
Coordination Chemistry inside Polymeric Nanoreactors: Metal Migration and Cross-Exchange in Amphiphilic Core-Shell Polymer Latexes


Abstract
:
1. Introduction


2. Materials and Methods
2.1. General
2.2. Characterization Techniques
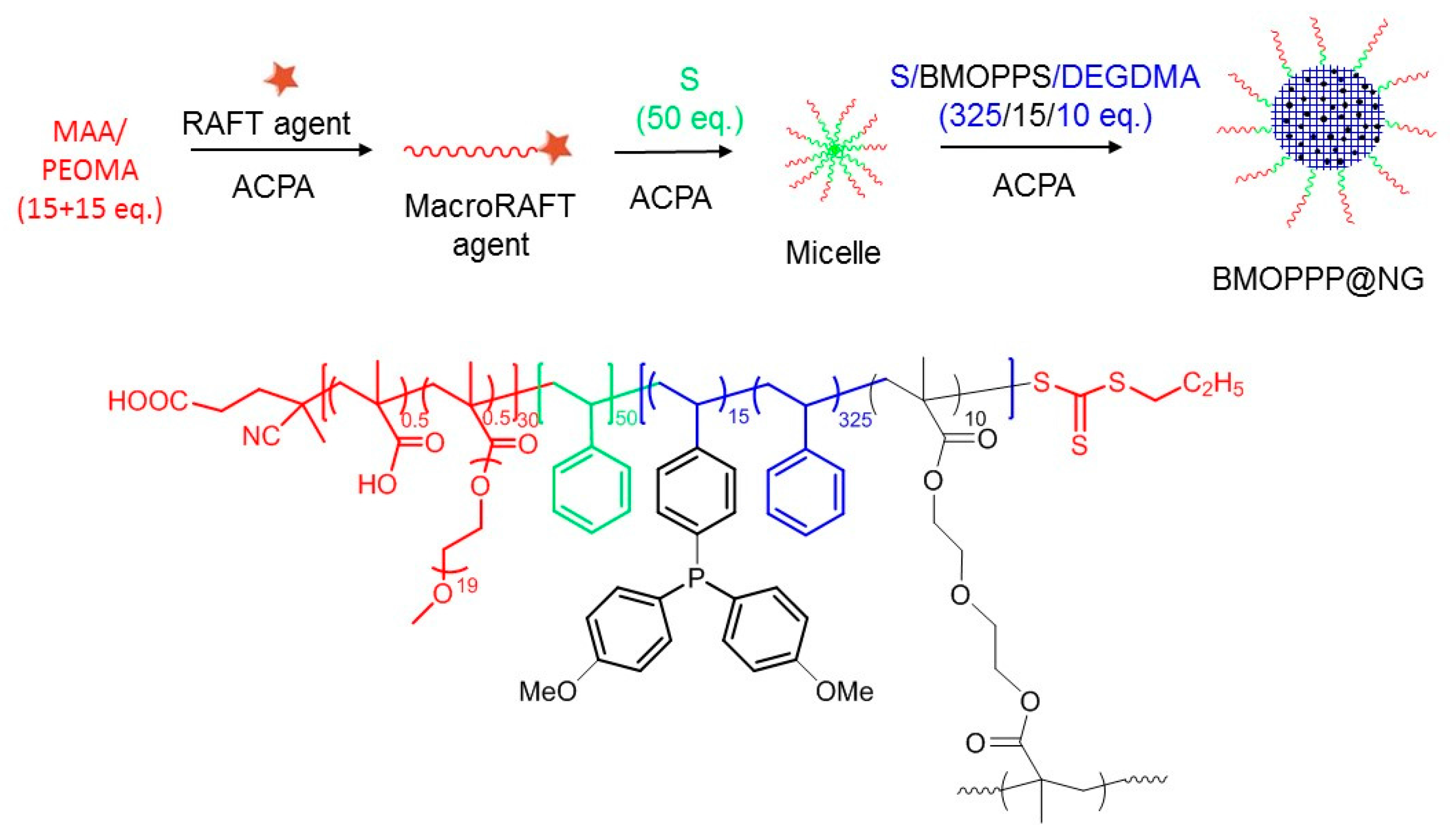
2.3. Preparation of BMOPPP@NG by One-Pot RAFT Polymerization in Water
2.3.1. Step 1: Preparation of the HOOCCH2CH2C(CN)(Me)-(MAA15-co-PEOMA15)-SC(S)SPr Macromolecular RAFT Agent in Water
2.3.2. Step 2: Preparation of the Nanogels
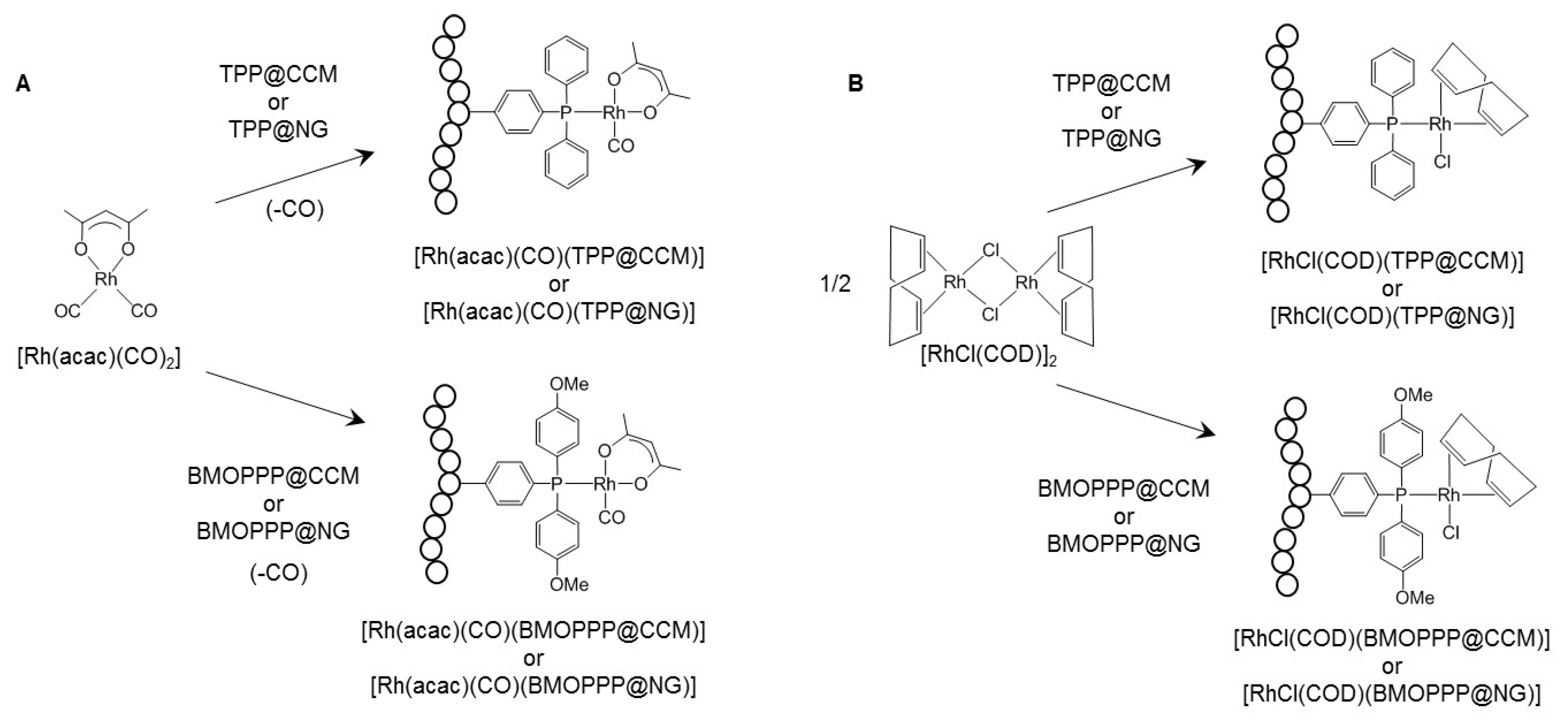
