
Tuning High and Low Temperature Foaming Behavior of Linear and Long-Chain Branched Polypropylene via Partial and Complete Melting


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
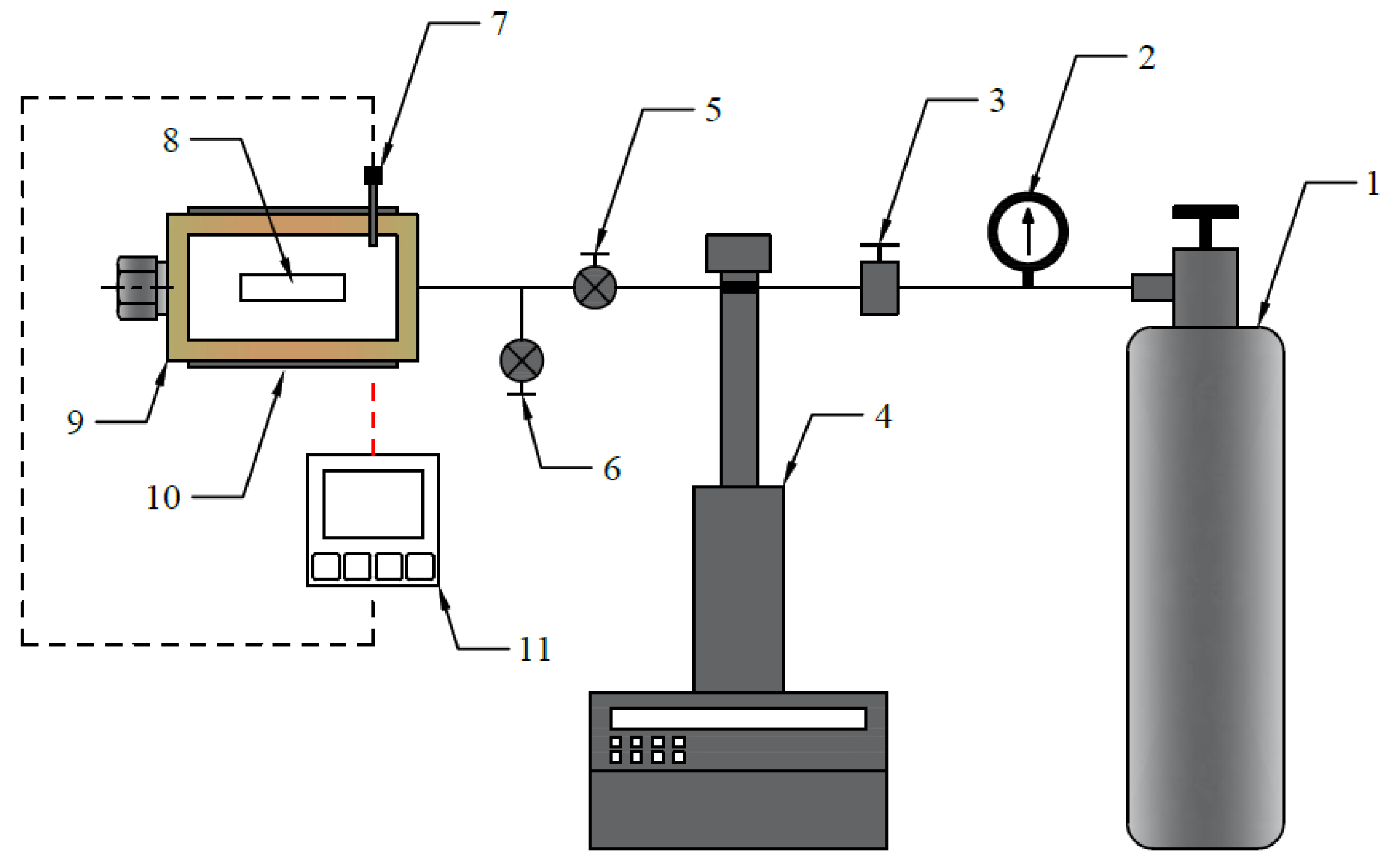
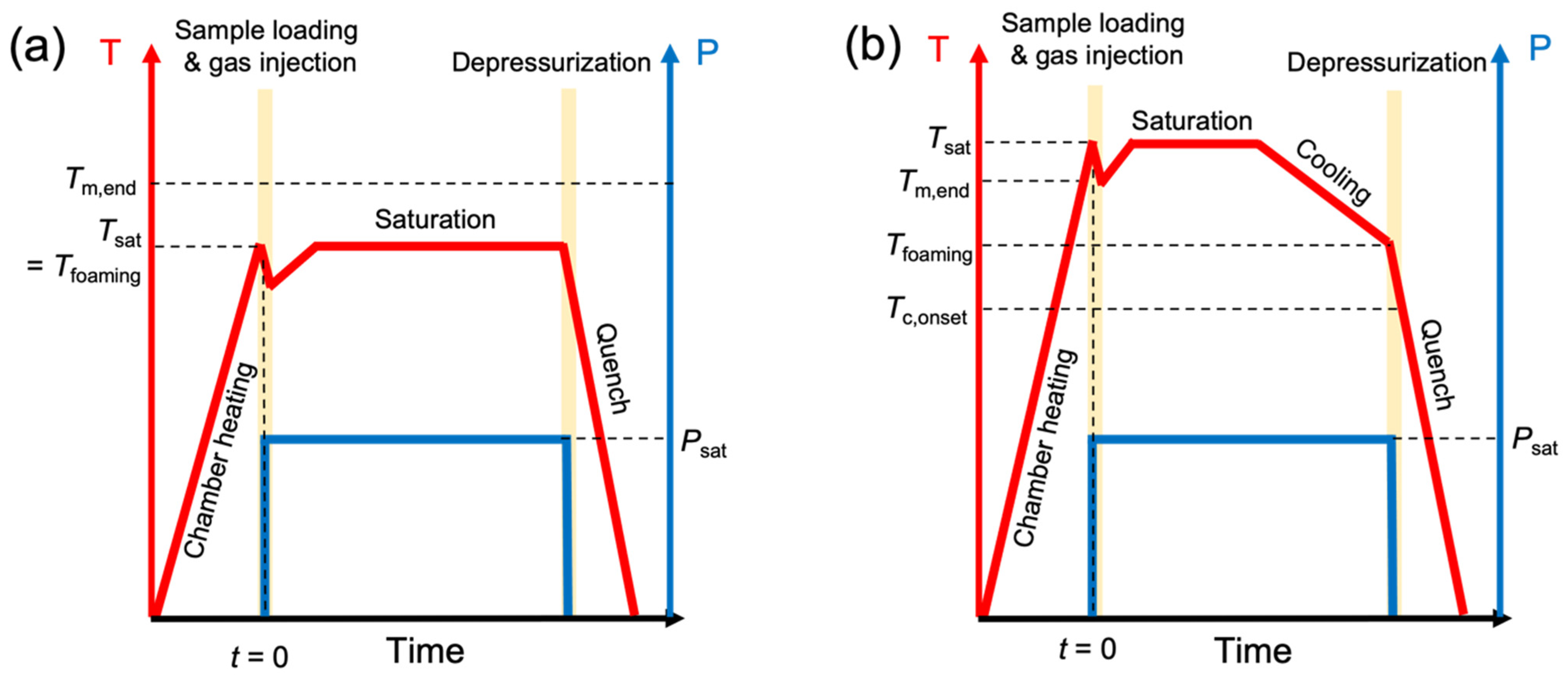
2.2. Batch Foaming Test Protocol
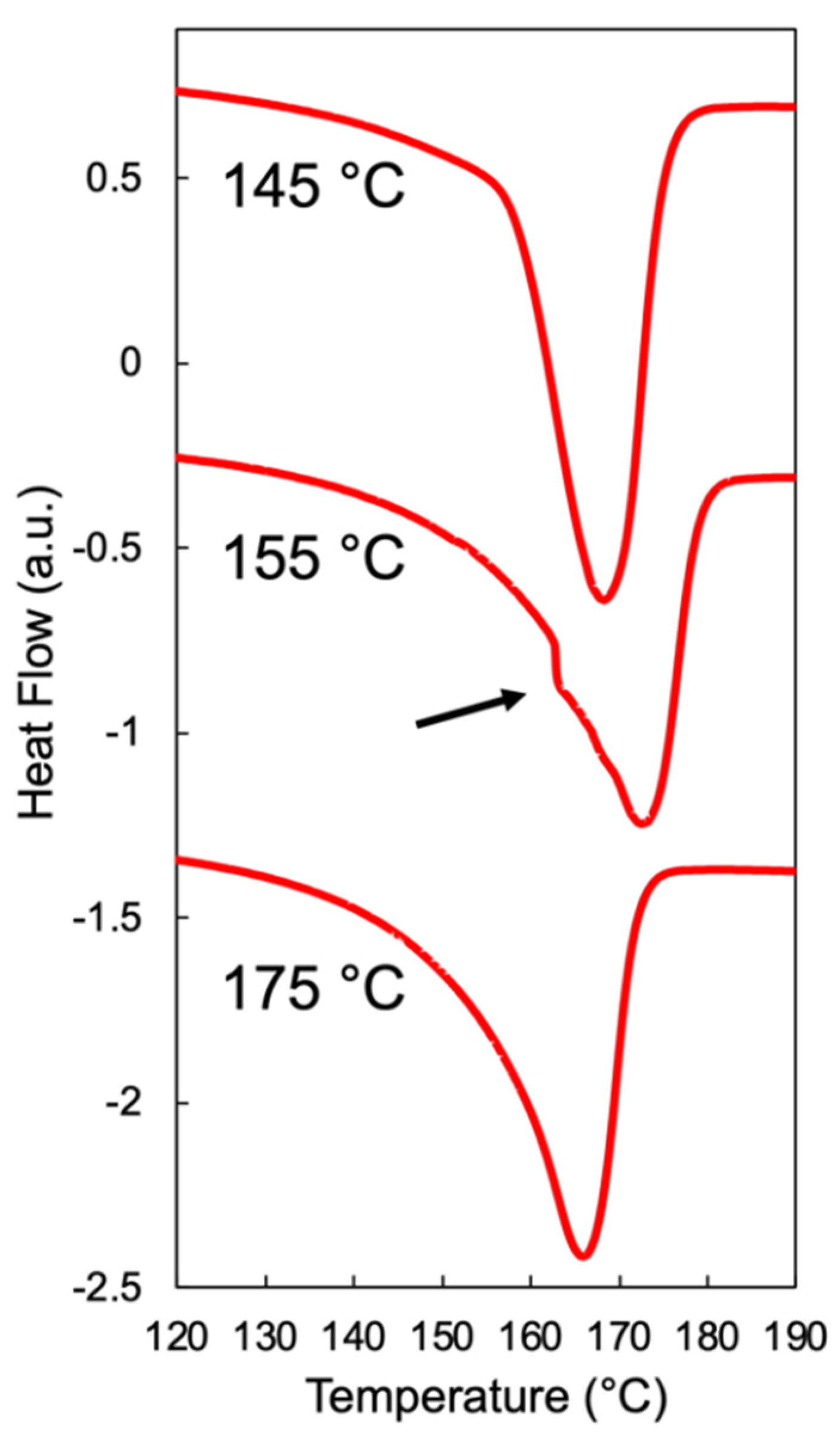
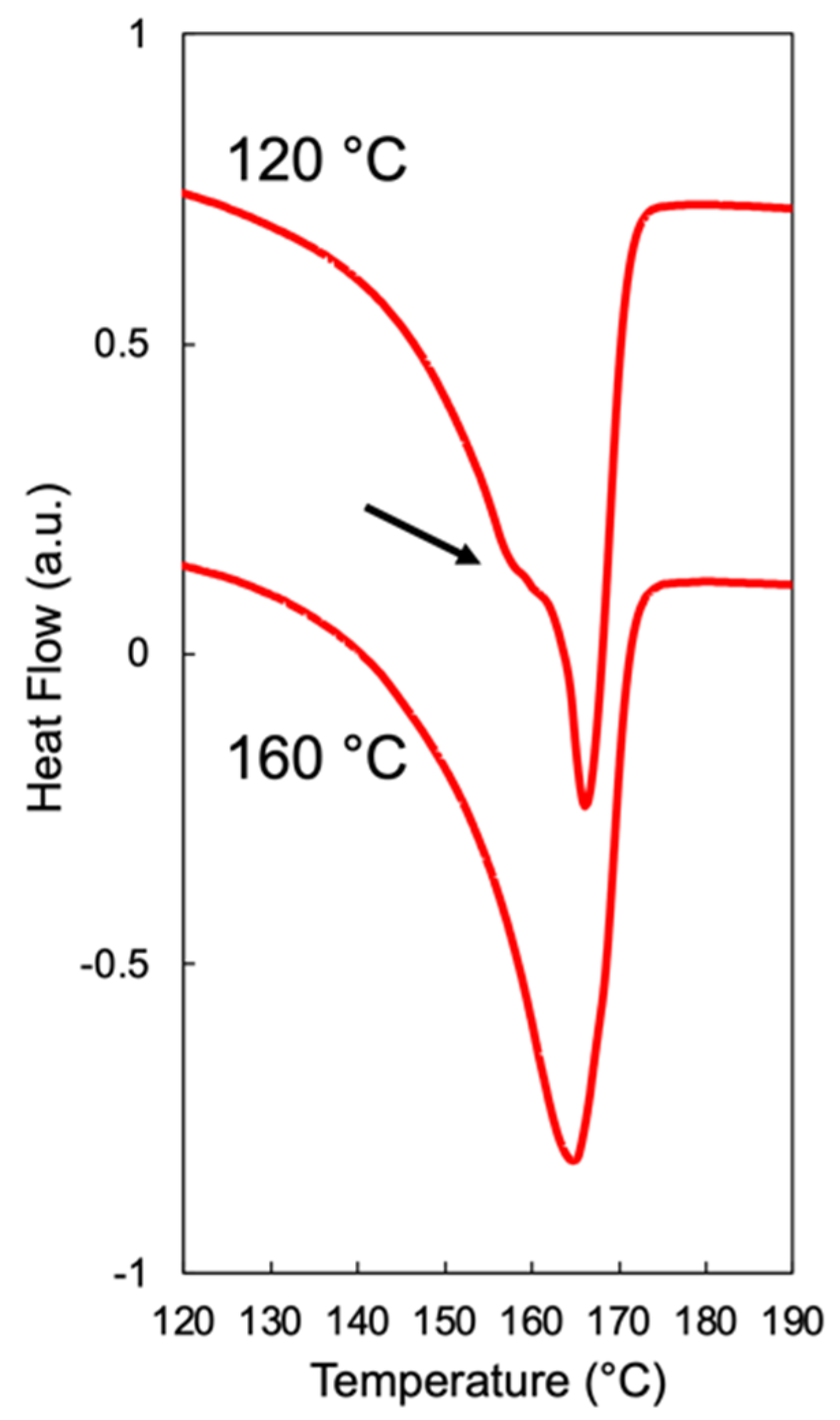
2.3. High-Pressure Differential Scanning Calorimetry
2.4. Foam Characterization
3. Results
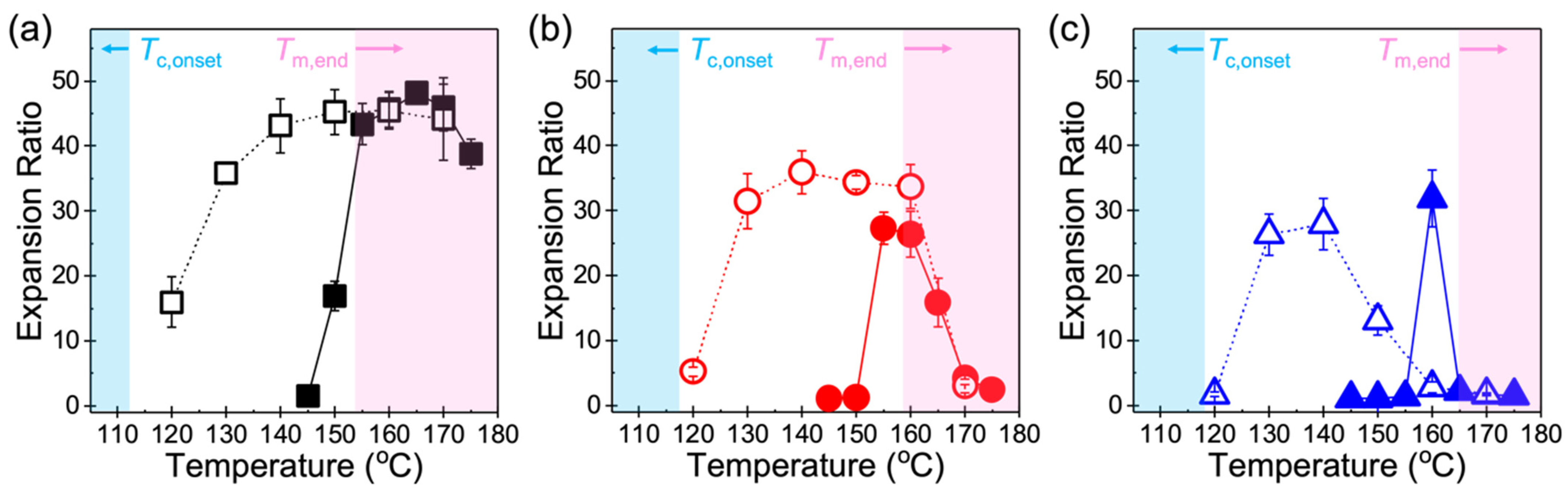
3.1. Volume Expansion Ratio
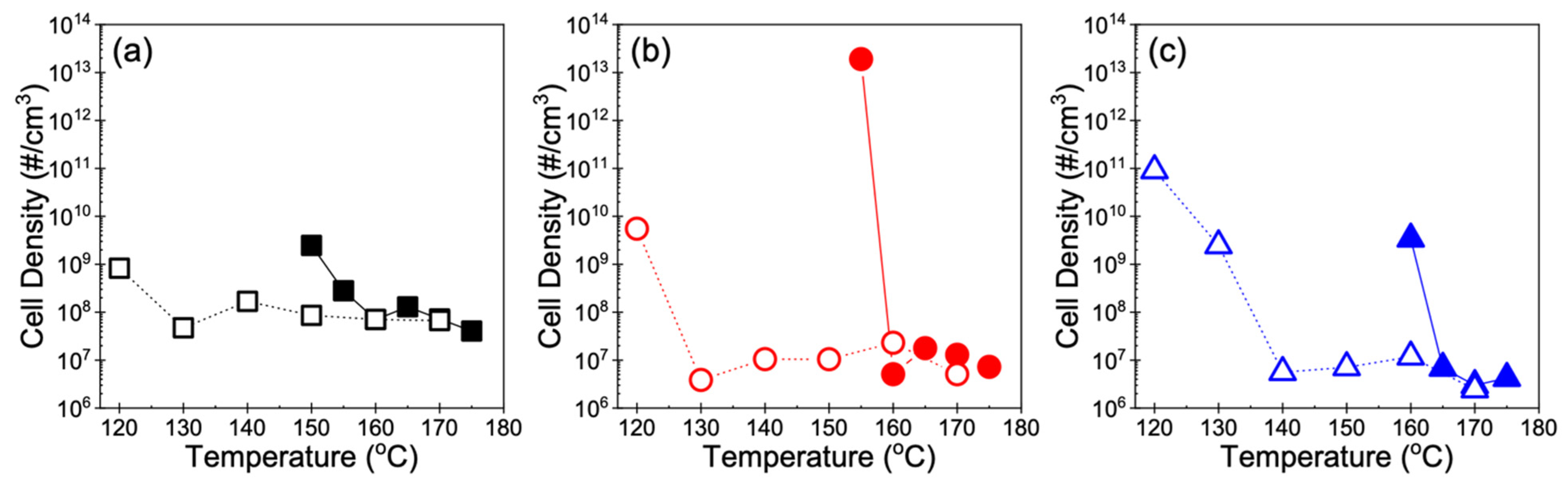
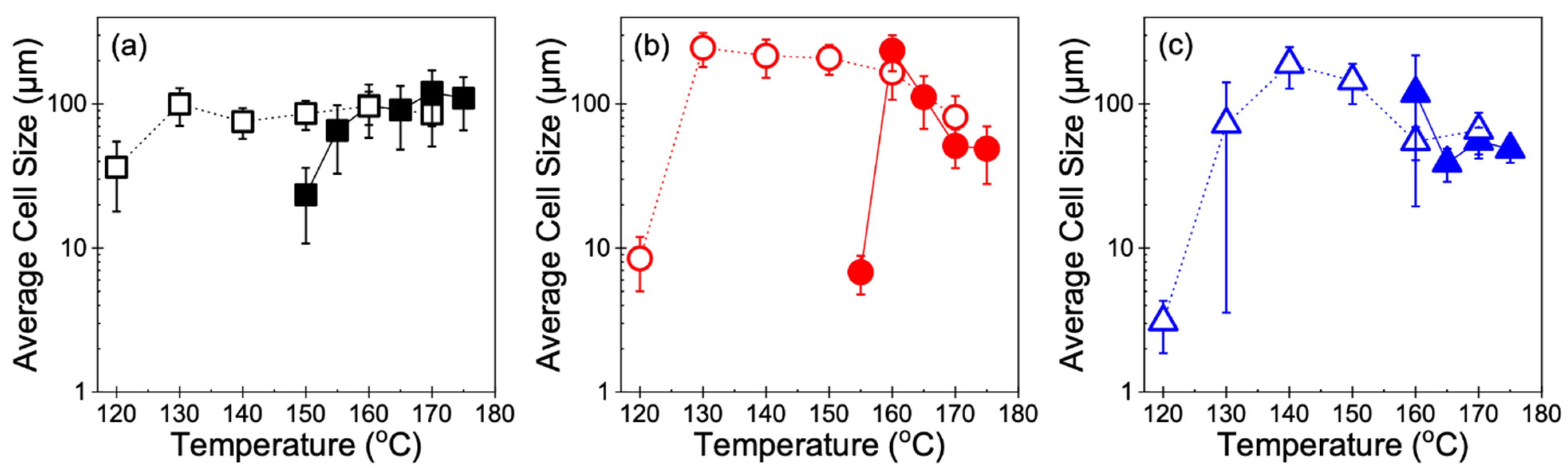
3.2. Cell Morphology
4. Discussion
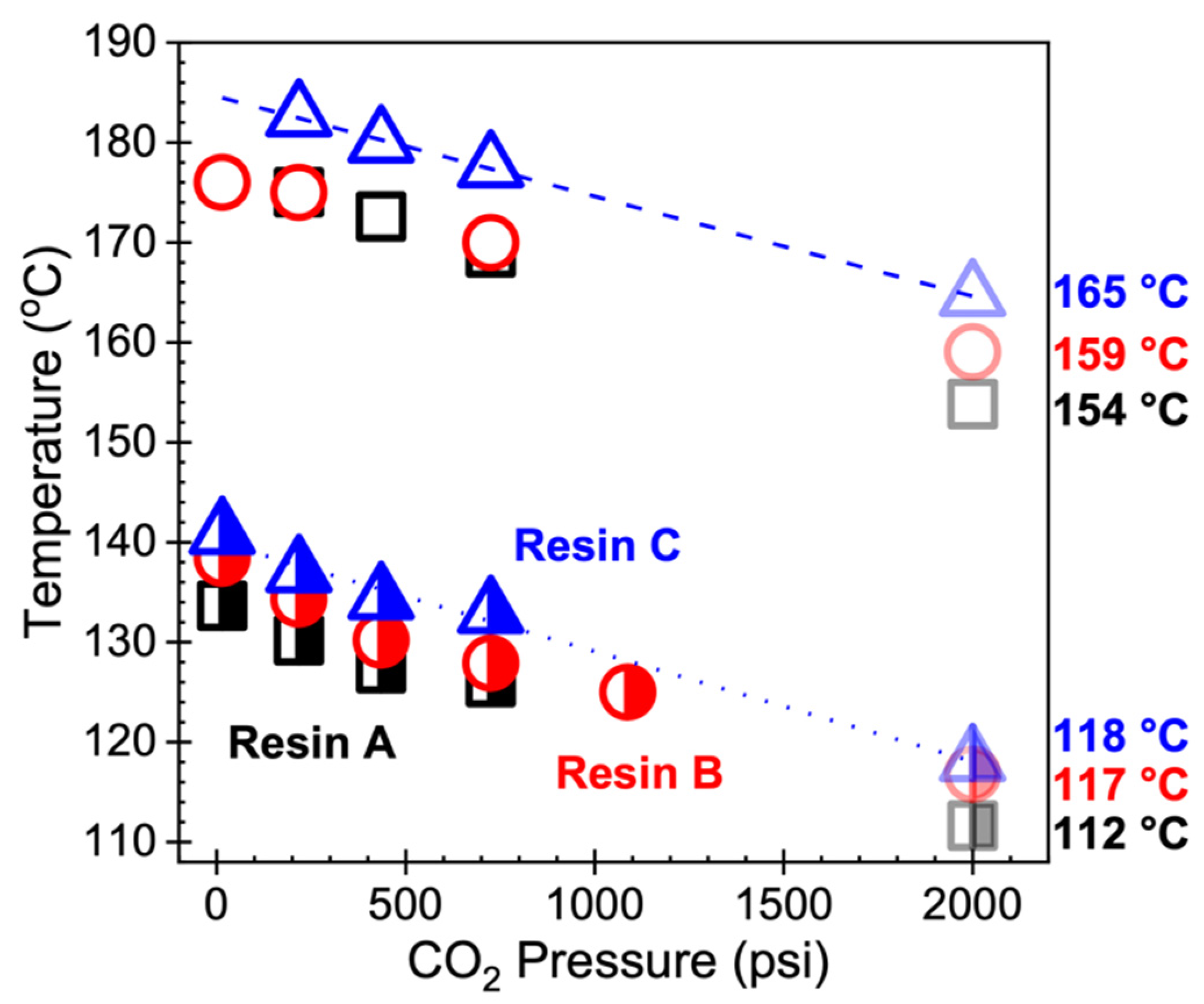
4.1. Foamability at Low Temperatures: Effect of Crystallization Onset
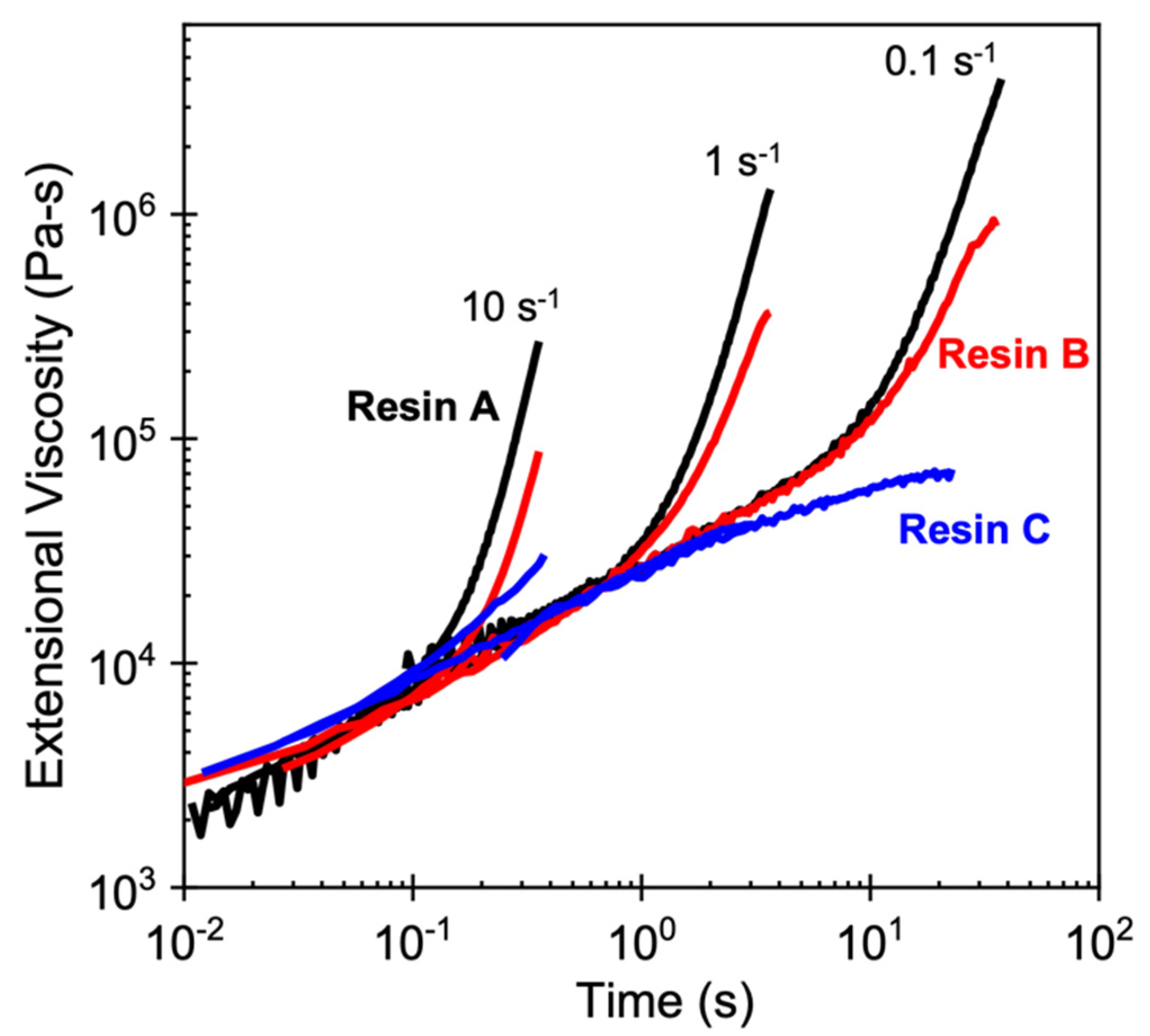
4.2. Foamability at High Temperatures: Effect of Strain Hardening
4.3. Other Perspectives on Foamability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rachtanapun, P.; Matuana, L.M.; Susan, E.S. Relationship Between Cell Morphology and Impact Strength of Microcellular Foamed High-Density Polyethylene/Polypropylene Blends. Polym. Eng. Sci. 2004, 44, 1551–1560. [Google Scholar] [CrossRef]
