2.3. Experimental Procedures
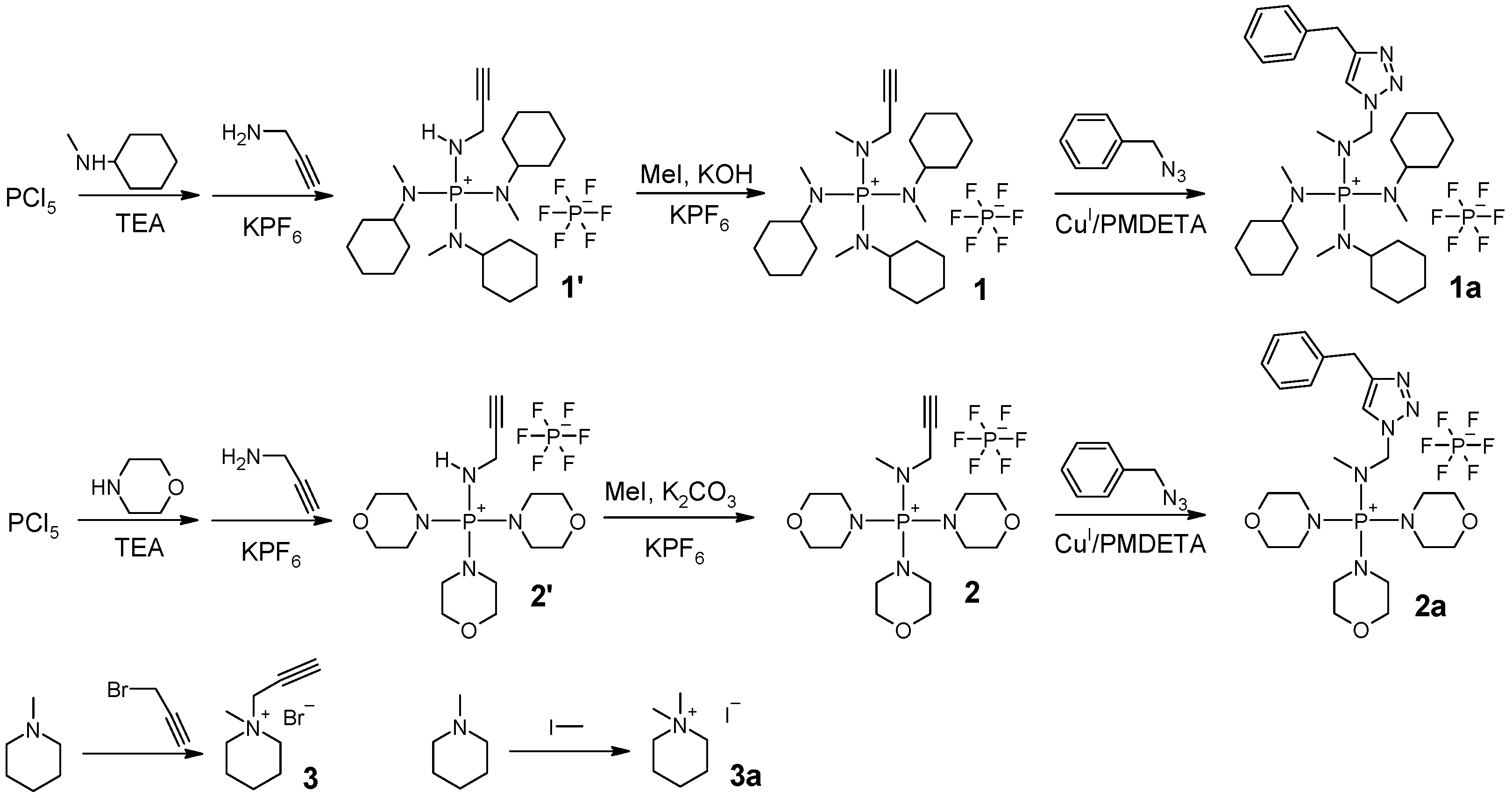
The ionic compounds used in this study were synthesized according to the procedures shown in
Scheme 2.
Synthesis of benzyl azide: Benzyl bromide ( mL, mmol) and mL of DMF were placed in a round-bottom flask. Sodium azide ( g, mmol) was added to the solution. The reaction mixture was stirred at room temperature overnight. The reaction mixture was added to 200 mL of water and extracted with toluene ( mL). The toluene solution of benzyl azide was dried with MgSO4, filtered, and used directly for the following reactions. The FTIR spectra showed strong absorption of the azide group at 2094 cm−1. The H NMR (400 MHz, CDCl3) spectra showed that the mole ratio between benzyl azide (−CH2−, 4.34 (s, 2H)) and toluene (−CH3, 2.36 (s, 3H)) was about 1:6.