2.4. General Procedure for the Phosphine Ligand Complexation to [Rh(acac)(CO)2] and [RhCl(COD)]2
2.4.1. Loading with [Rh(acac)(CO)2]
2.4.2. Loading with [RhCl(COD)]2
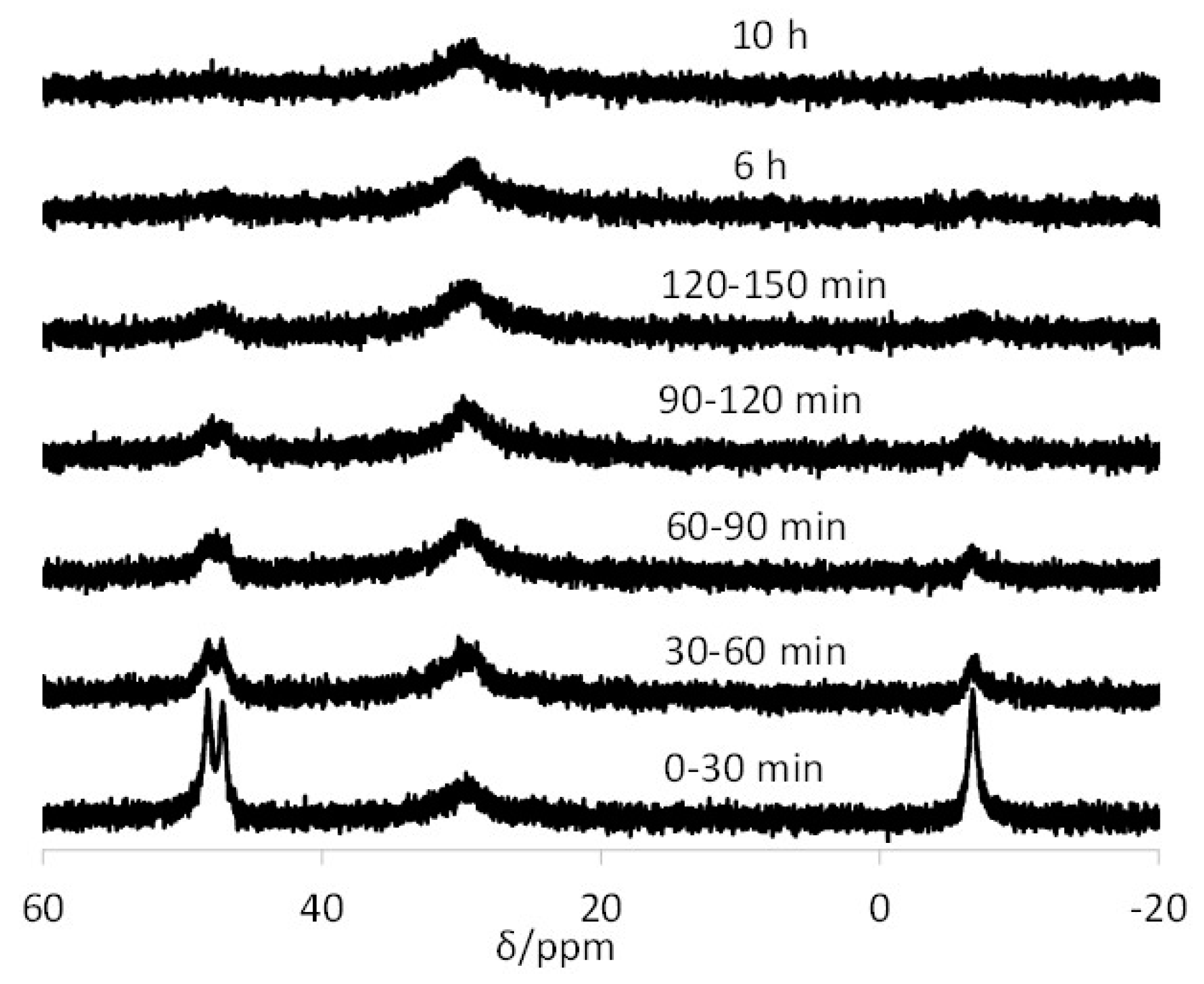
2.5. Interparticle Metal Exchange Study Involving 100% [Rh(acac)(CO)]-Loaded and Rh-Free TPP@NG
2.5.1. At the Natural pH
2.5.2. Under Basic Conditions
2.6. Interparticle Double Exchange Study Involving 100% [Rh(acac)(CO)]-Loaded BMOPPP-Functionalized Polymer Latex and 100% [RhCl(COD)]-Loaded TPP-Functionalized Polymer Latex
