
Polyamide 12 Materials Study of Morpho-Structural Changes during Laser Sintering of 3D Printing

 ,
,  , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
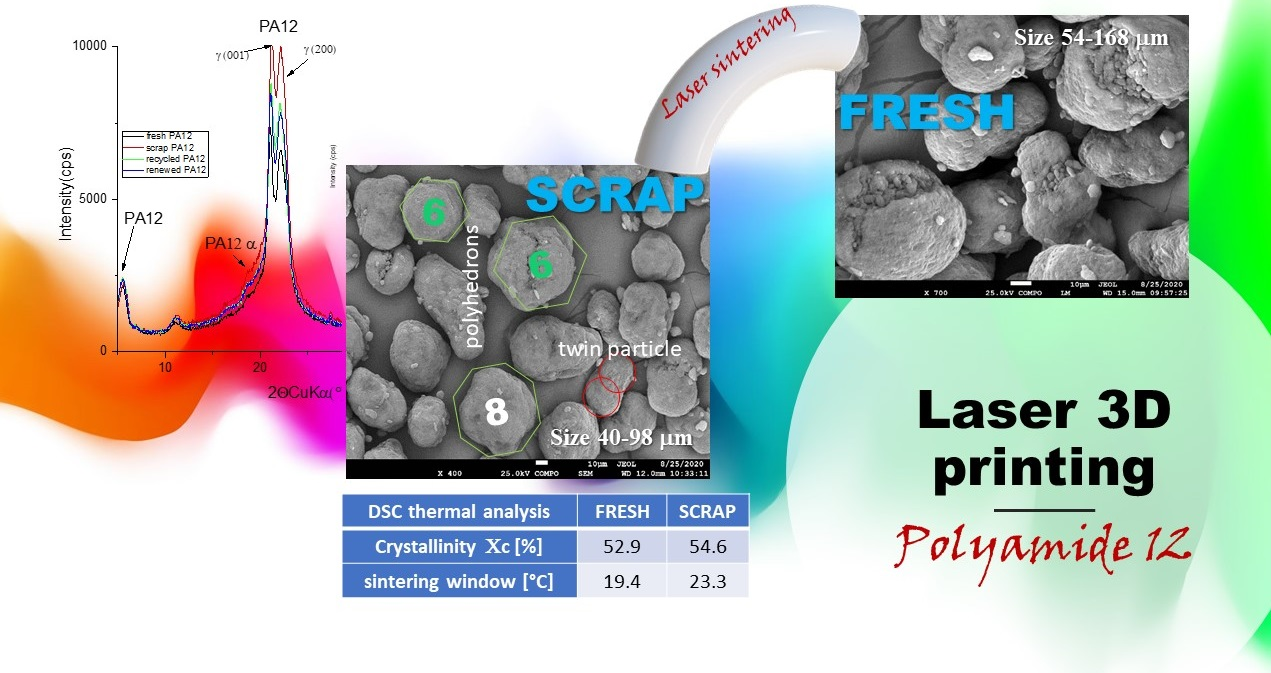
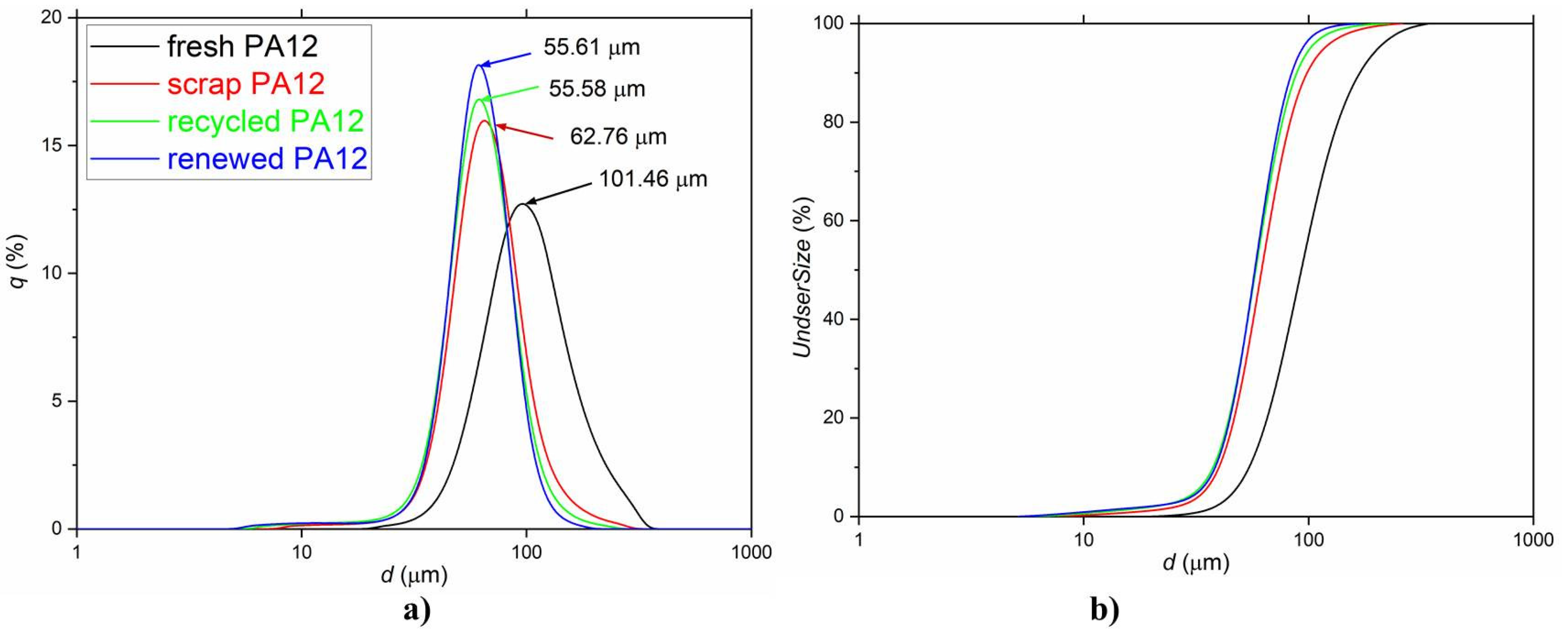
3.1. Morphology and Particle Size Observation
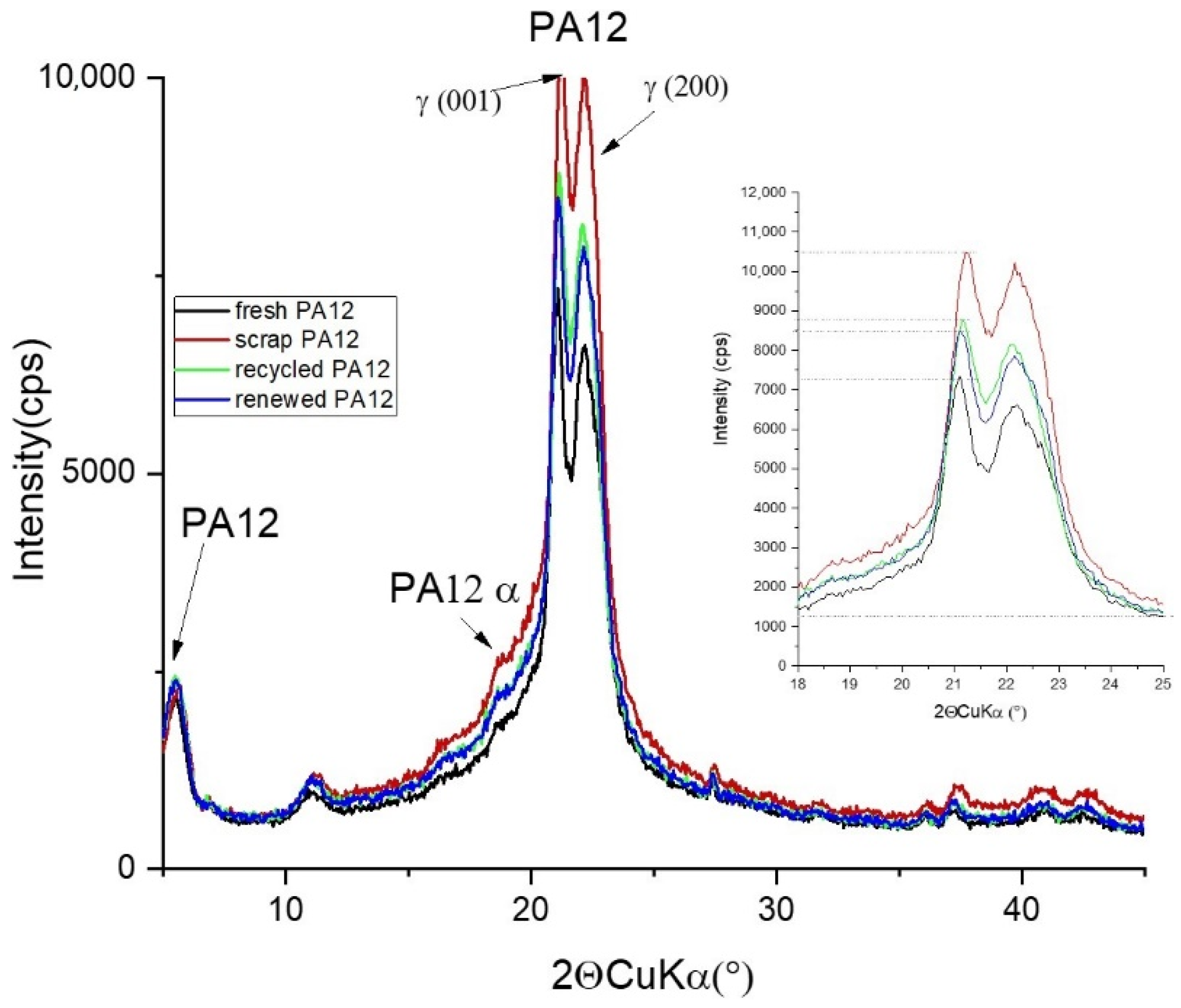
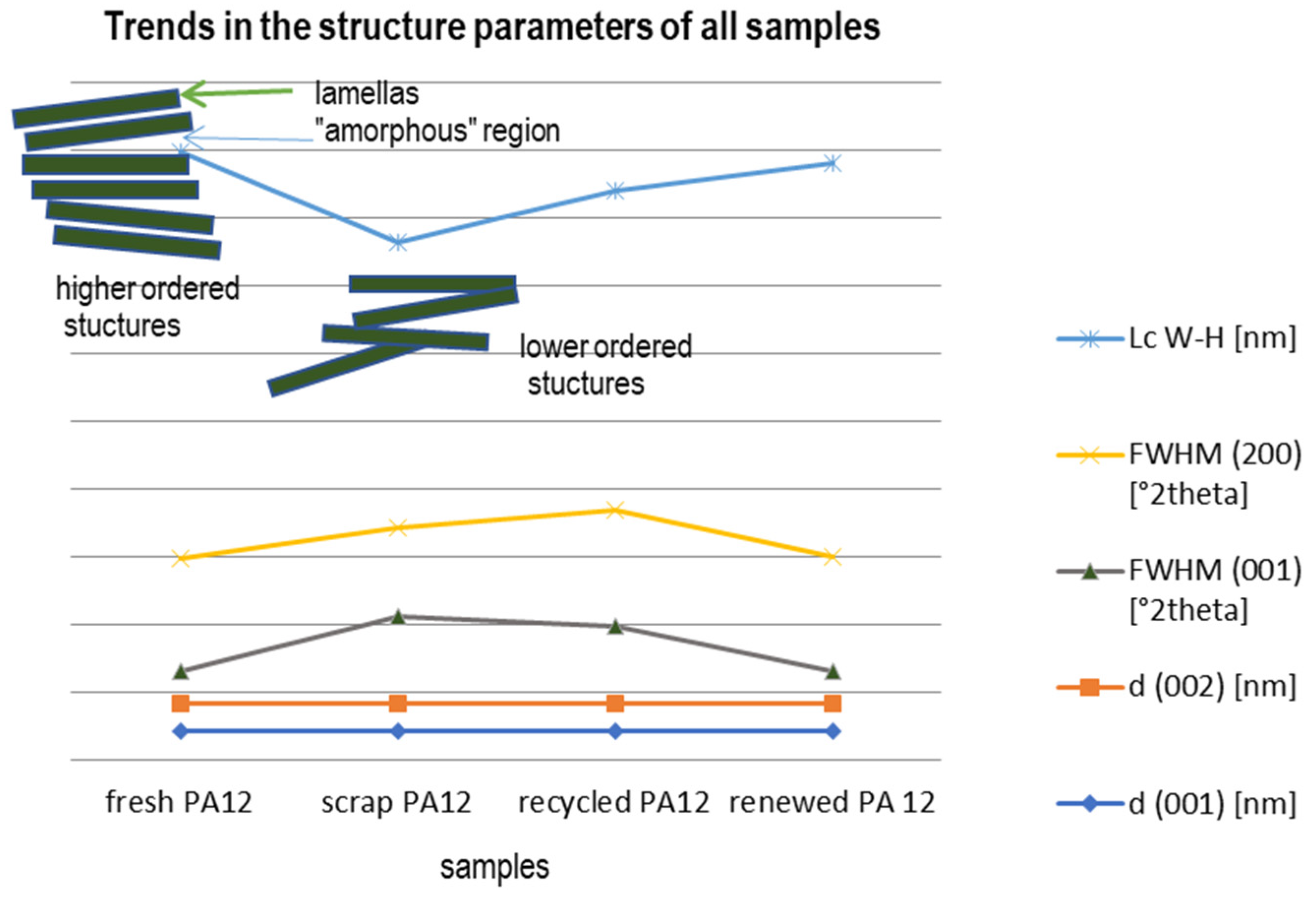
3.2. Phase Analysis and Structure Parameters
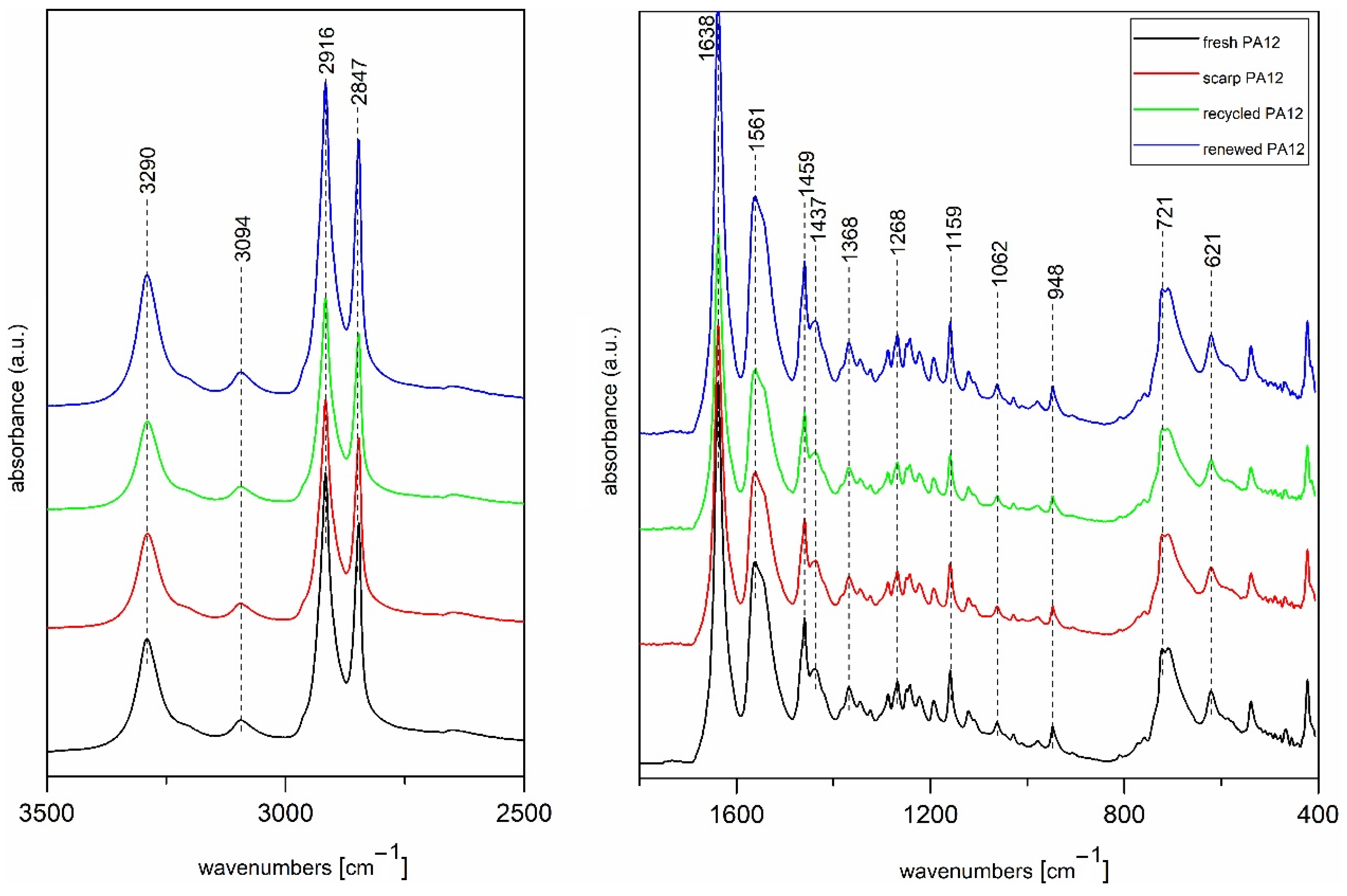
3.3. FT Infrared Spectroscopy Analysis
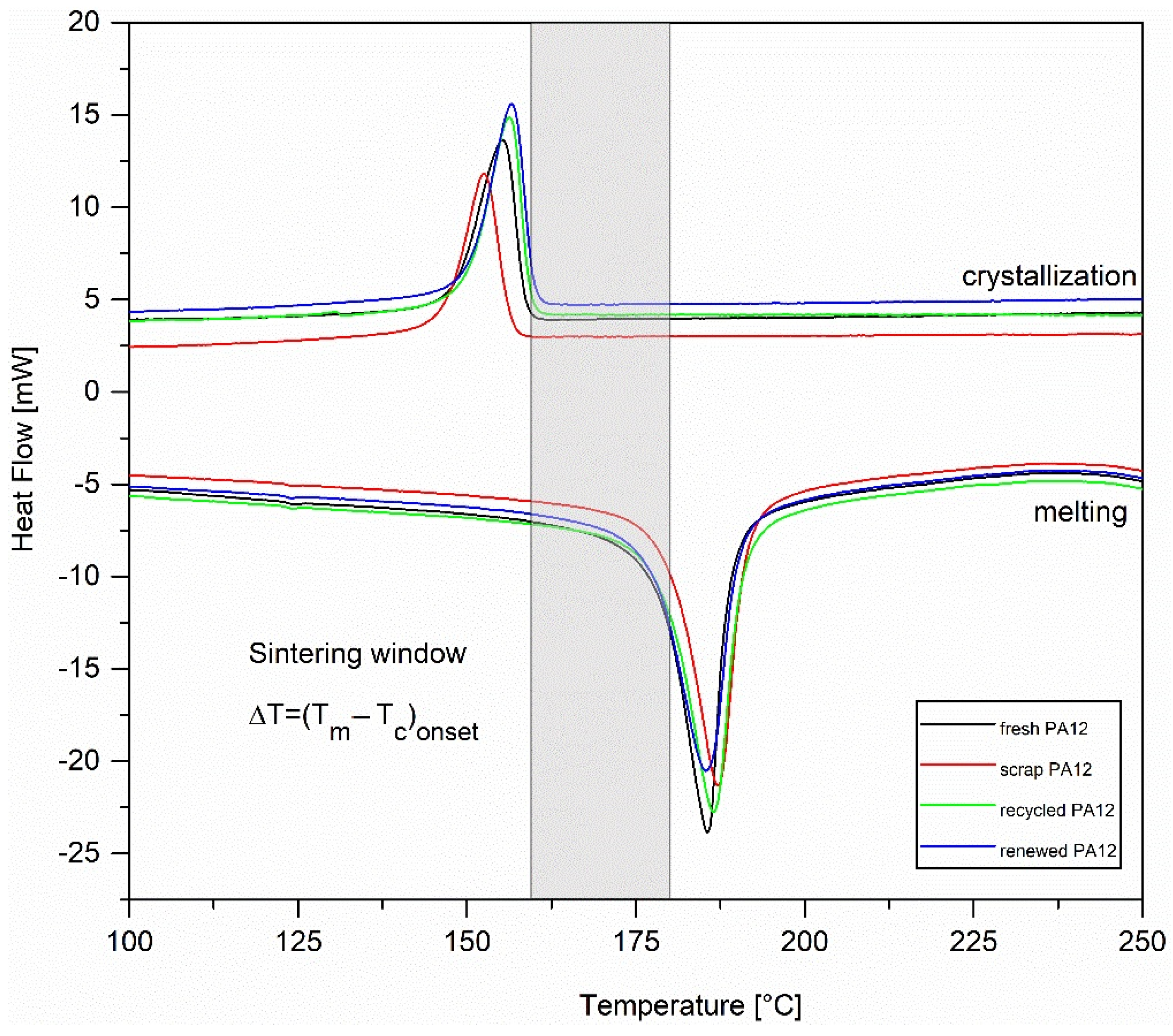
3.4. Thermal Characterization
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Fan, W.; Liu, T. Fused deposition modeling 3D printing of polyamide-based composites and its applications. Compos. Commun. 2020, 21, 100413. [Google Scholar] [CrossRef]
- Balemans, C.; Looijmans, S.F.S.P.; Grosso, G.; Hulsen, M.A.; Anderson, P.D. Numerical analysis of the crystallization kinetics in SLS. Addit. Manuf. 2020, 33, 101126. [Google Scholar] [CrossRef]
- Bandyopadhyay, A.; Bose, S. Additive Manufacturing; CRC Press: Boca Raton, FL, USA; Francis and Taylor: Abingdon, UK, 2016. [Google Scholar]
