
Microfibrillated Cellulose Grafted with Metacrylic Acid as a Modifier in Poly(3-hydroxybutyrate)

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Preparation of Microfibrillated Cellulose
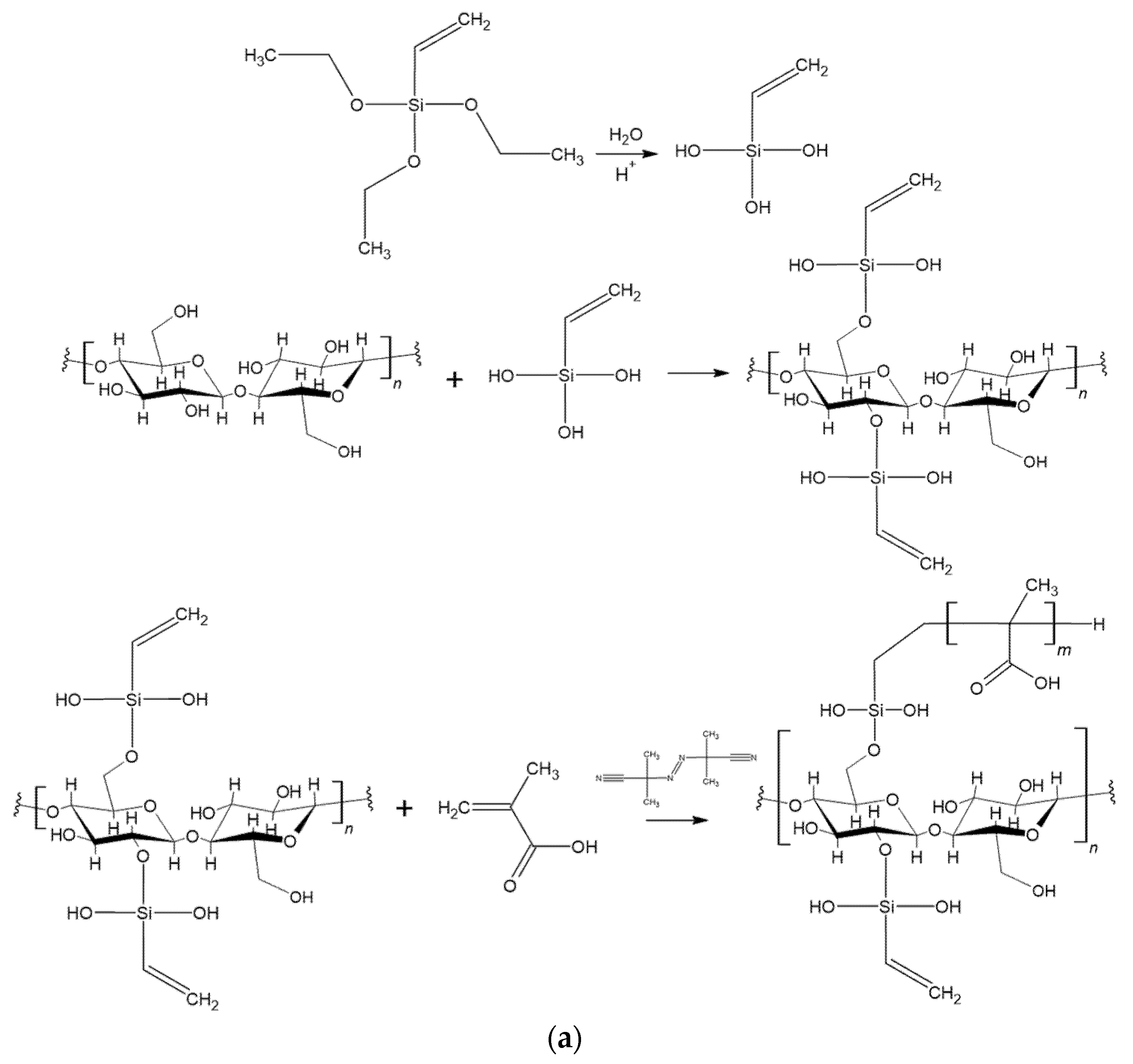
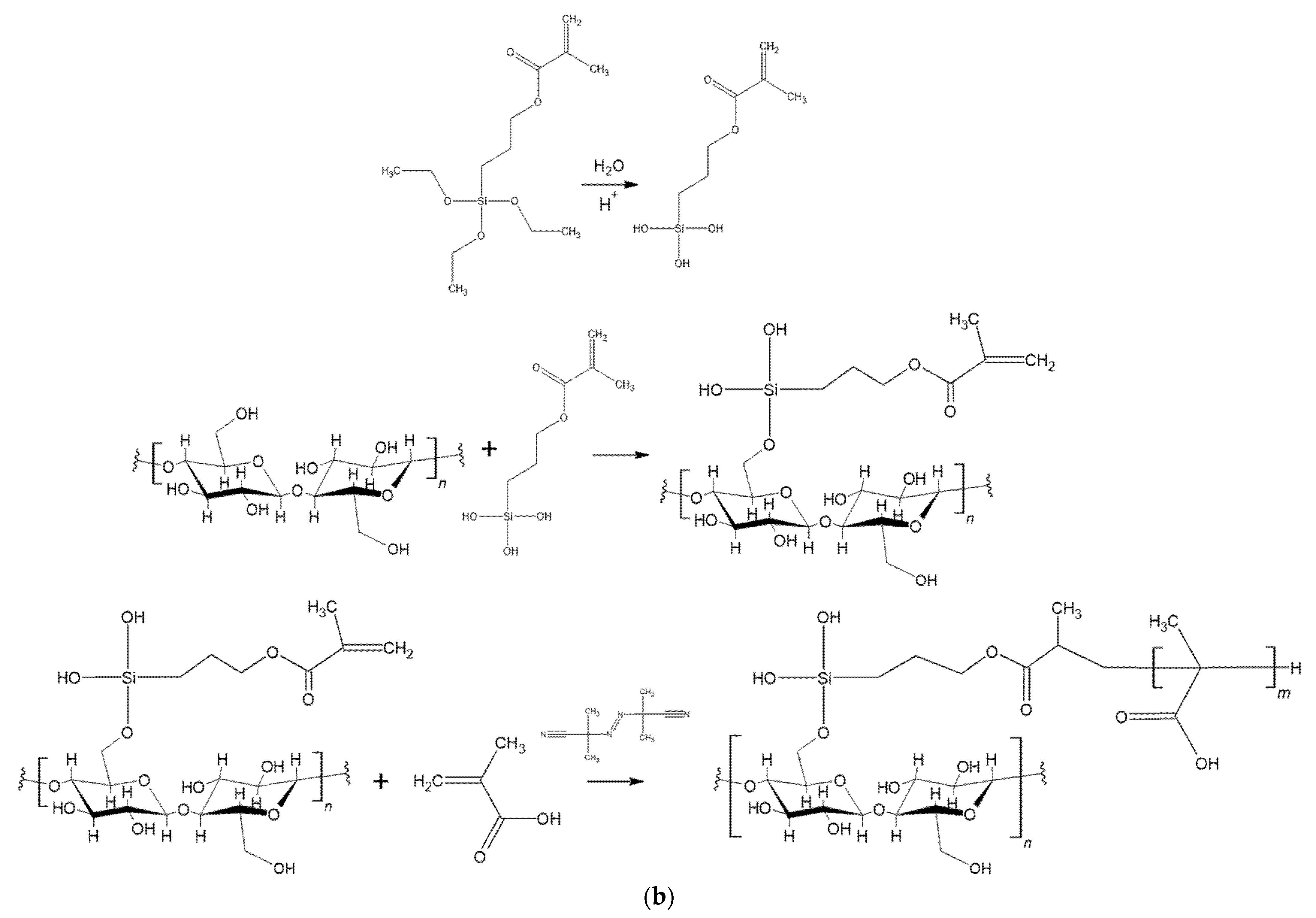
2.3. Silanization Reaction
2.4. Polymerization Reaction
2.5. Preparation of the PHB/Modified Cellulose Composite Films
2.6. Characterization
2.6.1. Fourier Transform Infrared Spectroscopy
2.6.2. Thermogravimetric Analysis
2.6.3. Differential Scanning Calorimetry (DSC)
2.6.4. Dynamic Mechanical Analysis (DMA)
2.6.5. Tensile Properties
2.6.6. Scanning Electron Microscopy (SEM)
2.6.7. Polarized Light Optical Microscopy
3. Results and Discussion
3.1. Morphological Investigation of MC Powder
3.2. Characterization of Modified Celluloses
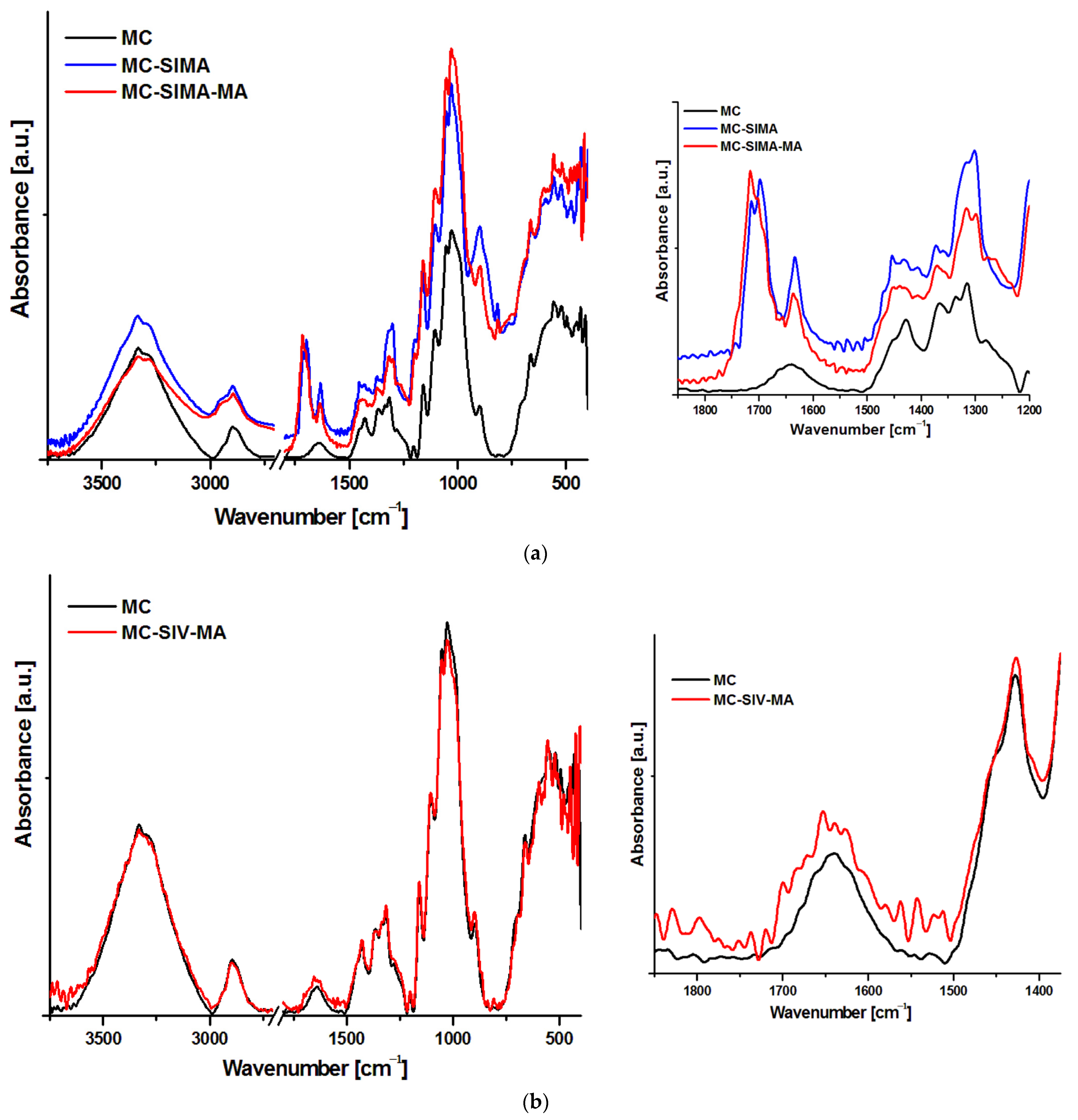
3.2.1. FTIR Analysis
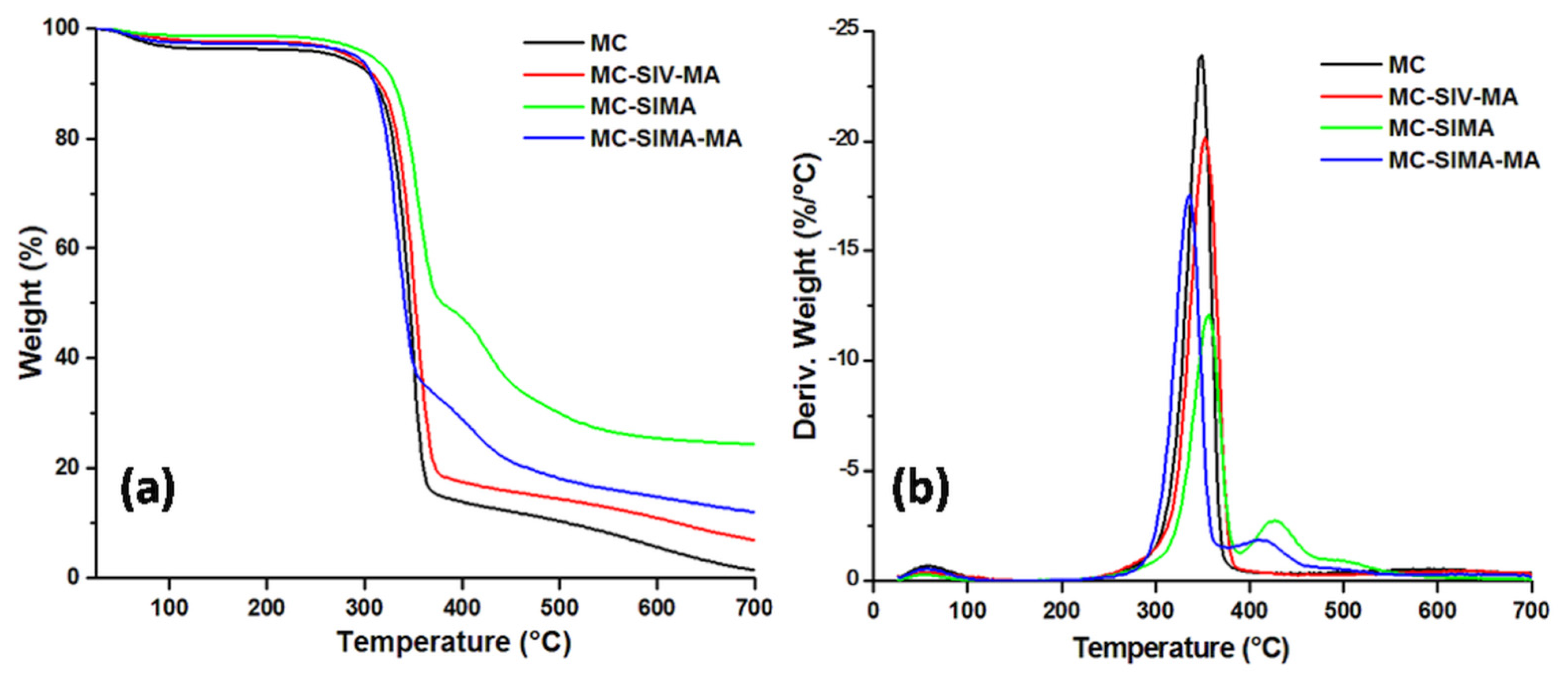
3.2.2. Thermogravimetric Analysis of Modified Celluloses
3.3. Characterization of Composites
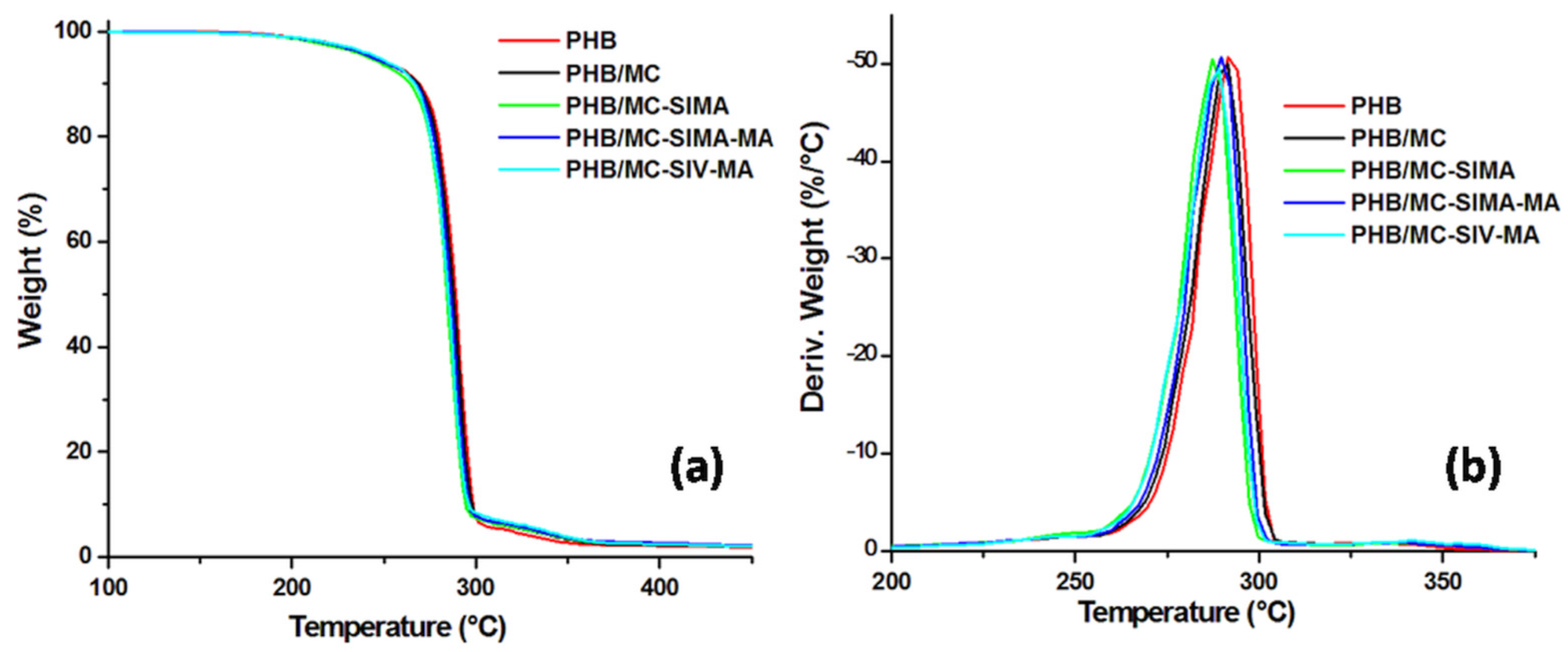
3.3.1. TGA Analysis
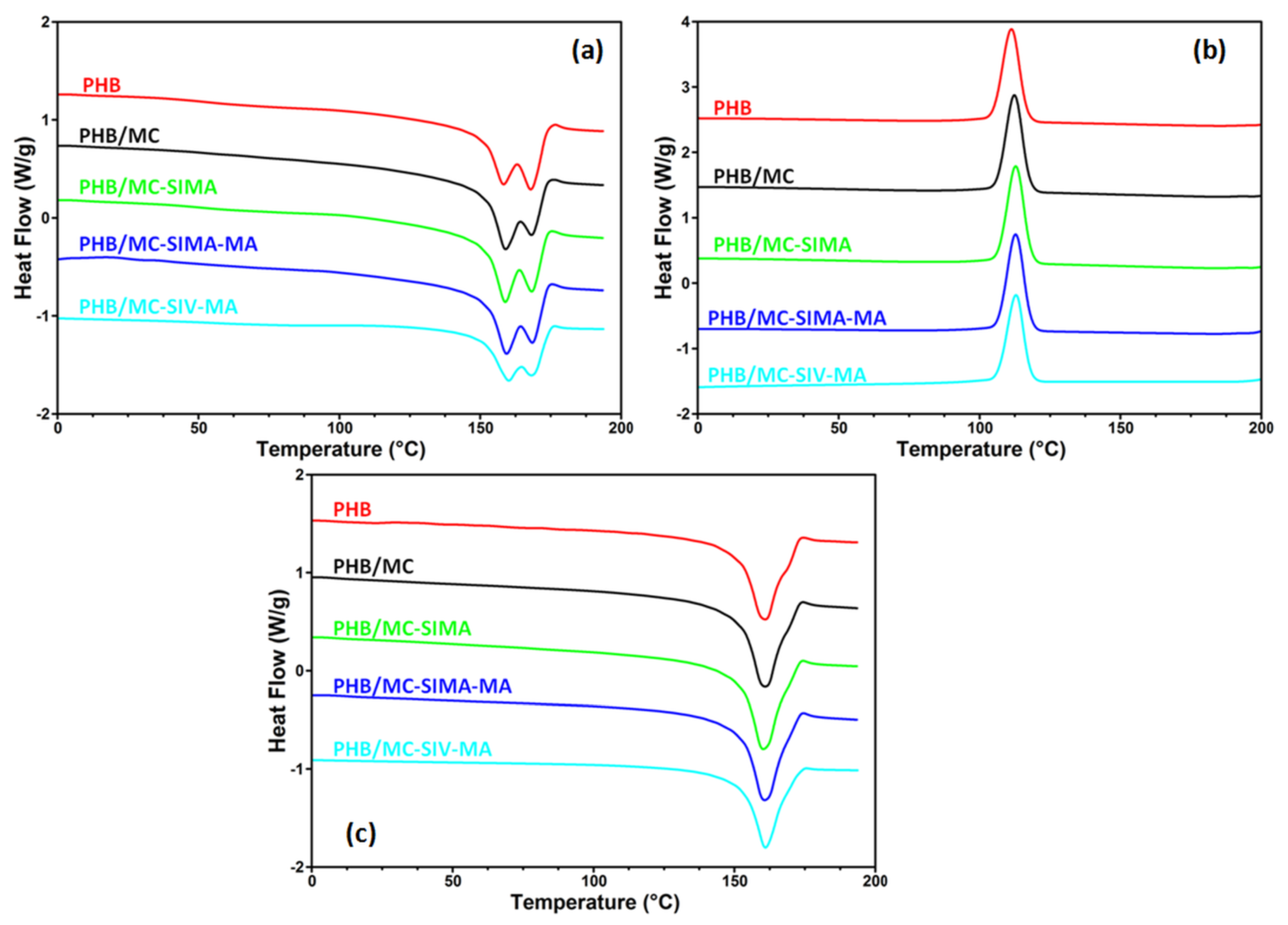
3.3.2. Differential Scanning Calorimetry
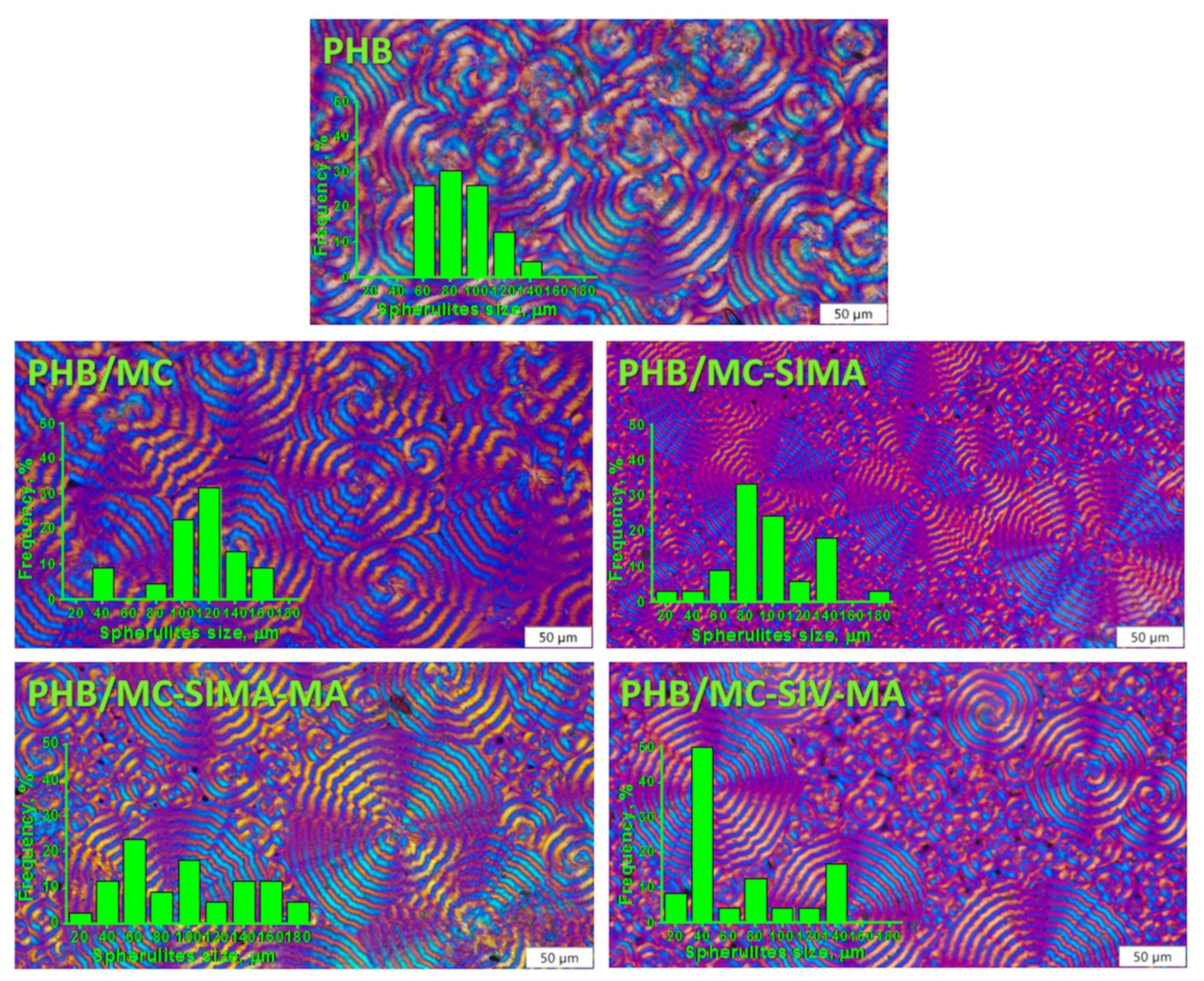
3.3.3. Polarized Optical Microscopy
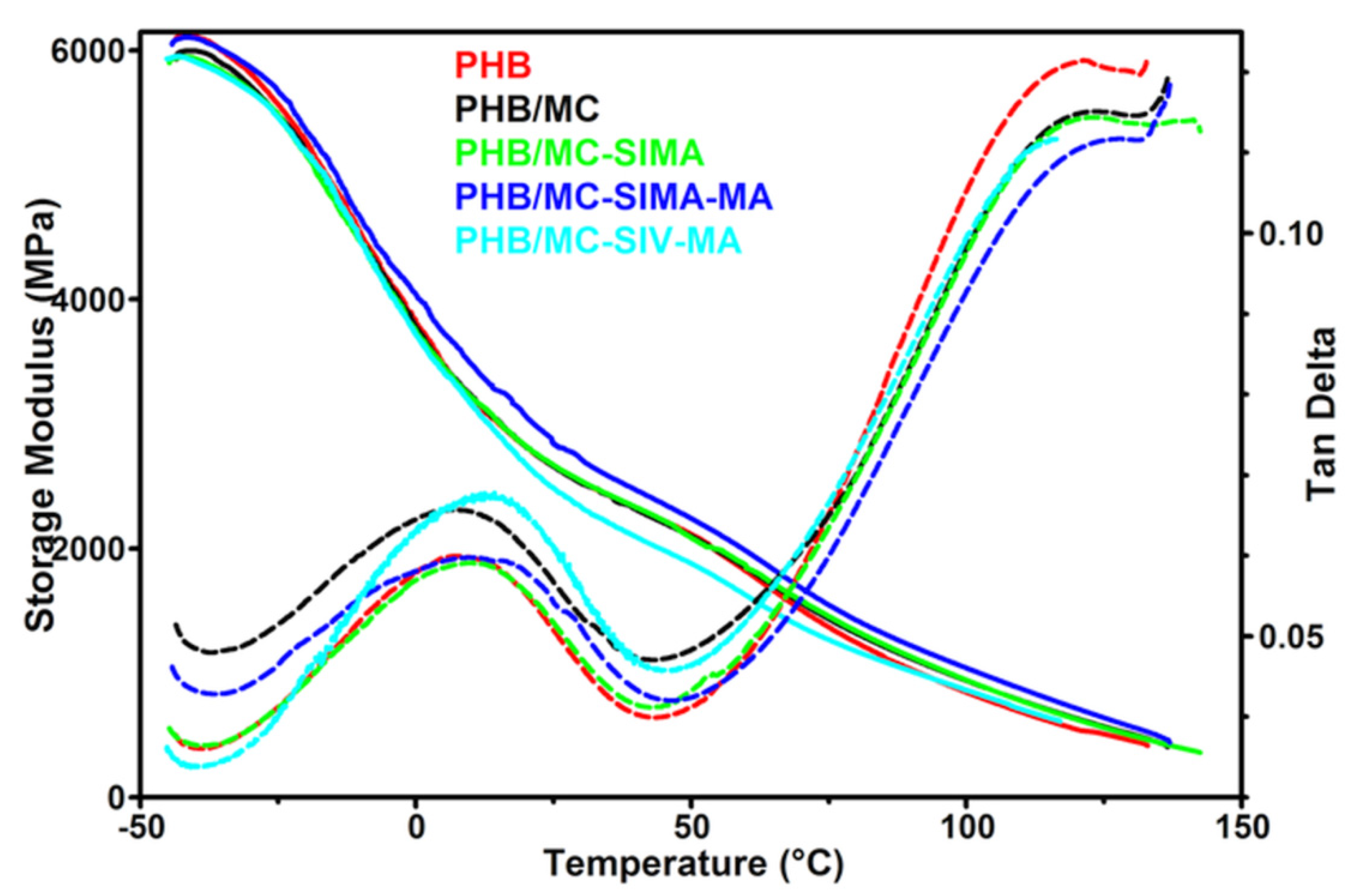
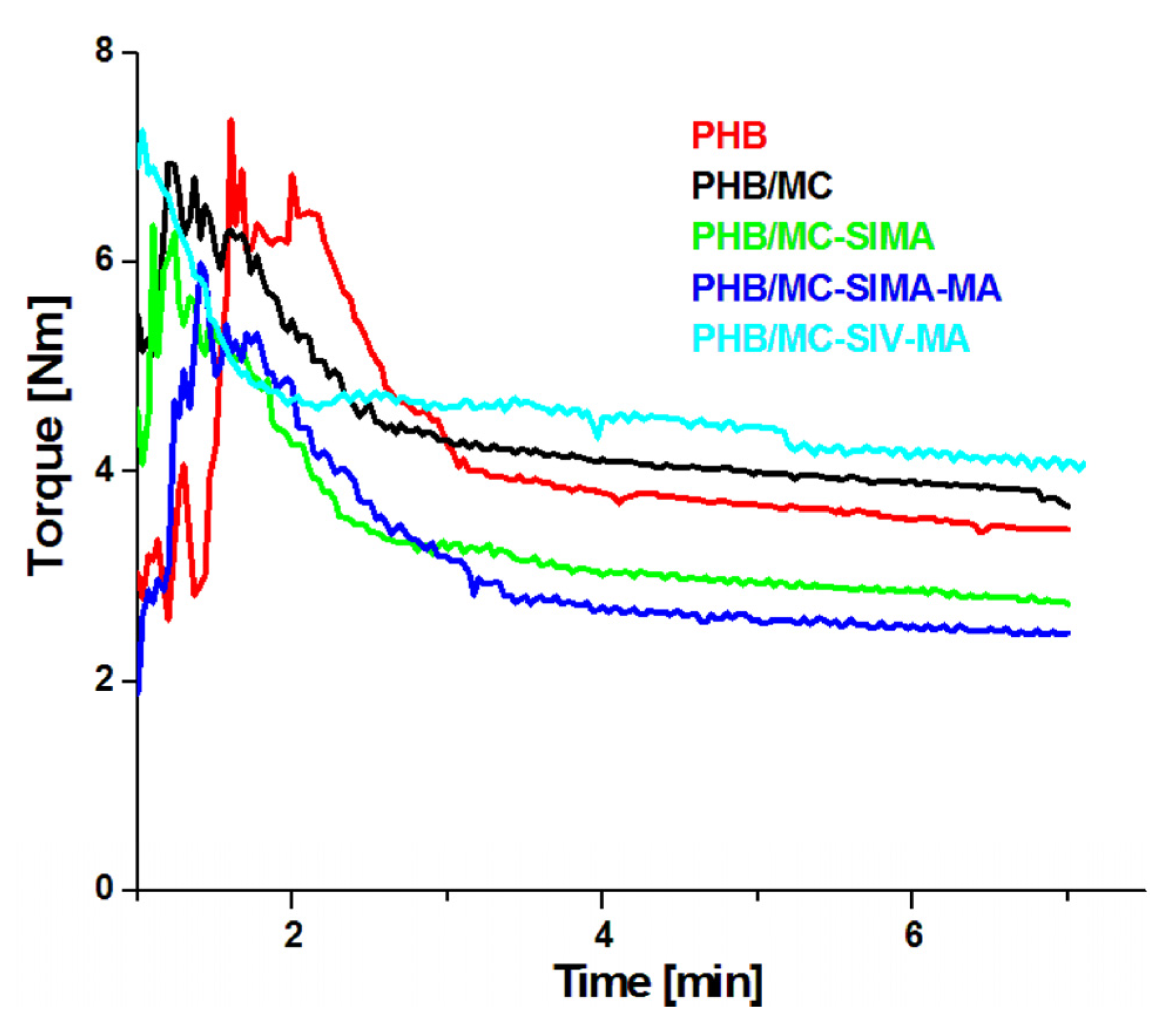
3.3.4. Dynamical Mechanical Analysis
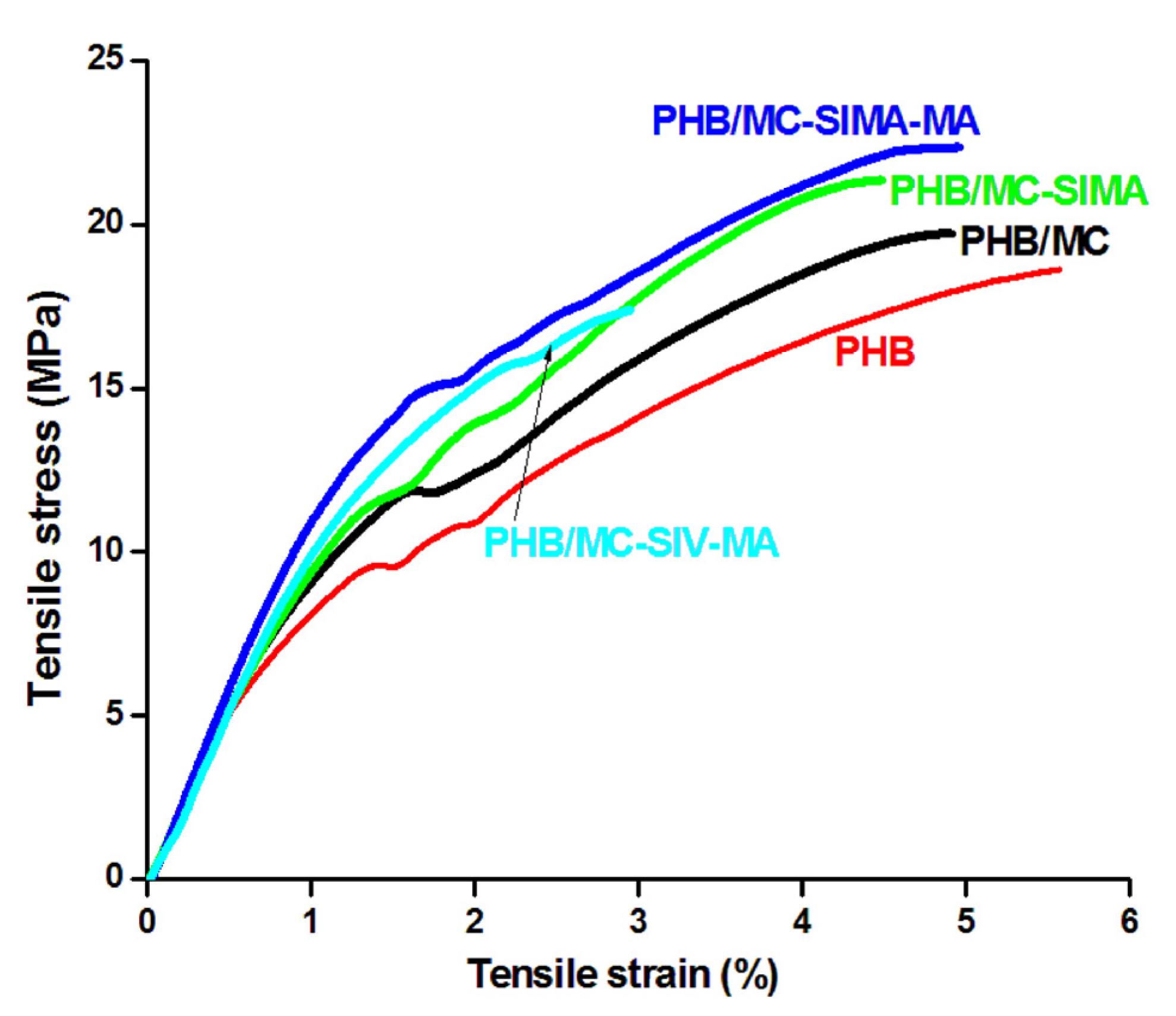
3.3.5. Tensile Properties of the Composites
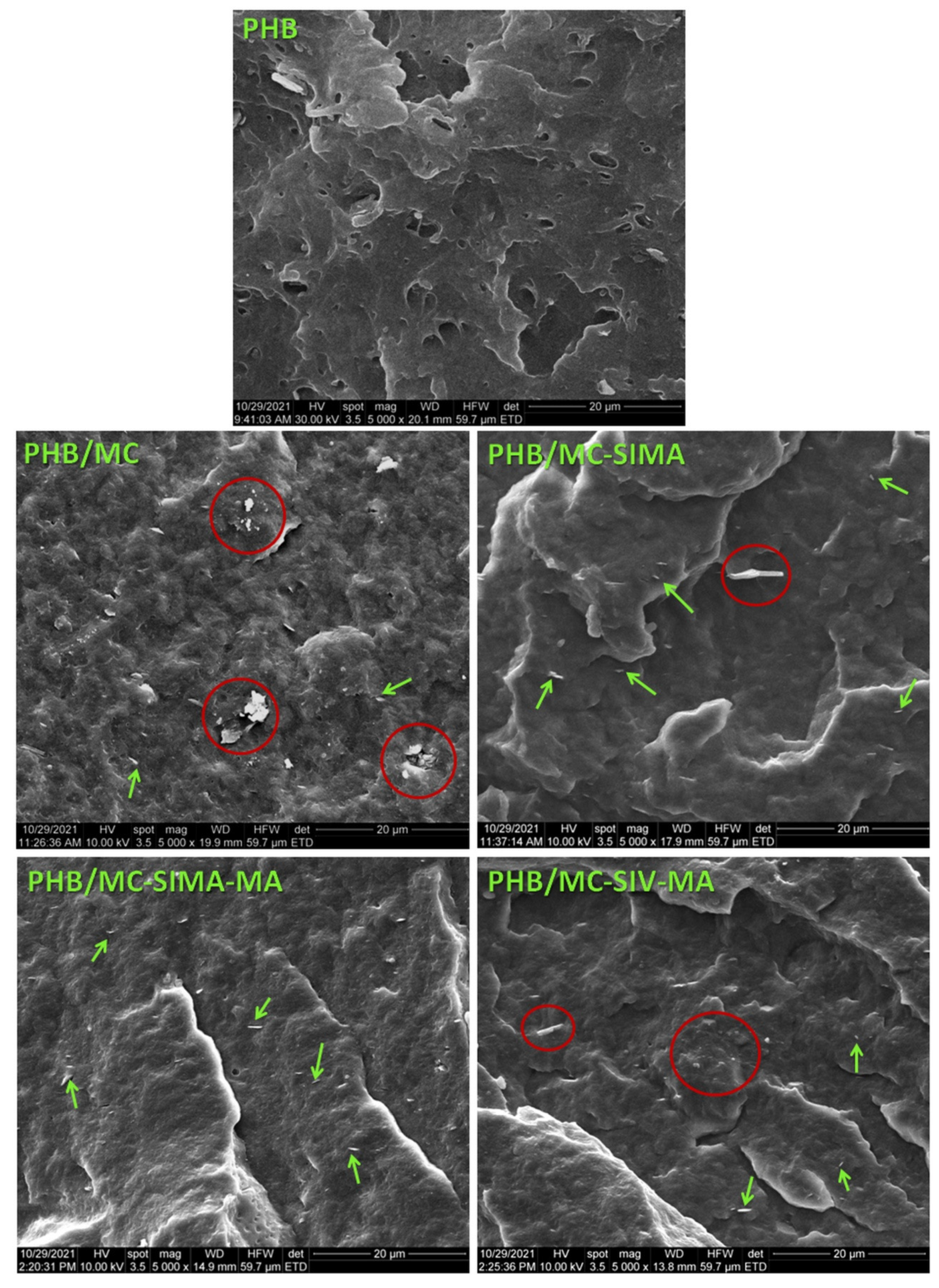
3.3.6. Morphological Investigation of Composites
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Friedlingstein, P.; Grasso, M.; Staiano, G.; Martorelli, M. Global carbon budget. Earth Syst. Sci. Data 2019, 11, 1783–1838. [Google Scholar] [CrossRef] [Green Version]