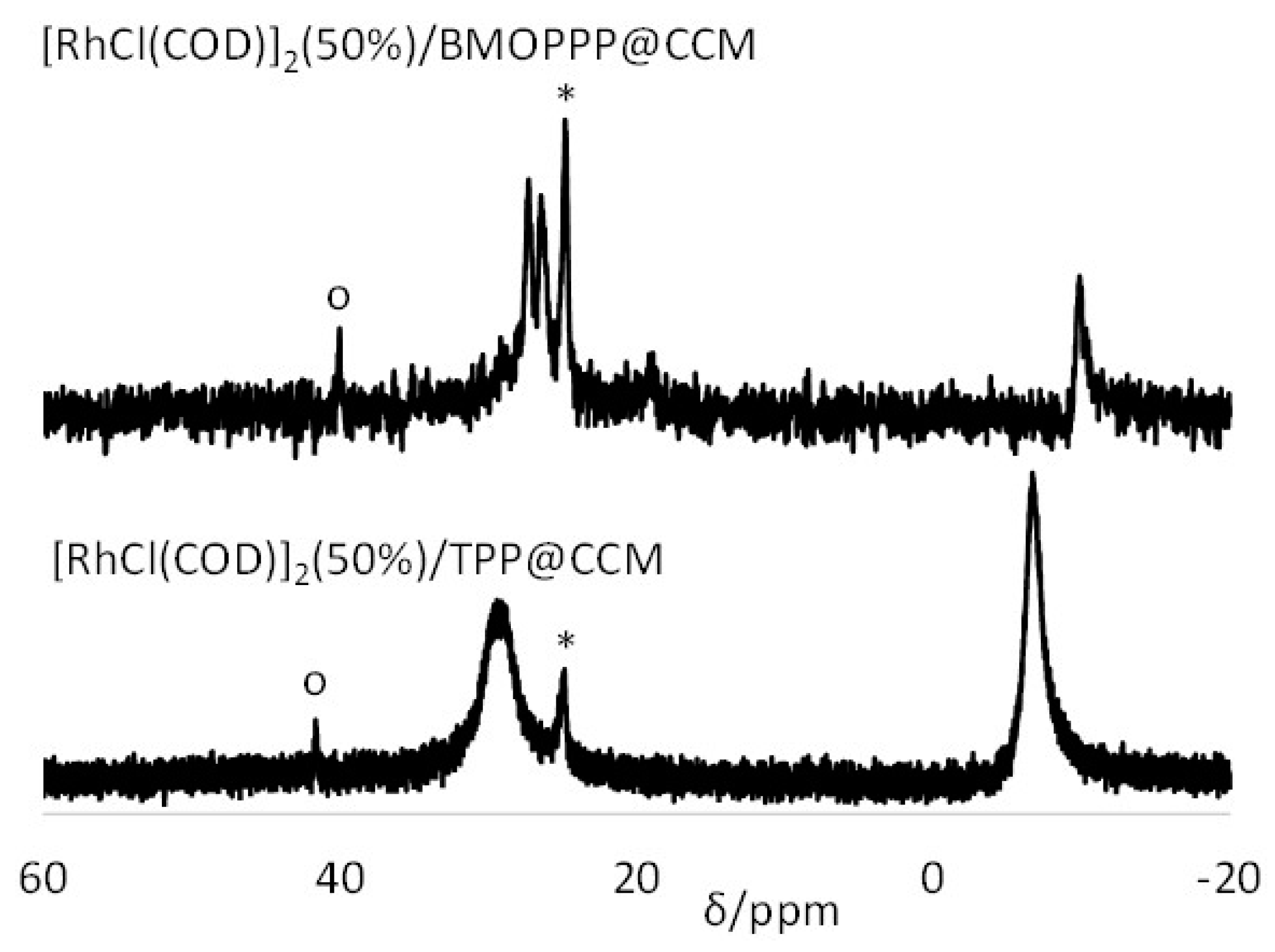
2.6.1. Using the CCM Particles
2.6.2. Using the NG particles.
3. Results and Discussion
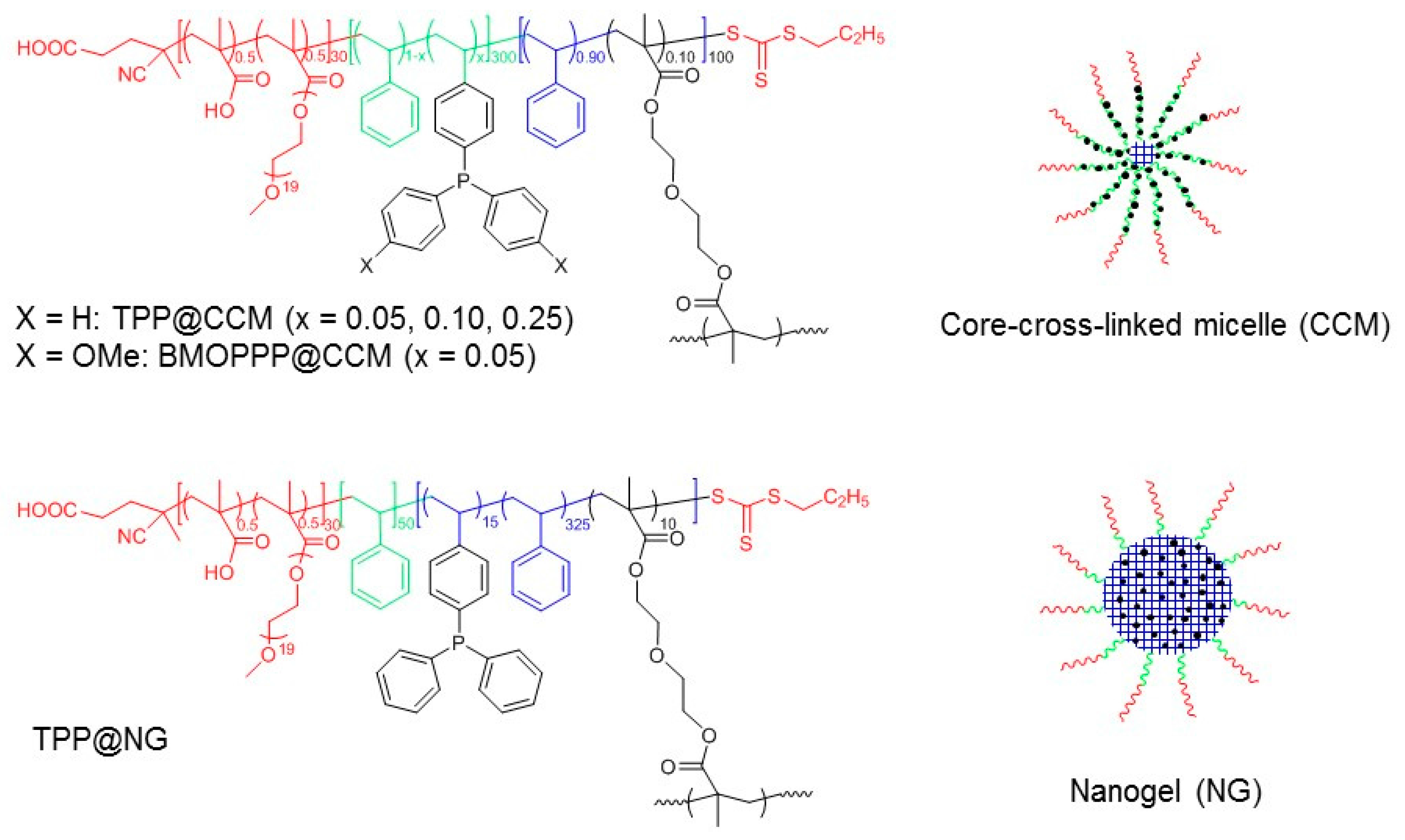
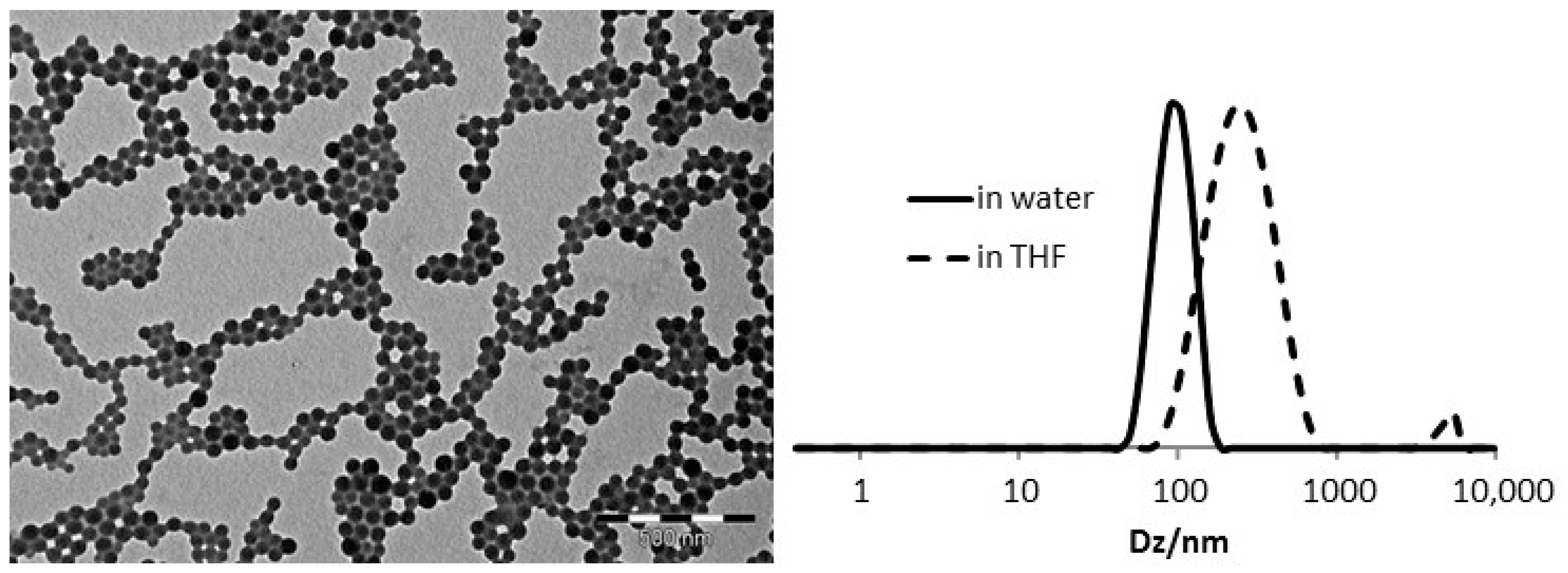
3.1. Synthesis and Characterization of BMOPP@NG


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | FS b | Dz (nm)/PDI | Solid (%) | [FS] (mmol/mL) | Reference | |
|---|---|---|---|---|---|---|
| H2O | THF | |||||
| TPP@CCM | DPPS | 100/0.28 | 163/0.07 | 25.6 | 0.060 | [7] |
| BMOPPP@CCM | BMOPPS | 81/0.16 | 216/0.20 | 25 | 0.058 | [8] |
| TPP@NG | DPPS | 86/0.20 | 188/0.15 | 27.8 | 0.068 | [42] |
| BMOPPP@NG | BMOPPS | 99/0.23 | 236/0.23 | 27.9 | 0.058 | This work |

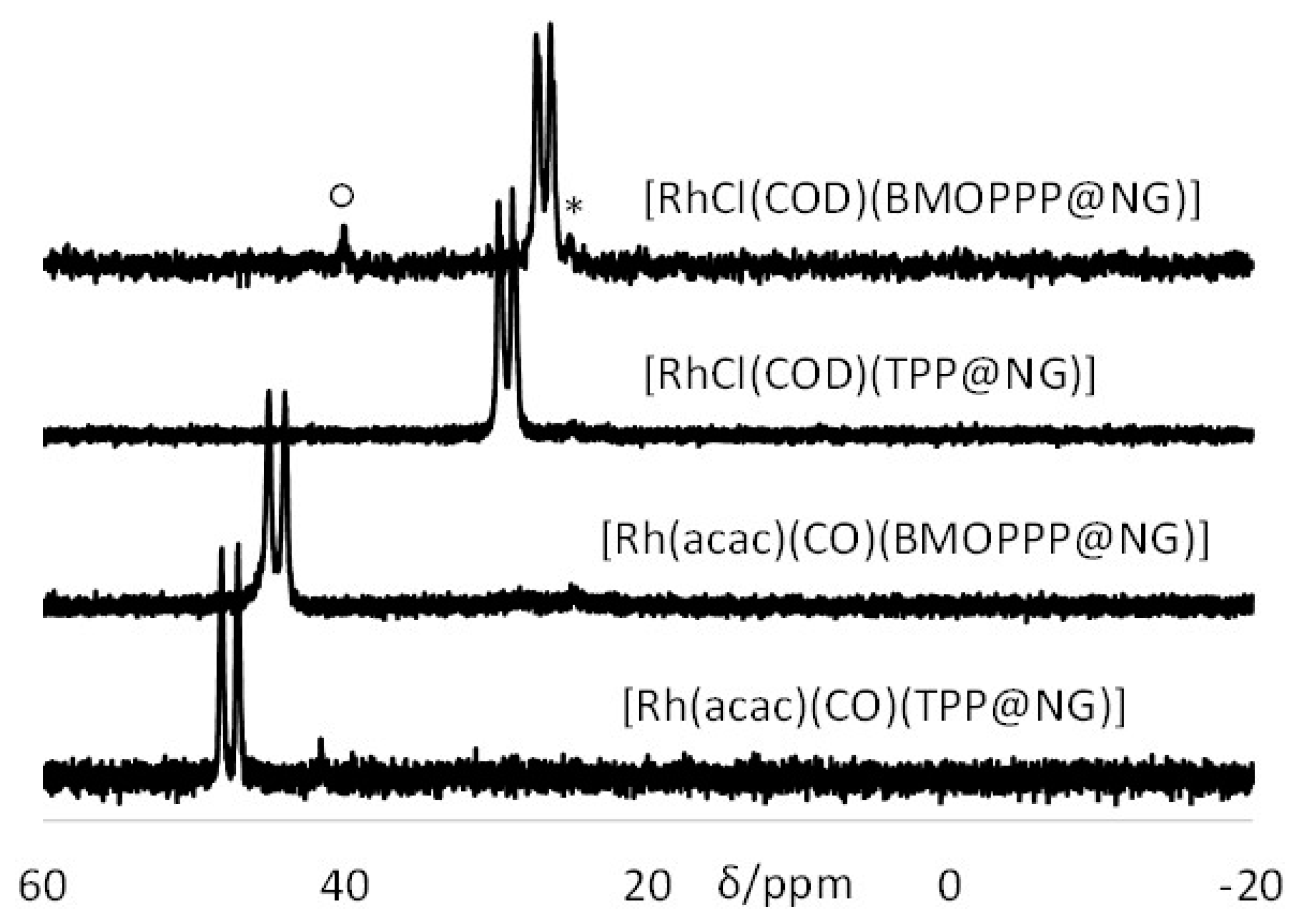
3.2. Metal Coordination inside the Nanoreactors

| Polymer | M = Rh(acac)(CO) | M = RhCl(COD) | ||
|---|---|---|---|---|
| δ/ppm (J/Hz) | Reference | δ/ppm (J/Hz) | Reference | |
| [M(TPP@CCM)] | 47.5 (175) | [7] | 29.3 (149) | This work |
| [M(BMOPPP@CCM)] | 44.5 (176) | [8] | 26.8 (151) | This work |
| [M(TPP@NG)] | 47.6 (175) | [42] | 29.3 (150) | This work |
| [M(BMOPPP@NG)] | 44.5 (172) | This work | 26.8 (150) | This work |

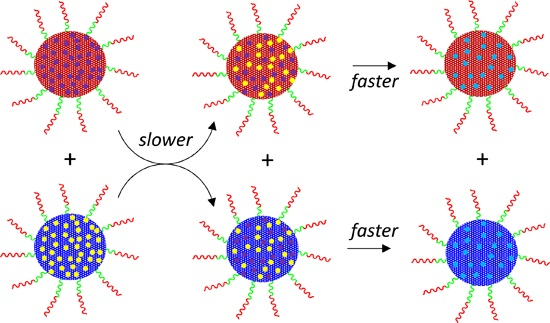
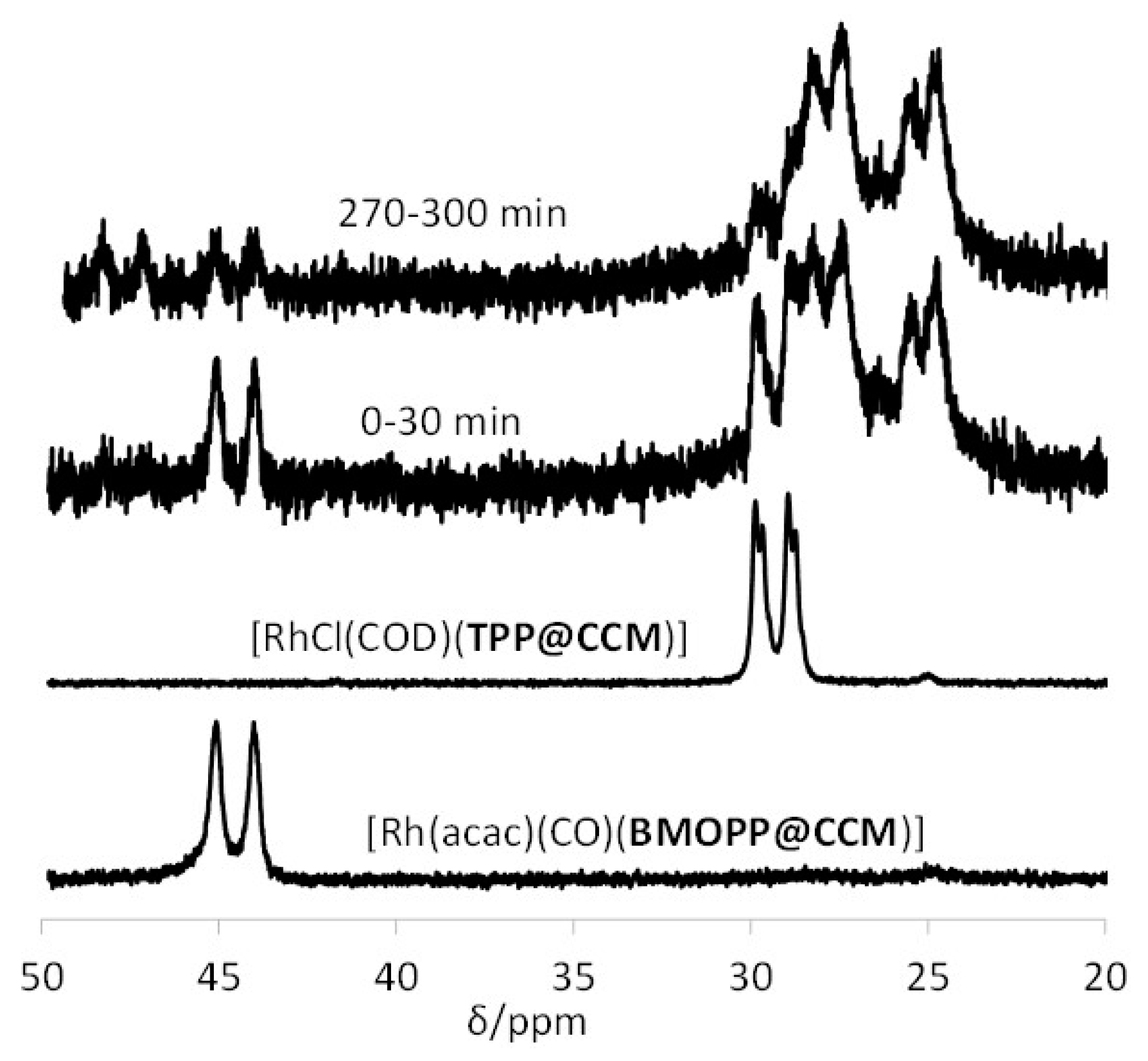
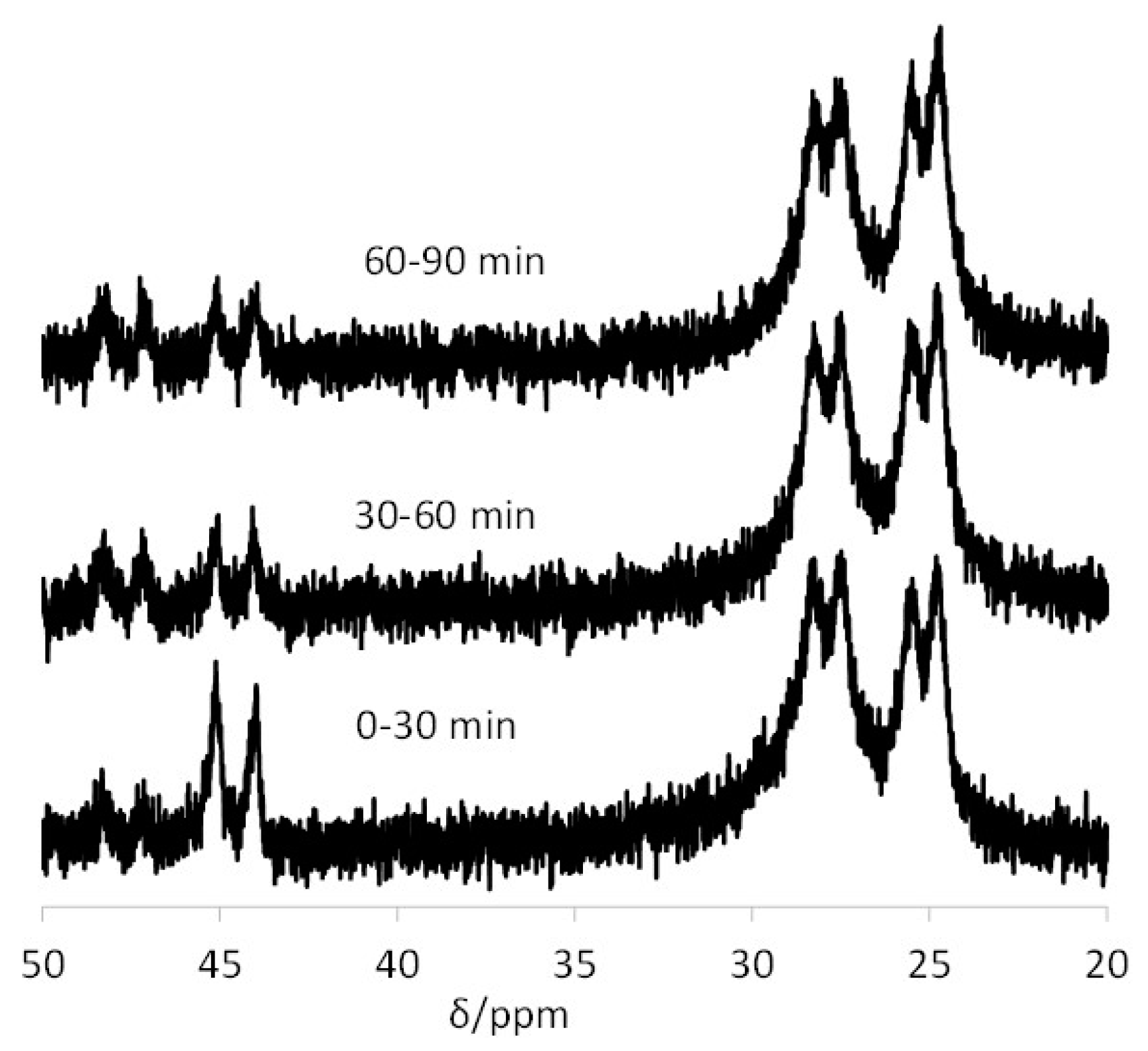
3.3. Interparticle Metal Migration for [Rh(acac)(CO)]-Loaded TPP@NG

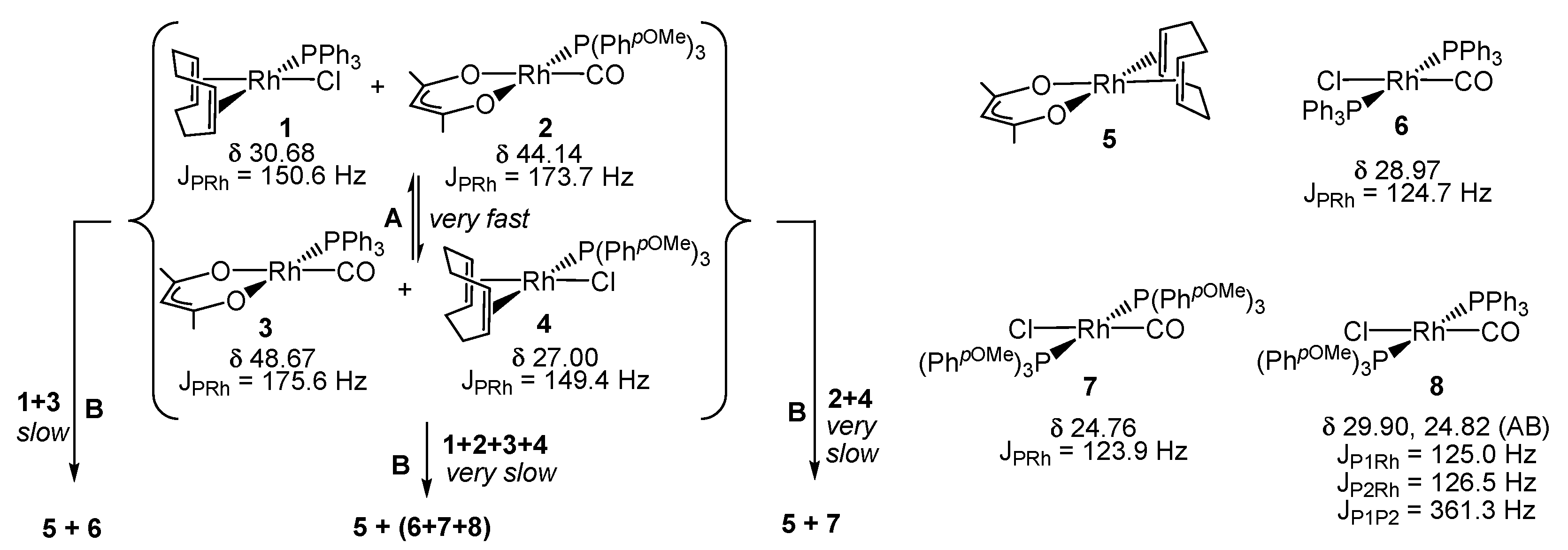
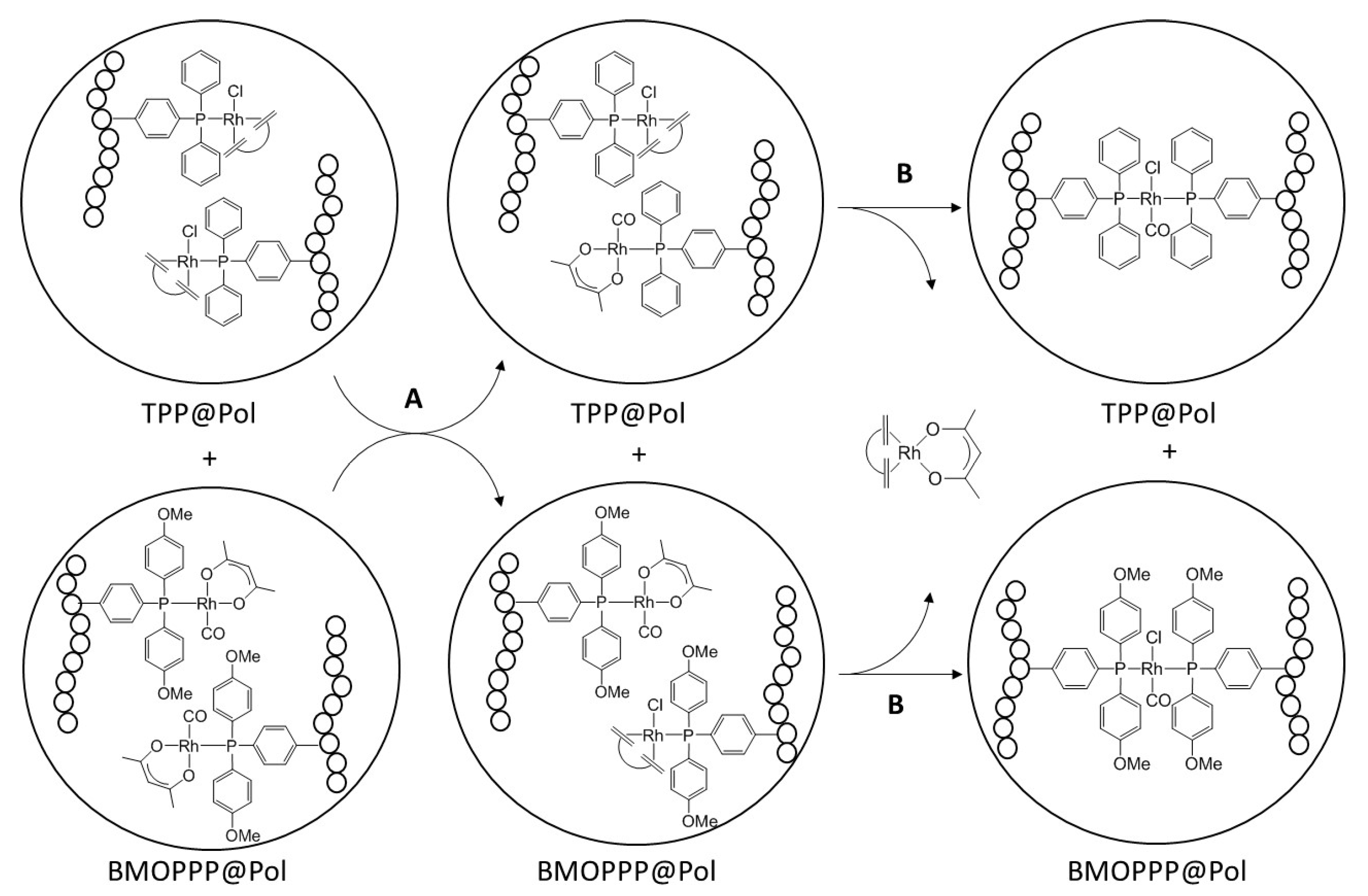
3.4. Interparticle Cross-Migration




4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| acac | acetylacetonato ligand |
| BMOPPP | bis(p-methoxyphenyl)phenylphosphine |
| CCM | core-cross-linked micelle |
| COD | η4-1,5-cyclooctadiene ligand |
| NG | nanogel |
| TPP | triphenylphosphine |
References
- Cotanda, P.; Petzetakis, N.; O’Reilly, R.K. Catalytic polymeric nanoreactors: More than a solid supported catalyst. MRS Commun. 2012, 2, 119–126. [Google Scholar] [CrossRef]
- Lu, A.; O’Reilly, R.K. Advances in nanoreactor technology using polymeric nanostructures. Curr. Opin. Biotech. 2013, 24, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Levins, A.D.; Wang, X.F.; Moughton, A.O.; Skey, J.; O’Reilly, R.K. Synthesis of core functionalized polymer micelles and shell cross-linked nanoparticles. Macromolecules 2008, 41, 2998–3006. [Google Scholar]