- Tofail, S.A.M.; Koumoulos, E.P.; Bandyopadhyay, A.; Bose, S.; O’Donoghue, L.; Charitidis, C. Additive manufacturing: Scientific and technological challenges, market uptake and opportunities. Mater. Today 2018, 21, 22–37. [Google Scholar] [CrossRef]
- Awad, A.; Fina, F.; Goyanes, A.; Gaisford, S.; Basit, A.W. 3D printing: Principles and pharmaceutical applications of selective laser sintering. Intern. J. Pharm. 2020, 586, 119594. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.J.; Zhu, W.; Sagar, K.; Zhou, K. Comparative study on the selective laser sintering of polypropylene homopolymer and copolymer: Processability, crystallization kinetics, crystal phases and mechanical properties. Addit. Manuf. 2020, 101610. [Google Scholar] [CrossRef]
- Mwania, F.M.; Maringa, M.; van der Walt, K. A Review of Methods Used to Reduce the Effects of High Temperature Associated with Polyamide 12 and Polypropylene Laser Sintering. Adv. Polym. Technol. 2020, 9497158. [Google Scholar] [CrossRef]
- Yang, F.; Jiang, T.; Lalier, G.; Bartolone, J.; Chen, X. A process control and interlayer heating approach to reuse polyamide 12 powders and create parts with improved mechanical properties in selective laser sintering. J. Manuf. Process. 2020, 57, 828–846. [Google Scholar] [CrossRef]
