Macromolecular Dyes by Chromophore-Initiated Ring Opening Polymerization of L-Lactide

 ,
, 
 ,
, 
 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Conventional Synthesis of Macromolecular Dyes (MDs)
2.3. Blending of Commercial PLA with MD (PLA/MD5s)
2.4. Blending of Commercial PLA with Chromophores (PLA/CHROs)
2.5. Migration Test
2.6. Instruments
3. Results and Discussion
3.1. Macromolecular Dyes (MDs)
3.1.1. Preparation, Structural Characterization, and Thermal Properties
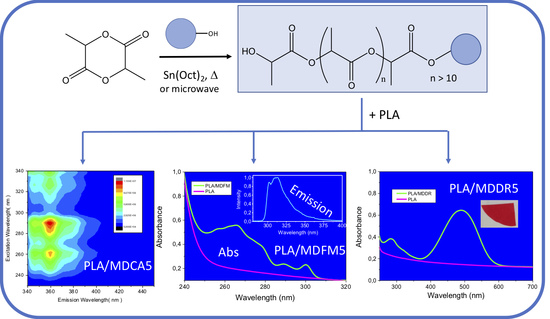
3.1.2. Photophysical Properties of MD5s
3.2. Blending of MDs with Commercial PLA
3.2.1. Preparation, Structural Characterization, and Thermal Properties of the Blends
3.2.2. Photophysical Properties of the Blends
3.3. Migration Tests
3.4. Microwave Assisted Polymerization of L-lactide in the Presence of FM as Co-initiator
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farah, S.; Anderson, D.G.; Langer, R. Physical and mechanical properties of PLA, and their functions in widespread applications—A comprehensive review. Adv. Drug Deliv. Rev. 2016, 107, 367–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Z.; Wang, X.; Chen, W.; Hua, E.; Liu, K. Synthesis, spectral and dyeing properties of phenylazopyrazolone-containing acylamide disperse dyes designed for poly(lactic acid). Color. Technol. 2012, 128, 283–289. [Google Scholar] [CrossRef]
- Hussain, T.; Tausif, M.; Ashraf, M. A review of progress in the dyeing of eco-friendly aliphatic polyester based polylactic acid fabrics. J. Clean. Prod. 2015, 108, 476–483. [Google Scholar] [CrossRef]
- Ge, F.; Ding, Y.; Yang, L.; Huang, Y.; Jiang, L.; Dan, Y. Effect of the content and distribution of ultraviolet absorbing groups on the UV protection and degradation of polylactide films. RSC Adv. 2015, 5, 70473–70481. [Google Scholar] [CrossRef]
- Hu, Y.; Daoud, W.A.; Cheuk, K.K.L.; Lin, C.S.K. Newly developed techniques on polycondensation, ring-opening polymerization and polymer modification: Focus on poly(lactic acid). Materials 2016, 9, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef]
- Gupta, A.P.; Kumar, V. New Emerging Trends in synthetic biodegradable polymers—Polylactide: A critique. Eur. Polym. J. 2007, 43, 4053–4074. [Google Scholar] [CrossRef]
- Williams, C.K. Synthesis of functionalized biodegradable polyesters. Chem. Soc. Rev. 2007, 36, 1573–1580. [Google Scholar] [CrossRef]
- Nguyen, C.A.; Allémann, E.; Schwach, G.; Doelker, E.; Gurny, R. Synthesis of a novel fluorescent poly(D,L-Lactide) end-capped with 1-pyrenebutanol used for the preparation of nanoparticles. Eur. J. Pharm. Sci. 2003, 20, 217–222. [Google Scholar] [CrossRef]
- Alwattar, A.; Haddad, A.; Zhou, Q.; Nascimento, T.; Greenhalgh, R.; Medeiros, E.; Blaker, J.; Parry, A.; Quaylea, P.; Yeatesa, S. Synthesis and characterisation of fluorescent pyrene-end-capped polylactide fibres. Polym. Int. 2019, 68, 360–368. [Google Scholar] [CrossRef]
- Yildirim, I.; Bus, T.; Sahn, M.; Yildirim, T.; Kalden, D.; Hoeppener, S.; Traeger, A.; Westerhausen, M.; Weber, C.; Schubert, U.S. Fluorescent amphiphilic heterografted comb polymers comprising Biocompatible PLA and PEtOx side chains. Polym. Chem. 2016, 7, 6064–6074. [Google Scholar] [CrossRef]