- Moughton, A.O.; O’Reilly, R.K. Noncovalently connected micelles, nanoparticles, and metal-functionalized nanocages using supramolecular self-assembly. J. Am. Chem. Soc. 2008, 130, 8714–8725. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Jia, Y.; Liu, L.; Chang, W.; Li, J. Novel organotin-containing shell-cross-linked knedel and core-cross-linked knedel synthesis and application in catalysis. J. Polym. Sci. Polym. Chem. 2010, 48, 5992–6002. [Google Scholar] [CrossRef]
- Liu, R.; Wang, S.; Yao, J.; Xu, W.; Li, H. Cross-linked reverse micelles with embedded water pools: A novel catalytic system based on amphiphilic block copolymers. RSC Adv. 2014, 4, 38234–38240. [Google Scholar] [CrossRef]
- Zhang, X.; Cardozo, A.F.; Chen, S.; Zhang, W.; Julcour, C.; Lansalot, M.; Blanco, J.F.; Gayet, F.; Delmas, H.; Charleux, B.; et al. Core-shell nanoreactors for efficient aqueous biphasic catalysis. Chem. Eur. J. 2014, 20, 15505–15517. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Cardozo, A.F.; Julcour, C.; Blanco, J.F.; Barthe, L.; Gayet, F.; Charleux, B.; Lansalot, M.; D’Agosto, F.; Delmas, H.; et al. Amphiphilic core-cross-linked micelles functionalized with bis(4-methoxyphenyl)phenylphosphine as catalytic nanoreactors for biphasic hydroformylation. Polymer 2015, 72, 327–335. [Google Scholar] [CrossRef]
- Cardozo, A.F.; Julcour, C.; Barthe, L.; Blanco, J.F.; Chen, S.; Gayet, F.; Manoury, E.; Zhang, X.; Lansalot, M.; Charleux, B.; et al. Aqueous phase homogeneous catalysis using core-shell nano-reactors: Application to rhodium catalyzed hydroformylation of 1-octene. J. Catal. 2015, 324, 1–8. [Google Scholar] [CrossRef]
- Poli, R.; Chen, S.; Zhang, X.; Cardozo, A.; Lansalot, M.; D’Agosto, F.; Charleux, B.; Manoury, E.; Gayet, F.; Julcour, C.; et al. One-pot raft synthesis of triphenylphosphine-functionalized amphiphilic core-shell polymers and application as catalytic nanoreactors in aqueous biphasic hydroformylation. ACS Symp. Ser. 2015, 1188, 203–220. [Google Scholar]