- Ligon, S.C.; Liska, R.; Stampf, J.; Gurr, M.; Mülhaupt, R. Polymers for 3D Printing and Customized Additive Manufacturing. Chem. Rev. 2017, 117, 10212–10290. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Wu, H.; Zhu, W.; Yang, L.; Li, Z.; Yan, C.; Wen, S.; Shi, Y. Investigation into the processability, recyclability and crystalline structure of selective laser sintered Polyamide 6 in comparison with Polyamide 12. Polym. Test. 2018, 69, 366–374. [Google Scholar] [CrossRef]
- Touris, A.; Turcios, A.; Mintz, E.; Pulugurtha, S.R.; Thor, P.; Jolly, M.; Jalgaonkar, U. Effect of molecular weight and hydration on the tensile properties of polyamide 12. Results Mater. 2020, 8, 100149. [Google Scholar] [CrossRef]
- Slíva, A.; Brázda, R.; Procházka, A.; Martynková, G.S. Study of the optimum arrangement of spherical particles in containers having different cross section shapes. J. Nanosci. Nanotechnol. 2019, 19, 2717–2722. [Google Scholar] [CrossRef] [PubMed]
- Slíva, A.; Samolejová, A.; Brázda, R.; Zegzulka, J.; Polák, J. Optical parameter adjustment for silica nano-and micro-particle size distribution measurement using Mastersizer 2000. In Microwave and Optical Technology 2003; Proceeding of International Society for Optics and Photonics 2004, Ostrava, Czech Republic, 7 April 2004; Photo-Optical Instrumentation Engineers (SPIE): Bellingham, WA, USA, 2004; Volume 5445, pp. 160–163. [Google Scholar] [CrossRef]
- Yang, F.; Jiang, T.; Lalier, G.; Bartolone, J.; Chen, X. Process control of surface quality and part microstructure in selective laser sintering involving highly degraded polyamide 12 materials. Polym. Test. 2021, 93, 106920. [Google Scholar] [CrossRef]
- Berretta, S.; Ghita, O.; Evans, K.E. Morphology of polymeric powders in Laser Sintering (LS): From Polyamide to new PEEK powders. Eur. Polym. J. 2014, 59, 218–229. [Google Scholar] [CrossRef]
- Feng, J.Q.; Hays, D.A. Relative importance of electrostatic forces on powder particles. Powder Technol. 2003, 135–136, 65–75. [Google Scholar] [CrossRef]
- Balemans, C.; Jaensson, N.O.; Hulsen, M.A.; Anderson, P.D. Temperature-dependent sintering of two viscous particles. Addit. Manuf. 2018, 24, 528–542. [Google Scholar] [CrossRef]