- Liu, S.; Duvigneau, J.; Vancso, G.J. Nanocellular polymer foams as promising high performance thermal insulation materials. Eur. Polym. J. 2015, 65, 33–45. [Google Scholar] [CrossRef]
- Sun, X.; Liang, W. Cellular structure control and sound absorption of polyolefin microlayer sheets. Compos. B Eng. 2015, 87, 21–26. [Google Scholar] [CrossRef]
- Mohebbi, A.; Mighri, F.; Ajji, A.; Rodrigue, D. Current Issues and Challenges in Polypropylene Foaming: A Review. Cell. Polym. 2015, 34, 299–338. [Google Scholar] [CrossRef]
- Doroudiani, S.; Park, C.B.; Kortschot, M.T. Effect of the Crystallinity and Morphology on the Microcellular Foam Structure of Semicrystalline Polymers. Polym. Eng. Sci. 1996, 36, 2645–2662. [Google Scholar] [CrossRef]
- Yeh, S.; Tsai, Y.-B.; Gebremedhin, K.F.; Chien, T.-Y.; Chang, R.-Y.; Tung, K.-L. Preparation of polypropylene/high-melt-strength PP open-cell foam for oil absorption. Polym. Eng. Sci. 2021, 61, 1139–1149. [Google Scholar] [CrossRef]
- Fu, L.; Shi, Q.; Ji, Y.; Wang, G.; Zhang, X.; Chen, J.; Shen, C.; Park, C.B. Improved cell nucleating effect of partially melted crystal structure to enhance the microcellular foaming and impact properties of isotactic polypropylene. J. Supercrit. Fluids 2020, 160, 104794. [Google Scholar] [CrossRef]