- Liu, Y.; Pinon, V.; Weck, M. Poly(norbornene) block copolymer-based shell cross-linked micelles with Co(III)-salen cores. Polym. Chem. 2011, 2, 1964–1975. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Wang, Y.F.; Lu, J.; Pinon, V.; Weck, M. Shell cross-linked micelle-based nanoreactors for the substrate-selective hydrolytic kinetic resolution of epoxides. J. Am. Chem. Soc. 2011, 133, 14260–14263. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Kamigaito, M.; Baek, K.Y.; Ando, T.; Sawamoto, M. Polymer catalysts from polymerization catalysts: Direct encapsulation of metal catalyst into star polymer core during metal-catalyzed living radical polymerization. J. Am. Chem. Soc. 2003, 125, 5288–5289. [Google Scholar] [CrossRef] [PubMed]
- Terashima, T.; Ouchi, M.; Ando, T.; Kamigaito, M.; Sawamoto, M. Metal-complex-bearing star polymers by metal-catalyzed living radical polymerization: Synthesis and characterization of poly(methyl methacrylate) star polymers with Ru(II)-embedded microgel cores. J. Polym. Sci., Polym. Chem. 2006, 44, 4966–4980. [Google Scholar] [CrossRef]
- Terashima, T.; Ouchi, M.; Ando, T.; Kamigaito, M.; Sawamoto, M. Amphiphilic, thermosensitive ruthenium(II)-bearing star polymer catalysts: One-pot synthesis of PEG armed star polymers with ruthenium(II)-enclosed microgel cores via metal-catalyzed living radical polymerization. Macromolecules 2007, 40, 3581–3588. [Google Scholar] [CrossRef]
- Terashima, T.; Ouchi, M.; Ando, T.; Sawamoto, M. Thermoregulated phase-transfer catalysis via PEG-armed Ru(II)-bearing microgel core star polymers: Efficient and reusable Ru(II) catalysts for aqueous transfer hydrogenation of ketones. J. Polym. Sci. Polym. Chem. 2010, 48, 373–379. [Google Scholar]
- Terashima, T.; Ouchi, M.; Ando, T.; Sawamoto, M. Transfer hydrogenation of ketones catalyzed by PEG-armed ruthenium-microgel star polymers: Microgel-core reaction space for active, versatile and recyclable catalysis. Polym. J. 2011, 43, 770–777. [Google Scholar]
- Terashima, T.; Ouchi, M.; Ando, T.; Sawamoto, M. Oxidation of sec-alcohols with Ru(II)-bearing microgel star polymer catalysts via hydrogen transfer reaction: Unique microgel-core catalysis. J. Polym. Sci. Polym. Chem. 2011, 49, 1061–1069. [Google Scholar] [CrossRef] [Green Version]