- Payne, J.; Mc Keown, P.; Jones, M.D. A circular economy approach to plastic waste. Polym. Degrad. Stab. 2019, 165, 170–181. [Google Scholar] [CrossRef]
- Khalid, M.Y.; Al Rashid, A.; Arif, Z.U.; Ahmed, W.; Arshad, H. Recent advances in nanocellulose-based different biomaterials: Types, properties, and emerging applications. J. Mater. Res. Technol. 2021, 14, 2601–2623. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Frone, A.N.; Ghiurea, M.; Chiulan, I. Influence of storage conditions on starch/PVA films containing cellulose nanofibers. Ind. Crops Prod. 2015, 70, 170–177. [Google Scholar] [CrossRef]
- Raza, Z.A.; Abid, S.; Banat, I.M. Polyhydroxyalkanoates: Characteristics, production, recent developments and applications. Int. Biodeter. Biodegr. 2018, 126, 45–56. [Google Scholar] [CrossRef]
- Kamravamanesh, D.; Pflügl, S.; Nischkauer, W.; Limbeck, A.; Lackner, M.; Herwig, C. Photosynthetic poly-β-hydroxybutyrate accumulation in unicellular cyanobacterium Synechocystis sp. PCC 6714. AMB Express 2017, 7, 143. [Google Scholar] [CrossRef]
- Yeo, J.C.C.; Muiruri, J.K.; Thitsartarn, W.; Li, Z.; He, C. Recent advances in the development of biodegradable PHB-based toughening materials: Approaches, advantages and applications. Mat. Sci. Eng. C 2018, 92, 1092–1116. [Google Scholar] [CrossRef]
- El-Hadi, A. Investigation of the effect of nanoclay type on the non-isothermal crystallization kinetics and morphology of poly(3(R)-hydroxybutyrate) PHB/clay nanocomposites. Polym. Bull. 2014, 71, 1449–1470. [Google Scholar] [CrossRef]