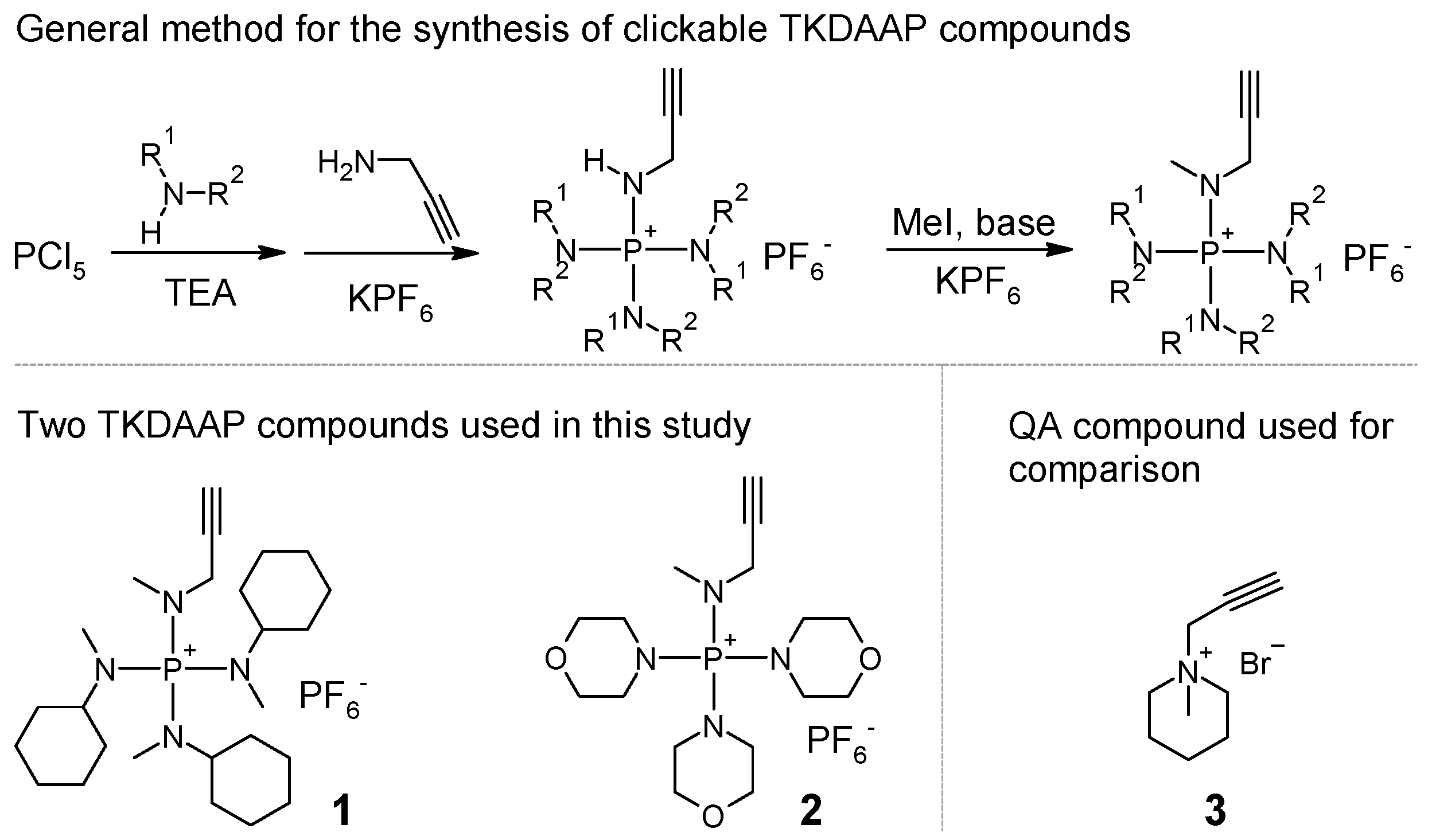
Synthesis of compound 1: To a Schlenk flask, g of PCl5 ( mmol) and 60 mL of chlorobenzene were added. The reaction mixture was bubbled with N2 for an hour, with stirring at 0 in an ice bath. Then, mL of N-methylcyclohexylamine ( mmol) was slowly added using a constant-pressure addition funnel for 10 min. After that, mL of triethylamine ( mmol) was added using a constant-pressure addition funnel for another 10 min. The reaction mixture was stirred overnight, while the temperature increased gradually to room temperature as the ice melted. Then, mL of propargylamine ( mmol) was added to the flask. The reaction mixture was heated to 45 , and stirring continued for another 24 h. The reaction mixture was poured into a beaker with 8 g of KPF6 and 150 mL of water. After having been stirred for about 10 min, the mixture was separated using a separatory funnel. The organic phase was saved. The water phase was extracted with cholorobenzene twice ( mL). The combined organic phases were washed with water once. The solvent was evaporated using a rotary evaporator at reduced pressure and dried under vacuum; intermediate product 1’ was obtained as a brown-colored semisolid ( g, 68.2% yield). H NMR (400 MHz, CDCl3) 4.47 (dt, 14.0, 7.1 Hz, 1H), 3.82–3.70 (m, 2H), 3.26–3.11 (m, 3H), 2.67 (d, Hz, 9H), 2.32 (t, Hz, 1H). P NMR (162 MHz, DMSO-) 41.05 (s), (sep, Hz). Both H NMR and P NMR spectra showed that the intermediate product contained impurities. However, no purification was needed at this stage. Intermediate product 1’ ( g, mmol) was dissolved in 30 mL of chlorobenzene (round-bottom flask), and 20 g of KOH water solution (50% w/w) was added. While stirring at room temperature, we added mL of iodomethane ( mmol). The reaction was stirred overnight. The mixture was poured into a beaker with 8 g of KPF6 and 150 mL of water. After stirring for about 10 min, we separated the mixture. The organic phase was collected. The water phase was extracted with cholorobenzene twice ( mL). The combined organic phases were washed with water once. The solution was dried with MgSO4 overnight. After filtering, the solution was concentrated using a rotary evaporator. The product was precipitated in diethyl ether and dried under vacuum. Solid powder with a slightly yellow color was collected ( g 46.2% yield). H NMR (400 MHz, DMSO-) 3.81 (dd, 11.4, 2.4 Hz, 2H), 3.56 (t, Hz, 1H), 3.05–2.91 (m, 3H), 2.73 (d, Hz, 3H), 2.63 (d, Hz, 9H), 1.84–0.99 (m, 30H). C NMR (101 MHz, DMSO-) 78.42, 76.30, 57.60, 54.71, 34.92, 29.61, 29.44, 25.37, 24.70. P NMR 44.91 (s), (sep, Hz). HRMS (ESI-TOF) (): [M] calculated for C25H48N4P+: 435.36111; found 435.36176. The MS spectra showed little impurity with 411.36175. This impurity did not react with benzyl azide and remained intact in 1a. Thus, the impurity could not be incorporated into the AEM.