- Bing, L.; Wu, Q.; Nanqiao, Z.; Baoshan, S. Batch Foam Processing of Polypropylene/Polydimethylsiloxane Blends. Int. J. Polym. Mater. 2010, 60, 51–61. [Google Scholar] [CrossRef]
- Peyda, S.; Morshedian, J.; Karbalaei-bagher, M.; Baharvand, H.; Khorasani, M.T. A novel technique in the foaming process of EPDM/PP via microwave radiation: The effect of blend compatibilization and additive encapsulation. R. Soc. Chem. 2016, 6, 81400–81407. [Google Scholar] [CrossRef]
- Doroudian, S.; Park, C.B.; Kortschot, M.T. Processing and Characterization of Microcellular Foamed High-Density Polyethylene/Isotactic Polypropylene Blends. Polym. Eng. Sci. 1998, 38, 1205–1215. [Google Scholar] [CrossRef]
- Shi, Q.; Fu, L.; Yang, Y.; Li, K.; Wang, Q.; Liu, B.; Zhang, X.; Chen, J. A facile methodology to effectively improve the melt strength and microcellular foamability of isotactic polypropylene. J. Polym. Res. 2020, 27, 1–13. [Google Scholar] [CrossRef]
- Jian, X.-L.; Liu, T.; Zhao, L.; Xu, Z.-M.; Yuan, W.-K. Effects of Blend Morphology on the FOaming of Polypropylene/Low-density Polyethylene Blends during a Batch Foaming Process. J. Cell. Plast. 2009, 45, 225–241. [Google Scholar] [CrossRef]