- Ge, F.; Huang, Y.; Luo, Y.; Jiang, L.; Dan, Y. Macromolecular chain structure design, synthesis and analysis of poly(L-lactide) linking ultraviolet absorbing groups. RSC Adv. 2014, 4, 63118–63127. [Google Scholar] [CrossRef]
- Ten Breteler, M.R.; Feijen, J.; Dijkstra, P.J.; Signori, F. Synthesis and thermal properties of hetero-bifunctional PLA oligomers and their stereocomplexes. React. Funct. Polym. 2013, 73, 30–38. [Google Scholar] [CrossRef]
- Burgos, N.; Tolaguera, D.; Fiori, S.; Jiménez, A. Synthesis and characterization of lactic acid oligomers: Evaluation of performance as poly(lactic acid) plasticizers. J. Polym. Environ. 2014, 22, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Báez, J.E.; Marcos-Fernández, Á.; Galindo-Iranzo, P. Exploring the effect of alkyl end group on poly(L-lactide) oligo-esters. Synthesis and characterization. J. Polym. Res. 2011, 18, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Giuntoli, G.; Rosi, L.; Frediani, M.; Sacchi, B.; Frediani, P. Fluoro-functionalized PLA polymers as potential water-repellent coating materials for protection of stone. J. Appl. Polym. Sci. 2012, 125, 3125–3133. [Google Scholar] [CrossRef]
- Fischer, E.W.; Sterzel, H.J.; Wegner, G. Investigation of the structure of solution grown crystals of lactide copolymers by means of chemical reactions. Kolloid Z. Z. Polym. 1973, 251, 980–990. [Google Scholar] [CrossRef]
- Inkinen, S.; Hakkaramen, M.; Albertsson, A.-C.; Södergård, A. From lactic acid to poly(lactic acid) (PLA): Characterization and analysis of PLA and its precursors. Biomacromolecules 2011, 12, 523–532. [Google Scholar] [CrossRef]
- Nikolic, L.; Ristic, I.; Adnadjevic, B.; Nikolic, V.; Jovanovic, J.; Stankovic, M. Novel microwave-assisted synthesis of poly(D,L-lactide): The influence of monomer/initiator molar ratio on the product properties. Sensors 2010, 10, 5063–5073. [Google Scholar] [CrossRef]
- Witzke, D.R.; Narayan, R. Reversible kinetics and thermodynamics of the homopolymerization of L-lactide with 2-ethylhexanoic acid tin(II) salt. Macromolecules 1997, 30, 7075–7085. [Google Scholar] [CrossRef]
- De Jong, S.J.; van Dijk-Wolthuis, W.N.E.; Kettenes-van den Bosch, J.J.; Schuyl, P.J.W.; Hennink, W.E. Monodisperse enantiomeric lactic acid oligomers: Preparation, characterization, and stereocomplex formation. Macromolecules 1998, 31, 6397–6402. [Google Scholar] [CrossRef]
- Ubeda, S.; Aznar, M.; Alfaro, P.; Nerín, C. Migration of oligomers from a food contact biopolymer based on polylactic acid (PLA) and polyester. Anal. Bioanal. Chem. 2019, 411, 3521–3532. [Google Scholar] [CrossRef] [PubMed]
- Penczek, S.; Duda, A.; Szymanski, R. Intra- and intermolecular chain transfer to macromolecules with chain scission, the case of cyclic esters. Macromol. Symp. 1998, 132, 441–449. [Google Scholar] [CrossRef]
- Yu, Y.; Storti, G.; Morbidelli, M. Ring-opening polymerization of L,L-lactide: Kinetic and modeling study. Macromolecules 2009, 42, 8187. [Google Scholar] [CrossRef]
- Kowalski, A.; Libiszowski, J.; Duda, A.; Penczek, S. Kinetics and mechanism of cyclic esters polymerization initiated with tin(II) octoate. 3 polymerization of L,L-dilactide. Macromolecules 2000, 33, 7359–7370. [Google Scholar] [CrossRef]