Synthesis of compound 1a: A piece of Cu(0) wire ( mm, cm) was placed in a Schlenk flask. Benzyl azide solution ( mL) in toluene (containing about g of mmol benzyl azide) was added to a round-bottom flask. Then, mL of DMF, g of compound 1 ( mmol), mg of CuBr2 ( mol), and L of PMDETA (34 mol) were added to the round-bottom flask. Both flasks were degassed with N2 going through for 1 h. The solution was then transferred to the Schlenk flask using a syringe. The reaction mixture was stirred at 50 for 24 h. The Cu catalyst was removed by filtering through a short column with Al2O3. The solvent was slowly evaporated in the air for three days when the product crystallized. The cubic crystals were washed with Et2O and dried under vacuum ( g, 42.3% yield). H NMR (400 MHz, DMSO-) 8.32 (s, 1H), 7.43–7.28 (m, 5H), 5.61 (s, 2H), 4.18 (d, Hz, 2H), 2.81 (d, Hz, 3H), 2.67 (d, Hz, 12H), 1.84–0.91 (m, 30H). C NMR (101 MHz, DMSO-) 142.00, 135.93, 128.71, 128.18, 127.96, 124.47, 54.73, 52.85, 42.95, 35.11, 29.59, 25.33, 24.68. P NMR 45.14 (s), (sep, Hz). HRMS (ESI-TOF) (): [M] calculated for C32H55N7P+: 568.42511; found 568.42605.
Synthesis of compound 2: To a Schlenk flask, g of PCl5 ( mmol) and 60 mL of chlorobenzene were added. The reaction mixture was bubbled with N2 for an hour, with stirring at 0 in an ice bath. Then, mL of morpholine ( mmol) was slowly added using a constant-pressure addition funnel for 10 min. After that, mL of triethylamine ( mmol) was added using a constant-pressure addition funnel for another 10 min. The reaction mixture was stirred overnight, while the temperature increased gradually to room temperature as the ice melted. Then, mL of propargylamine ( mmol) was added to the flask. The reaction mixture was heated to 45 and continued to be stirred for another 24 h. The reaction mixture was poured into a beaker with 8 g of KPF6 and 150 mL of water. After stirring for about 10 min, we filtered the mixture to collect the insoluble solids. The solids were washed with i-PrOH and further dried under vacuum, resulting in intermediate product 2’, white solid powder ( g, 85.0% yield). H NMR (400 MHz, DMSO-) 6.73 (dt, J = 13.9, 6.8 Hz, 1H), 3.83 (ddd, J = 14.3, 6.8, 2.5 Hz, 2H), 3.63 (m, 12H), 3.45 (t, J = 2.4 Hz, 1H), 3.16 (m, 12H). P NMR (162 MHz, DMSO-) 33.28 (s), (sep, Hz). Intermediate product 2’ ( g, mmol) was dissolved in 40 mL of DMSO in a round-bottom flask. Then, mL of iodomethane ( mmol) and g of K2CO3 ( mmol) were added. The mixture was stirred at room temperature overnight. The liquid was decanted into a beaker with 8 g of KPF6 and 150 mL of water. After stirring for about 10 min, we filtered the mixture. The solid was collected and washed with i-PrOH. After drying under vacuum, white solid powder was obtained ( g 65.1% yield). H NMR (400 MHz, DMSO-) 3.92 (dd, 11.6, 2.4 Hz, 2H), 3.72–3.55 (m, 12H), 3.52 (t, Hz, 1H), 3.24–3.08 (m, 12H), 2.81 (d, Hz, 3H). C NMR (101 MHz, DMSO-) 78.71, 76.44, 65.66, 45.13, 35.82. P NMR 36.61 (s), (sep, Hz). HRMS (ESI-TOF) (): [M] calculated for C16H30N4O3P+: 357.20500; found 357.20583.