- Wang, C.; Ying, S. Batch Foaming of Short Carbon Fiber Reinforced Polypropylene Composites. Fibers Polym. 2013, 14, 815–821. [Google Scholar] [CrossRef]
- Velasco, J.I.; Antunes, M.; Realinho, V.; Ardanuy, M. Characterization of Rigid Polypropylene-Based Microcellular Foams Produced by Batch Foaming Processes. Polym. Eng. Sci. 2011, 51, 2120–2128. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Gupta, R.K.; Jollands, M.; Bhattacharya, S.N. Foaming Behavior of High-Melt Strength Polypropylene/Clay Nanocomposites. Polym. Eng. Sci. 2009, 49, 2070–2084. [Google Scholar] [CrossRef]
- Keramati, M.; Ghasemi, I.; Karrabi, M.; Azizi, H. Microcellular foaming of PP/EPDM/organoclay nanocomposites: The effect of the distribution of nanoclay on foam morphology. Polym. J. 2012, 44, 433–438. [Google Scholar] [CrossRef]
- Li, M.; Qiu, J.; Xing, H.; Fan, D.; Wang, S.; Li, S.; Jiang, Z. In-situ cooling of adsorbed water to control cellular structure of polypropylene composite foam during CO2 batch foaming process. Polymer 2018, 155, 116–128. [Google Scholar] [CrossRef]
- Antunes, M.; Realinho, V.; Velasco, J.I. Foaming Behaviour, Structure, and Properties of Polypropylene Nanocomposites Foams. J. Nanomater. 2010, 2010, 306384. [Google Scholar] [CrossRef]