- Cai, Z.; Xiong, P.; He, S.; Zhu, C. Improved piezoelectric performances of highly orientated poly(β-hydroxybutyrate) electrospun nanofiber membrane scaffold blended with multiwalled carbon nanotubes. Mater. Lett. 2019, 240, 213–216. [Google Scholar] [CrossRef]
- Vadahanambi, S.; Lee, I.; Chun, H.; Park, H. Graphene reinforced biodegradable poly (3-hydroxybutyrate-co-4-hydroxybutyrate) nano-composites. Express Polym. Lett. 2013, 7, 320–328. [Google Scholar]
- Panaitescu, D.M.; Nicolae, C.A.; Gabor, A.R.; Trusca, R. Thermal and mechanical properties of poly(3-hydroxybutyrate) reinforced with cellulose fibers from wood waste. Ind. Crops Prod. 2020, 145, 112071. [Google Scholar] [CrossRef]
- Omran, A.A.B.; Mohammed, A.A.B.A.; Sapuan, S.M.; Ilyas, R.A.; Asyraf, M.R.M.; RahimianKoloor, S.S.; Petru, M. Micro- and nanocellulose in polymer composite materials: A review. Polymers 2021, 13, 231. [Google Scholar] [CrossRef] [PubMed]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose nanomaterials review: Structure, properties and nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef]
- Ten, E.; Turtle, J.; Bahr, D.; Jiang, L.; Wolcott, M. Thermal and mechanical properties of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/cellulose nanowhiskers composites. Polymer 2010, 51, 2652–2660. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Frone, A.N.; Chiulan, I.; Nicolae, C.A.; Trusca, R.; Ghiurea, M.; Gabor, A.R.; Mihailescu, M.; Casarica, A.; Lupescu, I. Role of bacterial cellulose and poly (3-hydroxyhexanoate-co-3-hydroxyoctanoate) in poly (3-hydroxybutyrate) blends and composites. Cellulose 2018, 25, 5569–5591. [Google Scholar] [CrossRef]
- Nagarajan, V.; Misra, M.; Mohanty, A.K. New engineered biocomposites from poly(3-hydroxybutyrate-co-3-hydroxyvalerate) (PHBV)/poly(butylene adipate-co-terephthalate) (PBAT) blends and switch grass: Fabrication and performance evaluation. Ind. Crops Prod. 2013, 42, 461–468. [Google Scholar] [CrossRef]