Synthesis of compound 2a: A piece of Cu(0) wire ( mm, cm) was placed in a Schlenk flask. mL of the benzyl azide solution in toluene (containing about g of mmol benzyl azide), mL of DMSO, g of compound 2 ( mmol), mg of CuBr2 (10 mol), and L of PMDETA (40 mol) were added to the round-bottom flask. Both flasks were degassed by letting N2 go through for 1 h. The solution was then transferred to the Schlenk flask using a syringe. The reaction mixture was stirred at 50 for 24 h. The Cu catalyst was removed by filtering through a short column with Al2O3. The solvent was slowly evaporated in the air for a week when the product crystallized. The crystals were washed with i-PrOH and dried under vacuum ( g, 50.6% yield). H NMR (400 MHz, DMSO-) 8.27 (s, 1H), 7.42–7.23 (m, 5H), 5.60 (s, 2H), 4.26 (d, Hz, 2H), 3.65–3.48 (m, 12H), 3.23–3.07 (m, 12H), 2.74 (d, Hz, 3H). C NMR (101 MHz, DMSO-) 142.25, 136.01, 128.80, 128.21, 127.86, 124.28, 65.68, 52.92, 45.23, 40.42, 36.18. P NMR 36.81 (s), (sep, Hz). HRMS (ESI-TOF) (): [M] calculated for C23H37N7O3P+: 490.26900; found 490.27003.
Synthesis of compound 3: A volume of mL of N-methylpiperidine ( mmol), g of propargyl bromide solution in toluene (80% by weight, containing 42 mmol propargyl bromide), and 15 mL of hexane were added to a round-bottom flask and stirred at 60 overnight. The liquid was decanted. The insoluble part was washed with THF and dried under vacuum. A light yellow colored solid was obtained ( g, 69.2% yield). H NMR (400 MHz, DMSO-) 4.48 (d, Hz 2H), 4.07 (t, Hz 1H), 3.45–3.39 (m, 4H), 3.10 (s, 3H), 1.87–1.73 (m, 4H), 1.61–1.42 (m, 2H). C NMR (101 MHz, DMSO-) 82.98, 72.15, 59.52, 52.36, 47.53, 20.49, 19.24.
Synthesis of compound 3a: N-methylpiperidine ( mL, mmol) and iodomethane ( mL, mmol) were dissolved in 10 mL of acetone and stirred at room temperature overnight. The white solid powder was collected, washed with acetone, and dried under vacuum. ( g, 75.1% yield) H NMR (400 MHz, DMSO-) 3.30 (t, Hz, 4H), 3.05 (s, 6H), 1.83–1.71 (m, 4H), 1.51 (p, Hz, 2H). C NMR (101 MHz, DMSO-) 61.51, 50.86, 20.53, 19.53.