- ISO. ISO 13320-1: 2009 Particle Size Analysis. Laser Diffraction Methods. Part 1: General Principles; ISO: Prague, Czech Republic, 2009. [Google Scholar]
- Chen, P.; Zhu, W.; Yang, L.; Wen, S.; Yan, C.; Ji, Z.; Nan, H.; Shi, Y. Systematical mechanism of Polyamide-12 aging and its micro-structural evolution during laser sintering. Polym. Test. 2018, 67, 370–379. [Google Scholar] [CrossRef]
- Inoue, K.; Hoshino, S. Crystal structure of nylon 12. J. Polym. Sci. Part. B 1973, 11, 1077–1089. [Google Scholar] [CrossRef]
- Li, L.; Koch, M.H.J.; de Jeu, W.H. Crystalline Structure and Morphology in Nylon-12: A Small- and Wide-Angle X-ray Scattering Study. Macromolecules 2003, 36, 1626–1632. [Google Scholar] [CrossRef]
- Socrates, G. Infrared and Raman Characteristic Group Frequencies, Tables and Charts, 3rd ed.; John Wiley & Sons: Chichester, UK, 2001. [Google Scholar]
- Dadbakhsh, S.; Verbelen, L.; Verkinderen, O.; Strobbe, D.; Van Puyveldeb, P.; Kruth, J.-P. Effect of PA12 powder reuse on coalescence behaviour and microstructure of SLS parts. Eur. Polym. J. 2017, 92, 250–262. [Google Scholar] [CrossRef]