- Ghasemlou, M.; Daver, F.; Ivanova, E.P.; Habibi, Y.; Adhikari, B. Surface modifications of nanocellulose: From synthesis to high-performance nanocomposites. Prog. Polym. Sci. 2021, 119, 101418. [Google Scholar] [CrossRef]
- Oprea, M.; Panaitescu, D.M.; Nicolae, C.A.; Gabor, A.R.; Frone, A.N.; Raditoiu, V.; Trusca, R.; Casarica, A. Nanocomposites from functionalized bacterial cellulose and poly(3-hydroxybutyrate-co-3-hydroxyvalerate). Polym. Degrad. Stab. 2020, 179, 109203. [Google Scholar] [CrossRef]
- Panaitescu, D.M.; Vizireanu, S.; Stoian, S.A.; Nicolae, C.-A.; Gabor, A.R.; Damian, C.M.; Trusca, R.; Carpen, L.G.; Dinescu, G. Poly(3-hydroxybutyrate) modified by plasma and TEMPO-oxidized celluloses. Polymers 2020, 12, 1510. [Google Scholar] [CrossRef]
- Sánchez-Safont, E.L.; Aldureid, A.; Lagarón, J.M.; Cabedo, L.; Gámez-Pérez, J. Study of the compatibilization effect of different reactive agents in PHB/natural fiber-based composites. Polymers 2020, 12, 1967. [Google Scholar] [CrossRef]
- Chiu, H.J.; Chen, H.L.; Lin, T.L.; Lin, J.S. Phase structure of poly(3-hydroxy butyrate)/poly(vinyl acetate) blends probed by small-angle X-ray scattering. Macromolecules 1999, 32, 4969–4974. [Google Scholar] [CrossRef]
- An, Y.; Dong, L.; Li, G.; Mo, Z.; Feng, Z. Miscibility, crystallizationkinetics, and morphology of poly(β-hydroxybutyrate) andpoly(methylacrylate) blends. J. Polym. Sci. B Polym. Phys. 2000, 38, 1860–1867. [Google Scholar] [CrossRef]