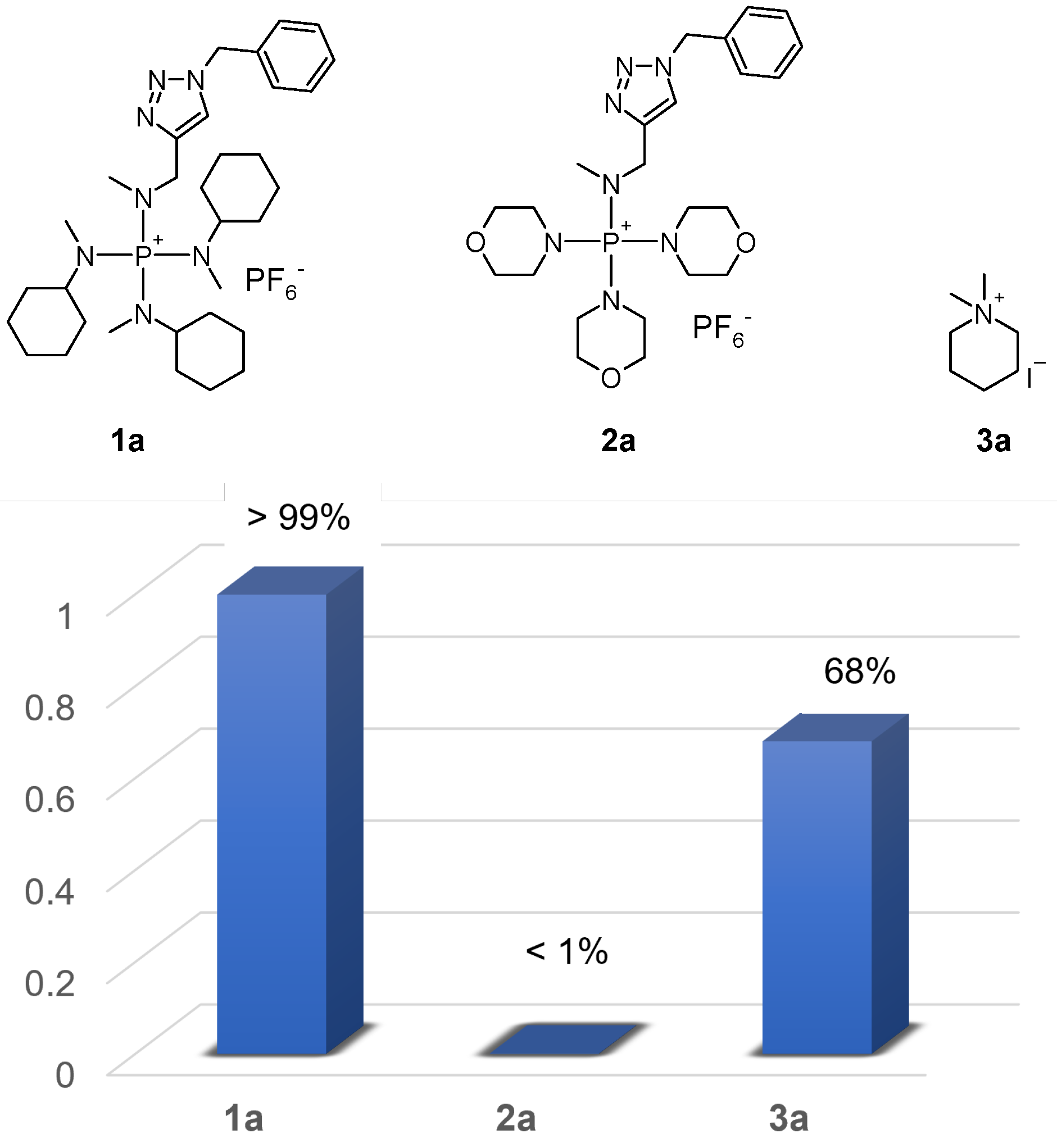
Evaluation of the alkaline stability of model compounds 1a, 2a, and 3a: KOH was dissolved in CD3OH to make a M solution. Compounds 1a, 2a, and 3a, 10 mg each, were dissolved in mL of regular DMSO individually and placed in Norell Inc. 5MM, 400MHZ 7IN NMR tubes. To each sample, L of 2-trimethylsilylethanol was added as internal standard. Then, mL of KOH/CD3OH solution was added to each sample to obtain a final KOH concentration of M. The NMR tubes were flame-sealed and heated to 80 in an oil bath. H NMR analyses in solvent suppression mode were performed every 3 or 4 days for 30 days. Solvent suppression was applied to the −OH signal in CD3OH and the −CH3 signal in DMSO.
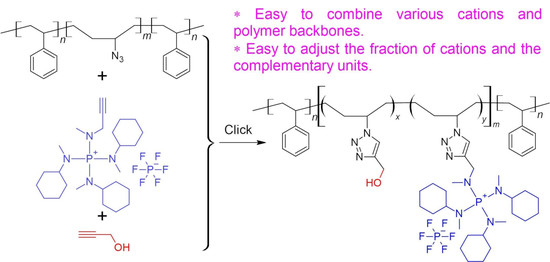
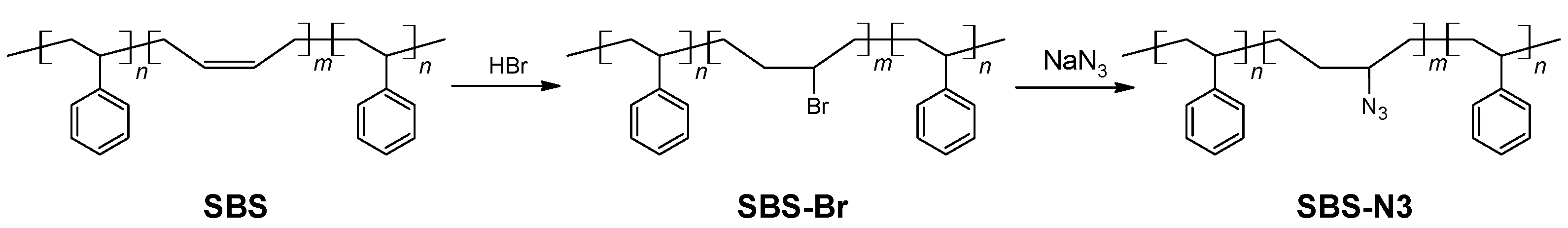
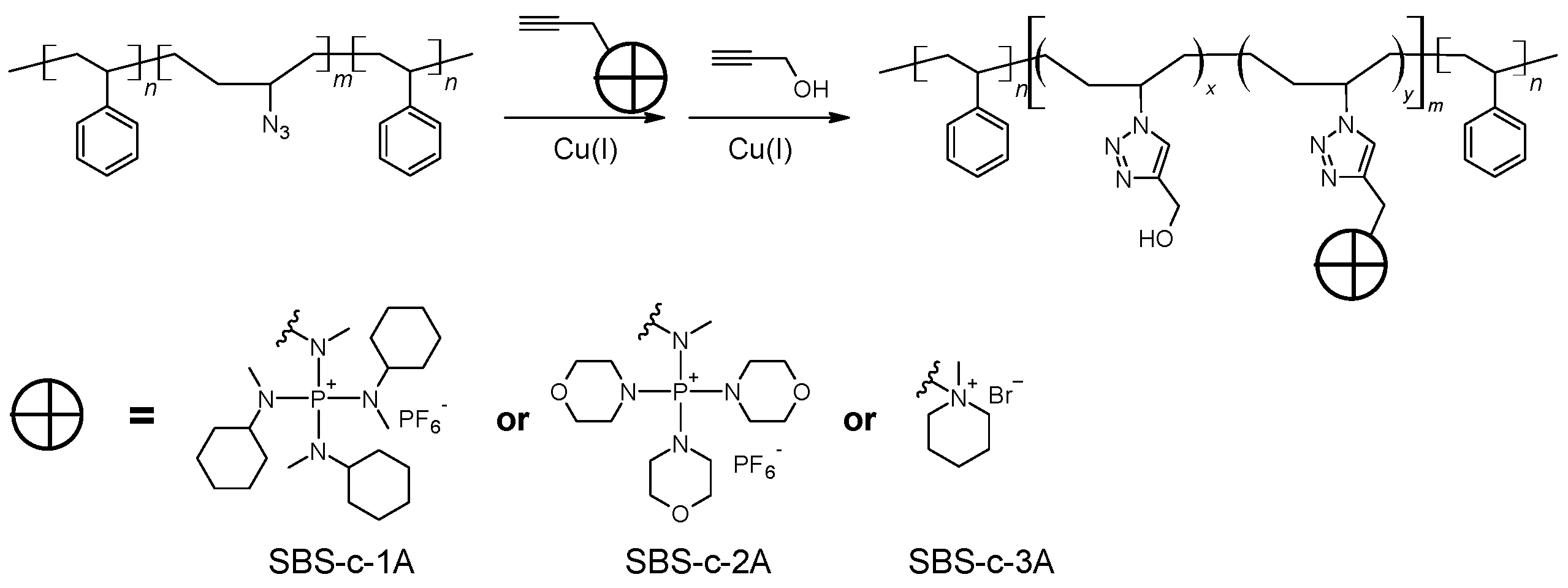
Synthesis of azide-modified triblock copolymer (SBS-N3).
The procedure for the synthesis of SBS-N3 is shown in
Scheme 3. In total,
g of SBS containing about
mmol butadiene units was dissolved in 200 mL of toluene in a round-bottom flask and stirred at 0
in an ice bath. A HBr solution in glacial acetic acid (
mL containing about 64 mmol HBr) was slowly added to the flask. The reaction mixture was stirred for 24 h, while the temperature increased gradually to room temperature as the ice melted. The conversion of the double bonds was confirmed with
H NMR analysis: the signal at 5.4 ppm completely disappeared. The resulting solution was washed with water 3 times to remove the majority of the acid. Then, the solution was concentrated using a rotary vaporizator. The bromide-modified polymer (SBS-Br) was obtained after precipitation in MeOH and drying under vacuum (
g, 93.7% yield). SBS-Br (
g containing about
mmol bromide-modified butadiene units) was dissolved in 100 mL of THF/DMF = 4/1 (
v/
v) in a round-bottom flask. To the solution,
g of 18-crown-6 (
mmol) and
g of NaN
3 (
mmol) were added. The reaction mixture was stirred at 50
for 24 h. The conversion of the bromide groups to the azide was confirmed with
H NMR analysis: the signal at 4.05 ppm completely disappeared. Insoluble solids were removed by filtering. The azide-modified polymer (SBS-N3) was precipitated in MeOH and dried under vacuum (
g, 79.6% yield). The FTIR spectra showed strong absorption at 2087 cm
−1, indicating the presence of azide groups.