- Terashima, T.; Sawamoto, M. Microgel-core star polymers as functional compartments for catalysis and molecular recognition. In Progress in Controlled Radical Polymerization: Materials and Applications; Matyjaszewski, K., Sumerlin, B.S., Tsarevsky, N.V., Eds.; American Chemical Society: Washington, D.C., USA, 2012; Volume 1101, pp. 65–80. [Google Scholar]
- Terashima, T. Polymer microgels for catalysis. In Encyclopedia of Polymer Science and Technology, 4th ed.; Mark, H.F., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Behr, A. Thermomorphic solvent systems. In Multiphase Homogeneous Catalysis; Cornils, B., Herrmann, W.A., Horvath, I.T., Leitner, W., Mecking, S., Olivier-Bourbigou, H., Vogt, D., Eds.; Wiley-VCH: Weinheim, Germany, 2005; Volume 1, pp. 327–329. [Google Scholar]
- Behr, A.; Henze, G.; Schomaecker, R. Thermoregulated liquid/liquid catalyst separation and recycling. Adv. Synth. Catal. 2006, 348, 1485–1495. [Google Scholar] [CrossRef]
- Oehme, G. Micellar catalysis. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook, 2nd ed.; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH: Weinheim, Germany, 2002; Volume 2, pp. 835–841. [Google Scholar]
- Nuyken, O.; Weberskirch, R.; Kotre, T.; Schoenfelder, D.; Woerndle, A. Polymers for micellar catalysis. In Polymeric Materials in Organic Synthesis and Catalysis; Buchmeiser, M.R., Ed.; Wiley-VCH: Weinheim, Germany, 2003; pp. 277–304. [Google Scholar]
- Reinsborough, V.C. Micellar Catalysis. In Interfacial Catalysis; Volkov, A.G., Ed.; Marcel Dekker: New York, NY, USA, 2003; pp. 377–390. [Google Scholar]
- Oehme, G. Micellar Systems. In Aqueous-Phase Organometallic Catalysis: Concepts and Applications, 2nd ed.; Cornils, B., Herrmann, W.A., Eds.; Wiley-VCH: Weinheim, Germany, 2004; pp. 256–271. [Google Scholar]
- Khan, M.N. Micellar Catalysis; CRC Press: Boca Raton, FL, USA, 2006; p. 464. [Google Scholar]
- Chiefari, J.; Chong, Y.K.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.; Mayadunne, R.T.A.; Meijs, G.F.; Moad, C.L.; Moad, G.; et al. Living free-radical polymerization by reversible addition-fragmentation chain transfer: The RAFT process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Moad, G.; Chiefari, J.; Chong, Y.K.; Krstina, J.; Mayadunne, R.T.A.; Postma, A.; Rizzardo, E.; Thang, S.H. Living free radical polymerization with reversible addition-fragmentation chain transfer (the life of RAFT). Polym. Int. 2000, 49, 993–1001. [Google Scholar] [CrossRef]