- Vitta, S.B.; Stahel, E.P.; Stannett, V.T. The preparation and properties of acrylic and methacrylic acid grafted cellulose prepared by ceric ion initiation. Part I. Preparation of the grafted cellulose. J. Macromol. Sci. A 1985, 22, 579–590. [Google Scholar] [CrossRef]
- Eldin, M.S.M.; Rahman, S.A.; Fawal, G.F.E. Preparationandcharacterization of grafted cellophane membranes for affinity separation of His-tagChitinase. Adv. Polym. Technol. 2011, 30, 191–202. [Google Scholar] [CrossRef]
- Barham, P.J.; Keller, A.; Otun, E.L.; Holmer, P.A. Crystallization and morphology of a bacterial thermoplastic: Poly-3-hydroxybutyrate. J. Mater. Sci. 1984, 19, 2781. [Google Scholar] [CrossRef]
- Ovalle-Serrano, S.A.; Gómez, F.N.; Blanco-Tirado, C.; Combariza, M.Y. Isolation and characterization of cellulose nanofibrils from Colombian Fique decortication by-products. Carbohydr. Polym. 2018, 189, 169–177. [Google Scholar] [CrossRef]
- Pantoja, M.; Díaz-Benito, B.; Velasco, F.; Abenojar, J.; del Real, J.C. Analysis of hydrolysis process of γ-methacryloxypropyltrimethoxysilane and its influence on the formation of silane coatings on 6063 aluminum alloy. Appl. Surf. Sci. 2009, 255, 6386–6390. [Google Scholar] [CrossRef]
- Abdelmouleh, M.; Boufi, S.; Belgacem, M.N.; Dufresne, A. Short natural-fibre reinforced polyethylene and natural rubber composites: Effect of silane coupling agents and fibres loading. Compos. Sci. Technol. 2007, 67, 1627–1639. [Google Scholar] [CrossRef]
- Lumbreras-Aguayo, A.; Meléndez-Ortiz, H.I.; Puente-Urbina, B.; Alvarado-Canché, C.; Ledezma, A.; Romero-García, J.; Betancourt-Galindo, R. Poly(methacrylic acid)-modified medical cotton gauzes with antimicrobial and drug delivery properties for their use as wound dressings. Carbohydr. Polym. 2019, 205, 203–210. [Google Scholar] [CrossRef]