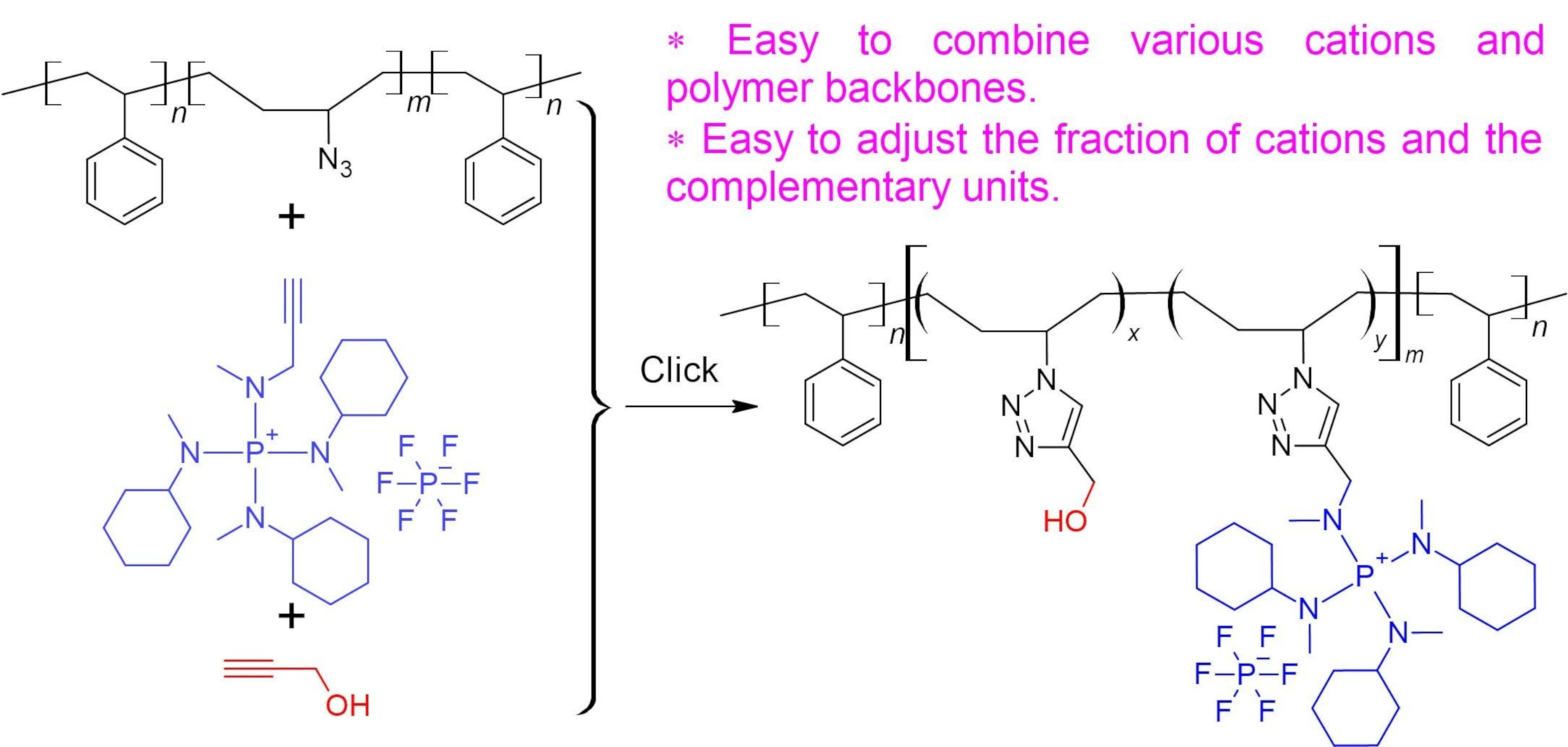
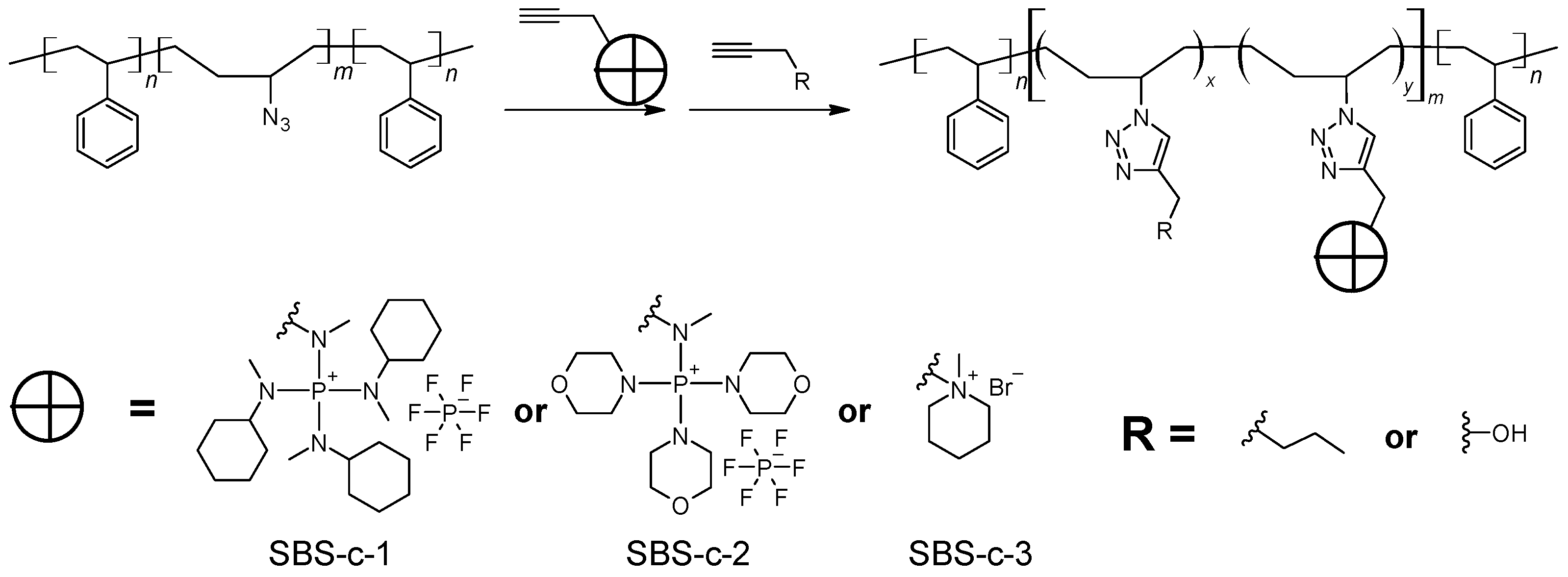
General procedure for the synthesis of polyelectrolytes via click chemistry.