- Liu, Z.; Qiu, J.; Shi, Z.; Zhang, S.; Xing, H.; Li, M.; Shi, Y.; Tang, T. Adjusting cell structure of polypropylene composite foams by controlling the size and dispersed state of NaCl particles during CO2 batch foaming process. Polymer 2020, 194, 122406. [Google Scholar] [CrossRef]
- Zhao, C.; Mark, L.H.; Chang, E.; Chu, R.K.M.; Lee, P.C.; Park, C.B. Highly expanded, highly insulating polypropylene/polybutylene-terephthalate composite foams manufactured by nano-fibrillation technology. Mater. Des. 2020, 188, 108450. [Google Scholar] [CrossRef]
- Li, Y.; Yao, Z.; Chen, Z.; Qiu, S.; Zeng, C.; Cao, K. High melt strength polypropylene by ionic modification: Preparation, rheological properties and foaming behaviors. Polymer 2015, 70, 207–214. [Google Scholar] [CrossRef]
- Zhai, W.; Wang, H.; Yu, J.; Dong, J.; He, J. Foaming behavior of isotactic polypropylene in supercritical CO2 influenced by phase morphology via chain grafting. Polymer 2008, 49, 3146–3156. [Google Scholar] [CrossRef]
- Xu, Z.; Jiang, X.; Liu, T.; Hu, G.; Zhao, L.; Zhu, Z.-N.; Yuan, W.-K. Foaming of polypropylene with supercritical carbon dioxide. J. Supercrit. Fluids. 2007, 41, 299–310. [Google Scholar] [CrossRef]