- Kakko, T.; King, A.W.T.; Kilpeläinen, I. Homogenous esterification of cellulose pulp in DBNH.OAc. Cellulose 2017, 24, 5341–5354. [Google Scholar] [CrossRef] [Green Version]
- Frone, A.N.; Panaitescu, D.M.; Chiulan, I.; Nicolae, C.A.; Casarica, A.; Gabor, A.R.; Trusca, R.; Damian, C.M.; Purcar, V.; Alexandrescu, E.; et al. Surface Treatment of Bacterial Cellulose in Mild, Eco-Friendly Conditions. Coatings 2018, 8, 221. [Google Scholar] [CrossRef] [Green Version]
- Castro Cabrera, I.; Berlioz, S.; Fahs, A.; Louarn, G.; Carriere, P. Chemical functionalization of nano fibrillated cellulose by glycidyl silane coupling agents: A grafted silane network characterization study. Int. J. Biol. Macromol. 2020, 165, 1773–1782. [Google Scholar] [CrossRef]
- Sarmento, V.H.V.; Schiavetto, M.G.; Hammer, P.; Benedetti, A.V.; Fugivara, C.S.; Suegama, P.H.; Pulcinelli, S.H.; Santilli, C.V. Corrosion protection of stainless steel by polysiloxane hybrid coatings prepared using the sol-gel process. Surf. Coat. Technol. 2010, 204, 2689–2701. [Google Scholar] [CrossRef]
- Srithep, Y.; Ellingham, T.; Peng, J.; Sabo, R.; Clemons, C.; Turng, L.S.; Pilla, S. Melt compounding of poly (3-hydroxybutyrate-co-3-hydroxyvalerate)/nanofibrillated cellulose nanocomposites. Polym. Degrad. Stab. 2013, 98, 1439–1449. [Google Scholar] [CrossRef]
- Saïdi, L.; Vilela, C.; Oliveira, H.; Silvestre, A.J.D.; Freire, C.S.R. Poly(N-methacryloyl glycine)/nanocellulose composites as pH-sensitive systems for controlled release of diclofenac. Carbohydr. Polym. 2017, 169, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Seoane, I.T.; Cerrutti, P.; Vazquez, A.; Cyras, V.P.; Manfredi, L.B. Ternary nanocomposites based on plasticized poly(3-hydroxybutyrate) and nanocellulose. Polym. Bull. 2019, 76, 967–988. [Google Scholar] [CrossRef]