- Ishikawa, T.; Nagai, S.; Kasai, N. Thermal behavior of alpha Nylon-12. J. Polym. Sci. Polym. Phys. 1980, 18, 1413–1419. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | d43 | d10 | d90 | SSAcal | SSABET |
|---|---|---|---|---|---|
| (µm) | (µm) | (µm) | (m2/cm3) | (m2/g) | |
| fresh PA 12 | 92.8 | 54.5 | 168.4 | 0.7010 | 6.7 |
| scrap PA 12 | 66.9 | 40.2 | 97.9 | 0.1057 | 0.7 |
| recycled PA 12 | 61.3 | 37.9 | 87.6 | 0.1158 | 0.4 |
| renewedPA 12 | 59.6 | 38.9 | 83.8 | 0.1183 | 2.2 |
| Samples | Width Mean (μm) | Standard Deviation | Length Mean (μm) | Standard Deviation |
|---|---|---|---|---|
| fresh PA12 | 43.2 | 16.14 | 60.07 | 21.43 |
| scrap PA12 | 38.91 | 14.05 | 48.93 | 18.71 |
| recycled PA 12 | 37.72 | 13.55 | 47.35 | 17.10 |
| renewed PA 12 | 40.35 | 13.38 | 53.06 | 17.47 |
| d (001) (nm) | d (200) (nm) | FWHM (001) (°2Θ) | FWHM (200) (°2Θ) | Intensity (001) | Lc W-H (nm) | |
|---|---|---|---|---|---|---|
| fresh PA12 | 0.4219 | 0.4004 | 0.48 | 1.66 | 6048 | 6.0 |
| scrap PA12 | 0.4206 | 0.4001 | 1.30 | 1.303 | 9275 | 4.2 |
| recycled PA12 | 0.4223 | 0.4006 | 1.15 | 1.72 | 7521 | 4.7 |