- Saeidlou, S.; Huneault, M.A.; Li, H.; Park, C.B. Poly(lactic acid) crystallization. Prog. Polym. Sci. 2012, 37, 1657–1677. [Google Scholar] [CrossRef]
- Rathi, S.; Kalish, J.P.; Coughlin, E.B.; Hsu, S.L. Utilization of oligo(lactic acid) for studies of chain conformation and chain packing in poly(lactic acid). Macromolecules 2011, 44, 3410–3415. [Google Scholar] [CrossRef]
- Pholharn, D.; Srithep, Y.; Morris, J. Effect of initiators on synthesis of poly(L-lactide) by ring opening polymerization. IOP Conf. Ser. Mater. Sci. Eng. 2017, 213, 012022. [Google Scholar] [CrossRef]
- Jin, X.; Chen, X.; Cheng, Q.; Zhang, N.; Caia, S.; Ren, J. Non-isothermal crystallization kinetics of ramie fiber-reinforced polylactic acid biocomposite. RSC Adv. 2017, 7, 46014–46021. [Google Scholar] [CrossRef] [Green Version]
- Lakowicz, J.R.; Hogen, D. Chlorinated hydrocarbon-cell membrane interactions studied by the fluorescence quenching of carbazole-labeled phospholipids: Probe synthesis and characterization of the quenching methodology. Chem. Phys. Lipids 1980, 26, 1–40. [Google Scholar] [CrossRef]
- Bonesi, S.M.; Erra-Balsells, R. Electronic spectroscopy of carbazole and N- and C-substituted carbazoles in homogeneous media and in solid matrix. J. Lumin. 2001, 93, 51–74. [Google Scholar] [CrossRef]
- Nkansah, M.A.; Christy, A.A.; Barth, T.; Francis, G.W. Preliminary photochemical studies of fluorene in various aqueous media. Am. J. Sci. Ind. Res. 2014, 5, 97–103. [Google Scholar] [CrossRef]
- Winnik, F.M. Association of hydrophobic polymers in water: Fluorescence studies with labeled (hydroxypropy1)cellulose. Macromolecules 1989, 22, 734–742. [Google Scholar] [CrossRef]
- Feng, G.; Qian, H.-F.; Bai, G.; Liu, Y.-C.; Hu, L.-L. Synthesis, characterization, and application of diester/diurethane tethered azo disperse dyes: A new strategy to improve dye’s fastness properties. Dyes Pigments 2016, 129, 54–59. [Google Scholar] [CrossRef]
- Martins, T.D.; Weiss, R.G.; Atvars, T.D.Z. Synthesis and photophysical properties of a poly(methyl methacrylate) polymer with carbazolyl side groups. J. Braz. Chem. Soc. 2008, 19, 1450–1461. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Zhou, J.; Zhang, L. Synthesis and fluorescent properties of carbazole-substituted hydroxyethylcelluloses. Macromol. Chem. Phys. 2012, 213, 57–63. [Google Scholar] [CrossRef]
- Heldt, J.; Heldt, J.R.; Redzimski, T.; Diehl, H.; Schultz, P. Spectroscopic studies of dimethylamino derivatives of fluorene. Z. Naturforsch. 2000, 55, 902–908. [Google Scholar] [CrossRef]
- Redzimski, T.; Heldt, J.R. Spectroscopic study of solvatochromic effects in solution of amino and hydroxy derivatives of fluorene. J. Fluoresc. 2003, 13, 393–401. [Google Scholar] [CrossRef]
- He, L.; Zhang, S.F.; Tang, B.T.; Wang, L.L.; Yang, J.Z. Dyes with high affinity for polylactide. Chin. Chem. Lett. 2007, 18, 1151–1153. [Google Scholar] [CrossRef]
- Ojanen, J.; Rantala, T.T. Electronic structure and absorption spectrum of disperse red 1: Comparison of computational approaches. Open Chem. Phys. J. 2009, 2, 37–46. [Google Scholar] [CrossRef]
