.; Patel, M.; Khan, S.; Bashir, A.; McConville, C. Development and characterisation of sustained release solid dispersion oral tablets containing the poorly water soluble drug disulfiram. Int. J. Pharm. 2016, 497, 3–11. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | CHI/% | PEG/% | PSD/nm | Polydispersity Index (PDI) | ζ Potential/mV | Entrapment Efficiency/% |
|---|---|---|---|---|---|---|
| Bulk | 0 | 0 | 278 ± 1.0 | 0.100 ± 0.033 | −17.2 ± 0.2 | - |
| 0.25 | 0 | 245 ± 2.4 | 0.156 ± 0.000 | 14.0 ± 0.9 | - | |
| 0.25 | 2 | 221 ± 4.1 | 0.028 ± 0.020 | 3.09 ± 0.2 | 16.5 ± 1.0 | |
| 0.25 | 4 | 143 ± 3.7 | 0.183 ± 0.013 | 20.3 ± 0.9 | 72.1 ± 0.9 | |
| 0.25 | 6 | 184 ± 2.6 | 0.189 ± 0.011 | 21.3 ± 3.5 | 72.6 ± 0.2 | |
| 0.25 | 8 | 162 ± 10 | 0.155 ± 0.037 | 11.5 ± 1.4 | 26.5 ± 0.4 | |
| Microfluidic | 0 | 0 | 95.8 ± 4.6 | 0.303 ± 0.031 | −6.92 ± 0.7 | - |
| 0.25 | 0 | 235 ± 1.9 | 0.292 ± 0.002 | 14.9 ± 1.0 | - | |
| 0.25 | 2 | 171 ± 14 | 0.284 ± 0.012 | 8.24 ± 1.8 | 29.9 ± 0.7 | |
| 0.25 | 4 | 220 ± 8.1 | 0.263 ± 0.001 | 19.2 ± 1.5 | 74.6 ± 1.0 | |
| 0.25 | 6 | 179 ± 2.5 | 0.247 ± 0.003 | 32.3 ± 0.1 | 78.7 ± 1.1 | |
| 0.25 | 8 | 204 ± 1.7 | 0.200 ± 0.006 | 18.3 ± 0.5 | 50.2 ± 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Essa, D.; Choonara, Y.E.; Kondiah, P.P.D.; Pillay, V. Comparative Nanofabrication of PLGA-Chitosan-PEG Systems Employing Microfluidics and Emulsification Solvent Evaporation Techniques. Polymers 2020, 12, 1882. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12091882
Essa D, Choonara YE, Kondiah PPD, Pillay V. Comparative Nanofabrication of PLGA-Chitosan-PEG Systems Employing Microfluidics and Emulsification Solvent Evaporation Techniques. Polymers. 2020; 12(9):1882. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12091882
Chicago/Turabian StyleEssa, Divesha, Yahya E. Choonara, Pierre P. D. Kondiah, and Viness Pillay. 2020. "Comparative Nanofabrication of PLGA-Chitosan-PEG Systems Employing Microfluidics and Emulsification Solvent Evaporation Techniques" Polymers 12, no. 9: 1882. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12091882