As shown in
Scheme 4, typically, a solution of SBS-N3 (
g) containing about
mmol azide units in 8 mL of THF was placed in a Schlenk flask. A piece of Cu(0) wire (diameter =
mm, length =
cm) and
L of PMDETA (
mmol) were added to the flask. The reaction mixture was bubbled with N
2 for 1 h. A total of
mg of CuBr
2 (28
mol) was dissolved in 2 mL of DMF and bubbled with N
2 for 1 h. The CuBr
2 solution was added to the Schlenk flask using a syringe. The reaction mixture was stirred at 45
. Ionic compound
1,
2, or
3 was dissolved in 5 mL of DMF or DMSO. The selection of solvents is summarized in
Table 1. After bubbling with N
2 for 1 h, the solution of the ionic compound was slowly added to the Schlenk flask using a gas-tight syringe. The reaction was continued at 45
for 24 h. Then,
mL of propargyl alcohol (
mmol), or
mL of 1-hexyne (
mmol), degassed by bubbling with N
2 for 1 h, was added to the Schlenk flask. The reaction was continued at 45
for another 24 h. The resulting solution was stored and used directly for the preparation of AEMs.
Preparation of AEMs: AEMs were prepared by drop-casting the polyelectrolyte solutions onto flat HDPE substrates. For samples SBS-c-1A, SBS-c-2A, and SBS-c-3A, the resulting membranes were peeled off from the substrates for analysis. However, the rest of the samples were too brittle to form stand-alone membranes or broke easily even if the membrane could be peeled off. One sample of SBS-c-1C was prepared on a glass microscope slide to evaluate the percentage of water uptake (WU%) and ion exchange capacity (IEC).
Evaluation of the IEC, WU%, and hydration number (
): The IEC of the AEMs was determined with back titration. Typically, the AEMs were added to
M NaOH solutions and allowed to soak for 24 h. After washing thoroughly with DI water, the AEMs were added to
mL of
M HCl solutions and allowed to soak for another 24 h. The HCl solutions were titrated with
M NaOH. The IEC was calculated by comparing with blank samples, i.e.,
mL of
M HCl.
where [NaOH] is the concentration of NaOH solution, i.e.,
M, used for titration;
is the volume of NaOH solution consumed by
mL of
M HCl solution;
is the volume of NaOH solution consumed by HCl solution with the sample; and
is the mass of the dried sample in hydroxide form.
WU% was evaluated by comparing the wet masses and dry masses of the samples in hydroxide form.
where
and
are the wet mass and dry mass of the sample, respectively. The wet mass was weighed after dabbing off excess surface water with a dry paper towel.
The hydration number (
) was calculated as
All measurements were performed at room temperature, i.e., 22 .
Measurement of ionic conductivity of the AEMs: The AEM was cut into a rectangular shape and treated in 1 M NaCl for 24 h; then, it was allowed to exchange with hydroxide ions in 1 M NaOH solution for another 24 h. The treated samples were washed with DI water and spread out on a homemade conductivity measurement setup with four parallel platinum strips as electrodes. The in-plane resistance (
) was obtained using electrochemical impedance spectroscopy (EIS). EIS was performed with a Biologic VMP3 multi-channel potentiostat in open circuit with an AC perturbation of 10 mV and a frequency range from
Hz to 50 kHz at room temperature, i.e., 22
. The conductivity (
) was calculated using the geometries of the AEM (
l is the distance between two voltage probes;
d is the thickness of the membrane; and
w is the width of the AEM).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}