- Barner-Kowollik, C. Handbook of RAFT Polymerization; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Moad, G.; Rizzardo, E.; Thang, S.H. Living radical polymerization by the raft process—A third update. Aust. J. Chem. 2012, 65, 985–1076. [Google Scholar] [CrossRef]
- Keddie, D.J.; Moad, G.; Rizzardo, E.; Thang, S.H. RAFT agent design and synthesis. Macromolecules 2012, 45, 5321–5342. [Google Scholar] [CrossRef]
- Keddie, D.J. A guide to the synthesis of block copolymers using reversible-addition fragmentation chain transfer (RAFT) polymerization. Chem. Soc. Rev. 2014, 43, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Charleux, B.; Delaittre, G.; Rieger, J.; D’Agosto, F. Polymerization-induced self-assembly: From soluble macromolecules to block copolymer nano-objects in one step. Macromolecules 2012, 45, 6753–6765. [Google Scholar] [CrossRef]
- Warren, N.J.; Armes, S.P. Polymerization-induced self-assembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization. J. Am. Chem. Soc. 2014, 136, 10174–10185. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Cornils, B.; Frohning, C.D.; Kohlpaintner, C.W. Progress in hydroformylation and carbonylation. J. Mol. Catal. A 1995, 104, 17–85. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A. Aqueous Phase Organometallic Catalysis; Wiley-VCH: Weinheim, Germany, 1997. [Google Scholar]
- Kohlpaintner, C.W.; Fischer, R.W.; Cornils, B. Aqueous biphasic catalysis: Ruhrchemie/Rhone-Poulenc oxo process. Appl. Catal. A 2001, 221, 219–225. [Google Scholar] [CrossRef]
- Steinborn, D. Fundamentals of Organometallic Catalysis; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Tudor, R.; Ashley, M. Enhancement of industrial hydroformylation processes by the adoption of rhodium-based catalyst: Part II. Key improvements to rhodium process, and use in non-propylene applications. Platin. Met. Rev. 2007, 51, 164–171. [Google Scholar] [CrossRef]
- Tudor, R.; Ashley, M. Enhancement of industrial hydroformylation processes by the adoption of rhodium-based catalyst: Part I. Development of the LP oxo process to the commercial stage. Platin. Met. Rev. 2007, 51, 116–126. [Google Scholar] [CrossRef]