| renewed PA 12 | 0.4223 | 0.4007 | 0.47 | 1.70 | 7238 | 5.8 |
| Vibrational Frequency [cm−1] | Assignments |
|---|---|
| 3290 | ν(N–H) stretching |
| 3094 | Fermi resonance of ν(N–H) stretching |
| 2916 | ν(CH2) asymmetric stretching |
| 2847 | ν(CH2) symmetric stretching |
| 1638 | Amide-I (ν(C=O) stretching and ν(C–N) stretching) |
| 1561 | Amide-II (δ(N–H) bending and ν(C–N) stretching) |
| 1459 | δ(CH2) scissoring |
| 1368 | δ(CH2) twisting |
| 1268 | Amide-III (ν(C–N) stretching and δ(C=O) in-plane bending) |
| 1159 | Skeletal motion CO–NH |
| 1062 | Skeletal motion CO–NH |
| 948 | δ(CO-NH) in-plane bending |
| 721 | ρ(CH2) rocking |
| 621 | Amide-IV (δ(N–H) out-of-plane bending) |
| Fresh PA12 | Scrap PA12 | Recycled PA12 | Renewed PA12 | |
|---|---|---|---|---|
| melting point Tm [°C] | 185.6 | 187.2 | 186.5 | 185.4 |
| melting onset Tmonset [°C] | 178.0 | 179.7 | 178.7 | 176.9 |
| melting enthalpy ∆Hm [J/g] | 110.7 | 114.3 | 120.2 | 117.9 |
| crystallization point Tc [°C] | 155.3 | 152.6 | 156.3 | 156.6 |
| crystallization onset Tconset [°C] | 158.6 | 156.4 | 159.3 | 159.8 |
| crystallization enthalpy ∆Hc [J/g] | −57.9 | −50.6 | −62.0 | −59.6 |
| Crystallinity Χc [%] | 52.9 | 54.6 | 57.4 | 56.3 |
| sintering window [°C] | 19.4 | 23.3 | 19.4 | 17.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simha Martynková, G.; Slíva, A.; Kratošová, G.; Čech Barabaszová, K.; Študentová, S.; Klusák, J.; Brožová, S.; Dokoupil, T.; Holešová, S. Polyamide 12 Materials Study of Morpho-Structural Changes during Laser Sintering of 3D Printing. Polymers 2021, 13, 810. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13050810
Simha Martynková G, Slíva A, Kratošová G, Čech Barabaszová K, Študentová S, Klusák J, Brožová S, Dokoupil T, Holešová S. Polyamide 12 Materials Study of Morpho-Structural Changes during Laser Sintering of 3D Printing. Polymers. 2021; 13(5):810. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13050810
Chicago/Turabian StyleSimha Martynková, Gražyna, Aleš Slíva, Gabriela Kratošová, Karla Čech Barabaszová, Soňa Študentová, Jan Klusák, Silvie Brožová, Tomáš Dokoupil, and Sylva Holešová. 2021. "Polyamide 12 Materials Study of Morpho-Structural Changes during Laser Sintering of 3D Printing" Polymers 13, no. 5: 810. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13050810