- Goel, S.K.; Beckman, E.J. Generation of Microcellular Polymeric Foams Using Supercritical Carbon Dioxide. II: Cell Growth and Skin Formation. Polym. Eng. Sci. 1994, 34, 1148–1156. [Google Scholar] [CrossRef]
- Goel, S.K.; Beckman, E.J. Generation of Microcellular Polymeric Foams Using Supercritical Carbon Dioxide. I: Effect of Pressure and Temperature on Nucleation. Polym. Eng. Sci. 1994, 34, 1137–1147. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, J.; Park, C.B. A Comparison of CO2 and N2 Foaming Behaviors of PP in a Visualization System. Int. Polym. Process. 2020, 35, 503–517. [Google Scholar] [CrossRef]
- Li, G.; Gunkel, F.; Wang, J.; Park, C.B.; Altstadt, V. Solubility Measurements of N2 and CO2 in Polypropylene and Ethene/Octene Copolymer. J. Appl. Polym. Sci. 2006, 103, 2945–2953. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, C.; Liu, T.; Hu, D.; Xu, Z.; Zhao, L. Application of a CO2 Pressure Swing Saturation Strategy in PP Semi-Solid-State Batch Foaming: Evaluation of Foamability by Experiments and Numerical Simulations. Ind. Eng. Chem. Res. 2020, 59, 4924–4935. [Google Scholar] [CrossRef]
- Fu, D.; Chen, F.; Kuang, T.; Li, D.; Peng, X.; Chiu, D.Y.; Shiang, C.; Ly, J.L. Supercritical CO2 foaming of pressure-induced-flow processed linear polypropylene. Mater. Des. 2016, 93, 509–513. [Google Scholar] [CrossRef]