- Iggui, K.; Le Moigne, N.; Kaci, M.; Cambe, S.; Degorce-Dumas, J.R.; Bergeret, A. A biodegradation study of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)/organoclay nanocomposites in various environmental conditions. Polym. Degrad. Stab. 2015, 119, 77–86. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | MC | MC-SIMA | MC-SIMA-MA | MC-SIV-MA |
|---|---|---|---|---|
| T5%, °C | 270.7 | 305.1 | 290.6 | 284.5 |
| Tmax, °C | 348.7 | 356.5 | 335.5 | 353.2 |
| WL200°C, % | 3.8 | 1.4 | 2.7 | 2.5 |
| Residue at 700 °C, % | 1.4 | 24.4 | 12.0 | 6.8 |
| Composites | PHB | PHB/MC | PHB/MC-SIMA | PHB/MC-SIMA-MA | PHB/MC-SIV-MA |
|---|---|---|---|---|---|
| T5%, °C | 246.1 | 245.5 | 242.4 | 244.5 | 246.8 |
| Tmax, °C | 292.4 | 290.9 | 287.9 | 290 | 288.6 |
| Residue at 700 °C, % | 1.3 | 1.3 | 1.7 | 2.0 | 1.3 |
| Composites | First Heating | Cooling | Second Heating | Xc (%) | |||
|---|---|---|---|---|---|---|---|
| Tm1(°C) | ΔHm1 (J/g) | Tc (°C) | ΔHc (J/g) | Tm2 (°C) | ΔHm2 (J/g) | ||
| PHB | 158.4/168.0 | 67.3 | 111.3 | 62.5 | 160.9 | 69.6 | 47.7 |
| PHB/MC | 159.0/168.2 | 67.3 | 112.3 | 64.2 | 160.9 | 72.3 | 50.3 |
| PHB/MC-SIMA | 158.9/168.2 | 69.1 | 112.8 | 64.9 | 160.2 | 73.6 | 51.4 |
| PHB/MC-SIMA-MA | 159.4/168.5 | 67.8 | 112.8 | 64.2 | 160.7 | 72.5 | 50.7 |
| PHB/MC-SIV-MA | 160.1/168.2 | 56.2 | 113.0 | 57.2 | 161.0 | 61.5 | 42.9 |
| Composites | PHB | PHB/MC | PHB/MC-SIMA | PHB/MC-SIMA-MA | PHB/MC-SIV-MA |
|---|---|---|---|---|---|
| E’−25°C, MPa | 5569 | 5484 | 5488 | 5674 | 5460 |
| E’0°C, MPa | 3817 | 3783 | 3741 | 4041 | 3722 |