- Airinei, A.; Homocianu, M.; Dorohoi, D.O. Changes induced by solvent polarity in electronic absorption spectra of some azo disperse dyes. J. Mol. Liq. 2010, 157, 13–17. [Google Scholar] [CrossRef]
- Priimagi, B.; Cattaneo, S.; Ras, R.H.A.; Valkama, S.; Ikkala, O.; Kauranen, M. Polymer-dye complexes: A facile method for high doping level and aggregation control of dye molecules. Chem. Mater. 2005, 17, 5798–5802. [Google Scholar] [CrossRef]
- Pérez Amaro, L.; Cicogna, F.; Passaglia, E.; Morici, E.; Oberhauser, W.; Al-Malaika, S.; Dintcheva, N.T.; Coiai, S. Thermo-oxidative stabilization of poly(lactic acid) with antioxidant intercalated layered double hydroxides. Polym. Degrad. Stab. 2016, 113, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Bertoldo, M.; Coltelli, M.B.; Miraglia, L.; Narducci, P.; Bronco, S. Surface energy inducing asymmetric phase distribution in films of a binary polymeric blend. Polymer 2005, 46, 11311–11321. [Google Scholar] [CrossRef]
- Stein, G.E.; Laws, T.S.; Verduzco, R. Tailoring the attraction of polymers toward surfaces. Macromolecules 2019, 52, 4787–4802. [Google Scholar] [CrossRef] [Green Version]
- Avinc, O.; Phillips, D.; Wilding, M. Influence of different finishing conditions on the wet fastness of selected disperse dyes on polylactic acid fabrics. Color. Technol. 2009, 125, 288–295. [Google Scholar] [CrossRef]
- Avinc, O.; Wilding, M.; Bone, J.; Phillips, D.; Farrington, D. Evaluation of color fastness and thermal migration in softened polylactic acid fabrics dyed with disperse dyes of differing hydrophobicity. Color. Technol. 2010, 126, 353–364. [Google Scholar] [CrossRef]
- Frediani, M.; Giachi, G.; Rosi, L.; Frediani, P. Synthesis and Processing of Biodegradable and Bio-Based Polymers by Microwave Irradiation; Usha, C., Ed.; InTech: London, UK, 2010; ISBN 978-953-307-573-0. Available online: http://www.intechopen.com/books/microwaveheating/synthesis-and-processing-of-biodegradable-and-bio-based-polymers-by-microwave-irradiation (accessed on 6 July 2019).
- Frediani, M.; Sémeril, D.; Matt, D.; Rosi, L.; Frediani, P.; Rizzolo, F.; Papini, A.M. Ring-opening polymerisation of rac -lactide using a calix[4]arene-based titanium (IV) complex. Int. J. Polym. Sci. 2010. [Google Scholar] [CrossRef] [Green Version]
- Hoogenboom, R.; Schubert, U.S. Microwave-assisted polymer synthesis: Recent developments in a rapidly expanding field of research. Macromol. Rapid Commun. 2007, 28, 368–386. [Google Scholar] [CrossRef]
- Sosnik, A.; Gotelli, G.; Abraham, G.A. Microwave-assisted polymer synthesis (MAPS) as a tool in biomaterials science: How new and how powerful. Prog. Polym. Sci. 2011, 36, 1050–1078. [Google Scholar] [CrossRef]
- Dubey, S.P.; Abhyankar, H.A.; Marchante, V.; Brighton, J.L.; Bergmann, B.; Trinhc, G.; David, C. Microwave energy assisted synthesis of poly lactic acid via continuous reactive extrusion: Modelling of reaction kinetics. RSC Adv. 2017, 7, 18529–18538. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhao, N.; Bai, W.; Chen, D.L.; Xiong, C.D. Microwave-assisted ring- opening polymerization of poly(glycolic acid-co-lactic acid) copolymers. e-Polymers 2010, 51, 1–6. [Google Scholar] [CrossRef]