- Ding, J.; Ma, W.; Song, F.; Zhong, Q. Foaming of Polypropylene with Supercritical Carbon Dioxide: An Experimental and Theoretical Study on a New Process. J. Appl. Polym. Sci. 2013, 130, 2877–2885. [Google Scholar] [CrossRef]
- Kumar, A.; Patham, B.; Mohanaty, S.; Nayak, S.K. Effect of temperature on thermal, mechanical and morphological properties of polypropylene foams prepared by single step and two step batch foaming process. J. Polym. Res. 2019, 26, 1–14. [Google Scholar] [CrossRef]
- Hou, J.; Zhao, G.; Zhang, L.; Wang, G.; Li, B. High-expansion polypropylene foam prepared in non-crystalline state and oil adsorption performance of open-cell foam. J. Colloid Interface Sci. 2019, 542, 233–242. [Google Scholar] [CrossRef]
- Romero-Diez, S.; Kweon, M.S.; Kim, E.S.; Gupta, A.; Yan, X.; Pehlert, G.; Park, C.B.; Lee, P.C. In situ visualization of crystal nucleation and growth behaviors of linear and long chain branched polypropylene under shear and CO2 pressure. Polymer 2021, 213, 123215. [Google Scholar] [CrossRef]
- ASTM Standard D792-13. Standard Test Methods for Density and Specific Gravity (Relative Density) of Plastics. ASTM International: West Conshohocken, PA, USA, 2013.