| E’25°C, MPa | 2660 | 2659 | 2671 | 2844 | 2486 |
| E’50°C, MPa | 2116 | 2099 | 2087 | 2242 | 1880 |
| E’100°C, MPa | 844 | 938 | 943 | 1040 | 863 |
| Tg, °C | 6.8 | 6.9 | 9.3 | 10.9 | 12.2 |
| tan δ | 0.060 | 0.066 | 0.059 | 0.059 | 0.067 |
| C | - | 0.886 | 0.882 | 0.827 | 0.959 |
| Composites | PHB | PHB/MC | PHB/MC-SIMA | PHB/MC-SIMA-MA | PHB/MC-SIV-MA |
|---|---|---|---|---|---|
| Elongation at break, % | 5.3 ± 0.6 | 4.9 ± 0.4 | 4.5 ± 0.6 | 5.0 ± 0.2 | 3.0 ± 0.5 |
| Tensile strength at break, MPa | 18.7 ± 1.9 | 19.8 ± 1.6 | 21.1 ± 0.7 | 22.0 ± 0.3 | 17.4 ± 2.0 |
| Young’s modulus, MPa | 868 ± 58 | 954 ± 42 | 946 ± 61 | 1116 ± 12 | 966 ± 17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popa, M.S.; Frone, A.N.; Radu, I.C.; Stanescu, P.O.; Truşcă, R.; Rădiţoiu, V.; Nicolae, C.A.; Gabor, A.R.; Panaitescu, D.M. Microfibrillated Cellulose Grafted with Metacrylic Acid as a Modifier in Poly(3-hydroxybutyrate). Polymers 2021, 13, 3970. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13223970
Popa MS, Frone AN, Radu IC, Stanescu PO, Truşcă R, Rădiţoiu V, Nicolae CA, Gabor AR, Panaitescu DM. Microfibrillated Cellulose Grafted with Metacrylic Acid as a Modifier in Poly(3-hydroxybutyrate). Polymers. 2021; 13(22):3970. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13223970
Chicago/Turabian StylePopa, Marius Stelian, Adriana Nicoleta Frone, Ionut Cristian Radu, Paul Octavian Stanescu, Roxana Truşcă, Valentin Rădiţoiu, Cristian Andi Nicolae, Augusta Raluca Gabor, and Denis Mihaela Panaitescu. 2021. "Microfibrillated Cellulose Grafted with Metacrylic Acid as a Modifier in Poly(3-hydroxybutyrate)" Polymers 13, no. 22: 3970. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13223970