- Zhang, C.; Liao, L.; Gong, S. Microwave-assisted synthesis of PLLA-PEG-PLLA triblock copolymers. Macromol. Rapid Commun. 2007, 28, 422–427. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conversion (%) 1 | PD (NMR) 2 | Mn (NMR) (g/mol) 3 | Mn (SEC) (g/mol) 4 | Mn (CAL) (g/mol) 5 | Đ (SEC) 6 |
|---|---|---|---|---|---|---|
| MDCA1 | 93 | 41 | 3166 | 12,702 | 13,616 | 1.5 |
| MDCA3 | 99 | 26 | 2085 | 5452 | 7346 | 1.4 |
| MDCA5 | 99 | 28 | 2229 | 4002 | 3065 | 1.3 |
| MDCA7 | 91 | 17 | 1436 | 2633 | 2085 | 1.4 |
| MDCA10 | 88 | 13 | 1148 | 2088 | 1480 | 1.3 |
| MDFM1 | 92 | 9 | 845 | 2262 | 13,457 | 1.2 |
| MDFM3 | 91 | 10 | 917 | 2146 | 4568 | 1.1 |
| MDFM5 | 99 | 26 | 2142 | 3822 | 3050 | 1.2 |
| MDFM7 | 99 | 19 | 1566 | 1682 | 2234 | 1.1 |
| MDFM10 | 99 | 4 | 485 | 1334 | 1623 | 1.1 |
| MDDR1 | 84 | 6 | 781 | 2320 | 12,456 | 1.2 |
| MDDR3 | 85 | 10 | 1069 | 2407 | 4432 | 1.1 |
| MDDR5 | 82 | 28 | 2367 | 2204 | 2713 | 1.1 |
| MDDR7 | 82 | 7 | 853 | 1218 | 2037 | 1.1 |
| MDDR10 | 82 | 4 | 637 | 754 | 1531 | 1.1 |
| Sample | Tg (°C) | Tcc (°C) | ΔHcc (J/g) | Tm (°C) | ΔHm (J/g) | Cryst.2 (%) |
|---|---|---|---|---|---|---|
| PLA | 57.5 | 113.7 | −2.2 | 149.5 | 3.4 | 3.7 |
| PLA/CA | 58.9 | 114.2 | −31.6 | 149.9 156.2 | 32.9 | 1.5 |
| PLA/FM | 59.1 | 114.9 | −32.6 | 150.2 156.6 | 32.4 | − |
| PLA/DR | 58.3 | 131.0 | −13.7 | 153.3 | 17.6 | 4.0 |
| PLA/MDCA5 | 58.8 | 119.5 | −32.9 | 151.2 | 32.7 | − |
| PLA/MDFM5 | 58.8 | 119.5 | −33.7 | 152.0 | 32.1 | − |
| PLA/MDDR5 | 59.2 | 120.1 | −30.9 | 151.3 | 37.6 | 7.2 |
| Chromophore | PLA/CHROs (wt.%) | PLA/MDs (wt.%) |
|---|---|---|
| CA | 40 | 9 |
| FM | 74 | 8 |
| DR | 28 | 4 |
| Entry | Conversion (%) 2 | PD (NMR) 3 | Mn (NMR) (g/mol) 4 | Mn (SEC) (g/mol) 5 | Mn (CAL) (g/mol) 6 | Đ (SEC) 7 |
|---|---|---|---|---|---|---|
| MDFMmw1 | 93 | 52 | 3940 | 3321 | 13,457 | 1.7 |
| MDFMmw3 | 90 | 38 | 2932 | 4389 | 4568 | 1.5 |
| MDFMmw5 | 88 | 29 | 2284 | 2981 | 3050 | 1.4 |
| MDFMmw7 | 77 | 27 | 2140 | 2006 | 2234 | 1.2 |
| MDFMmw10 | 79 | 16 | 1348 | 2105 | 1623 | 1.2 |
| MDFM1 | 92 | 9 | 845 | 2262 | 13,457 | 1.2 |
| MDFM3 | 91 | 10 | 917 | 2146 | 4568 | 1.1 |
| MDFM5 | 99 | 26 | 2142 | 3822 | 3050 | 1.2 |
| MDFM7 | 99 | 19 | 1566 | 1682 | 2234 | 1.1 |
| MDFM10 | 99 | 4 | 485 | 1334 | 1623 | 1.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicogna, F.; Giachi, G.; Rosi, L.; Passaglia, E.; Coiai, S.; Spiniello, R.; Prescimone, F.; Frediani, M. Macromolecular Dyes by Chromophore-Initiated Ring Opening Polymerization of L-Lactide. Polymers 2020, 12, 1979. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12091979
Cicogna F, Giachi G, Rosi L, Passaglia E, Coiai S, Spiniello R, Prescimone F, Frediani M. Macromolecular Dyes by Chromophore-Initiated Ring Opening Polymerization of L-Lactide. Polymers. 2020; 12(9):1979. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12091979
Chicago/Turabian StyleCicogna, Francesca, Guido Giachi, Luca Rosi, Elisa Passaglia, Serena Coiai, Roberto Spiniello, Federico Prescimone, and Marco Frediani. 2020. "Macromolecular Dyes by Chromophore-Initiated Ring Opening Polymerization of L-Lactide" Polymers 12, no. 9: 1979. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12091979