- Yang, C.; Zhao, Q.; Xing, Z.; Zhang, W.; Zhang, M.; Tan, H.; Wang, J.; Wu, G. Improving the Supercritical CO2 Foaming of Polypropylene by the Addition of Fluoroelastomer as a Nucleation Agent. Polymers 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shau-Tarng, L.; Park, C.B. Rheology of Thermoplastic Foam Extrusion Process. In Foam Extrusion: Principles and Practice. Encyclopedic Dictionary of Polymers; Taylor & Francis Group: London, UK, 2014. [Google Scholar] [CrossRef]
- Wong, A.; Park, C.B. The effects of extensional stresses on the foamability of polystyrene-talc composites blown with carbon dioxide. Chem. Eng. Sci. 2012, 75, 49–62. [Google Scholar] [CrossRef]
- Leung, S.N.; Wong, A.; Wang, L.C.; Park, C.B. Mechanism of extensional stress-induced cell formation in polymeric foaming processes with the presence of nucleating agents. J. Supercrit. Fluids 2012, 63, 187–198. [Google Scholar] [CrossRef]
- Kim, E.; Kweon, M.S.; Romero-Diez, S.; Gupta, A.; Yan, X.; Spofford, C.; Pehlert, G.; Lee, P.C. Effects of pressure drop rate and CO2 content on the foaming behavior of newly developed high-melt-strength polypropylene in continuous extrusion. J. Cell. Plast. 2021, 57, 413–432. [Google Scholar] [CrossRef]
- Hoffman, J.D. Thermodynamic driving force in nucleation and growth processes. J. Chem. Phys. 1958, 29, 1192–1193. [Google Scholar] [CrossRef]
- Yamada, K.; Hikosaka, M.; Toda, A.; Yamazaki, S.; Tagashira, K. Equilibrium melting temperature of isotactic polypropylene with high tacticity: 1. Determination by differential scanning calorimetry. Macromolecules 2003, 36, 4790–4801. [Google Scholar] [CrossRef]
- Ahmed, A.K.; Atiqullah, M.; Pradhan, D.R.; Al-Harthi, M.A. Crystallization and melting behavior of i-PP: A perspective from Flory’s thermodynamic equilibrium theory and DSC experiment. RSC Adv. 2017, 7, 42491–42504. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Yu, W.; Zhou, C. Crystallization kinetics of linear and long-chain branched polypropylene. J. Macromol. Sci. Part B Phys. 2006, 45, 969–985. [Google Scholar] [CrossRef]
- Marand, H.; Xu, J.; Srinivas, S. Determination of the Equilibrium Melting Temperature of Polymer Crystals: Linear and Nonlinear Hoffman-Weeks Extrapolations. Macromolecules 1998, 31, 8219–8229. [Google Scholar] [CrossRef]
- Wagner, M.H.; Bastian, H.; Hachmann, P.; Meissner, J.; Kurzbeck, S.; Münstedt, H.; Langouche, F. The strain-hardening behaviour of linear and long-chain-branched polyolefin melts in extensional flows. Rheol. Acta. 2000, 39, 97–109. [Google Scholar] [CrossRef]
- Weingart, N.; Raps, D.; Lu, M.; Endner, L.; Altstädt, V. Comparison of the Foamability of Linear and Long-Chain Branched Polypropylene-the Legend of Strain-Hardening as a Requirement for Good Foamability. Polymers 2020, 12, 725. [Google Scholar] [CrossRef] [Green Version]
- Nam, G.J.; Yoo, J.H.; Lee, J.W. Effect of Long-Chain Branches of Polypropylene on Rheological Properties and Foam-Extrusion Performances. J. Appl. Polym. Sci. 2005, 96, 1793–1800. [Google Scholar] [CrossRef]
- Stange, J.; Münstedt, H. Effect of Long-Chain Branching on the Foaming of Polypropylene with Azodicarbonamide. J. Cell. Plast. 2006, 42, 445–467. [Google Scholar] [CrossRef] [Green Version]
- Li, D.C.; Liu, T.; Zhao, L.; Yuan, W.K. Foaming of linear isotactic polypropylene based on its non-isothermal crystallization behaviors under compressed CO2. J. Supercrit. Fluids. 2011, 60, 89–97. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Resin A | Resin B | Resin C | |
|---|---|---|---|
| Melt Strength (190 °C) [cN] | 58 | 45 | 40 |
| Weight Average Molecular Weight [kg/mol] | 585 | 541 | 520 |
| Polydispersity Index | 8.7 | ~13 | ~13 |
| Resin | Tc,onset [°C] | SHR | Method | Foaming Temperature Range * [°C] | Cell Density Range ** [#/cm3] | |
|---|---|---|---|---|---|---|
| A | 112 | 13 | 1 | 48 ± 2 | 150–175 | 107–108 |
| 2 | 45 ± 3 | 120–170 | 107–108 | |||
| B | 117 | 5.6 | 1 | 27 ± 2 | 155–165 | 106–107 |
| 2 | 36 ± 3 | 130–160 | 106–107 | |||
| C | 118 | 1.0 | 1 | 32 ± 4 | 160 | 106–107 |
| 2 | 28 ± 4 | 130–150 | 106–107 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kweon, M.S.; Embabi, M.; Shivokhin, M.E.; Gupta, A.; Yan, X.; Pehlert, G.; Lee, P.C. Tuning High and Low Temperature Foaming Behavior of Linear and Long-Chain Branched Polypropylene via Partial and Complete Melting. Polymers 2022, 14, 44. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14010044
Kweon MS, Embabi M, Shivokhin ME, Gupta A, Yan X, Pehlert G, Lee PC. Tuning High and Low Temperature Foaming Behavior of Linear and Long-Chain Branched Polypropylene via Partial and Complete Melting. Polymers. 2022; 14(1):44. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14010044
Chicago/Turabian StyleKweon, Mu Sung, Mahmoud Embabi, Maksim E. Shivokhin, Anvit Gupta, Xuejia Yan, George Pehlert, and Patrick C. Lee. 2022. "Tuning High and Low Temperature Foaming Behavior of Linear and Long-Chain Branched Polypropylene via Partial and Complete Melting" Polymers 14, no. 1: 44. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14010044