- Lobry, E.; Cardozo, A.F.; Barthe, L.; Blanco, J.F.; Delmas, H.; Chen, S.; Gayet, F.; Zhang, X.; Lansalot, M.; D’Agosto, F.; et al. Core phosphine-functionalized amphiphilic nanogels as catalytic nanoreactors for aqueous biphasic hydroformylation. In preparation.
- Chen, S.; Gayet, F.; Manoury, E.; Joumaa, A.; Lansalot, M.; D’Agosto, F.; Poli, R. Coordination chemistry inside polymeric nanoreactors: Interparticle metal exchange processes and ionic compound vectorization in phosphine-functionalized amphiphilic polymer latexes. Submitted.
- Boursier, T.; Chaduc, I.; Rieger, J.; D’Agosto, F.; Lansalot, M.; Charleux, B. Controlled radical polymerization of styrene in miniemulsion mediated by peo-based trithiocarbonate macromolecular raft agents. Polym. Chem. 2011, 2, 355–362. [Google Scholar] [CrossRef]
- Pruchnik, F.P.; Smolenski, P.; Wajda-Hermanowicz, K. Rhodium(I) acetylacetonato complexes with functionalized phosphines. J. Organomet. Chem. 1998, 570, 63–69. [Google Scholar] [CrossRef]
- Naaktgeboren, A.J.; Nolte, R.J.M.; Drenth, W. P-31 nuclear magnetic-resonance studies of polymer-anchored rhodium(I) complexes. J. Am. Chem. Soc. 1980, 102, 3350–3354. [Google Scholar] [CrossRef]
- Chen, S.; Manoury, E.; Poli, R. Slow exchange of bidentate ligands between rhodium(I) complexes: Evidence of both neutral and anionic ligand exchange. Eur. J. Inorg. Chem. 2014, 2014, 5820–5826. [Google Scholar] [CrossRef]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Manoury, E.; Gayet, F.; Poli, R. Coordination Chemistry inside Polymeric Nanoreactors: Metal Migration and Cross-Exchange in Amphiphilic Core-Shell Polymer Latexes. Polymers 2016, 8, 26. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8020026
Chen S, Manoury E, Gayet F, Poli R. Coordination Chemistry inside Polymeric Nanoreactors: Metal Migration and Cross-Exchange in Amphiphilic Core-Shell Polymer Latexes. Polymers. 2016; 8(2):26. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8020026
Chicago/Turabian StyleChen, Si, Eric Manoury, Florence Gayet, and Rinaldo Poli. 2016. "Coordination Chemistry inside Polymeric Nanoreactors: Metal Migration and Cross-Exchange in Amphiphilic Core-Shell Polymer Latexes" Polymers 8, no. 2: 26. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8020026