
Analysis of the Heterogeneities of First and Second Order of Cellulose Derivatives: A Complex Challenge


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
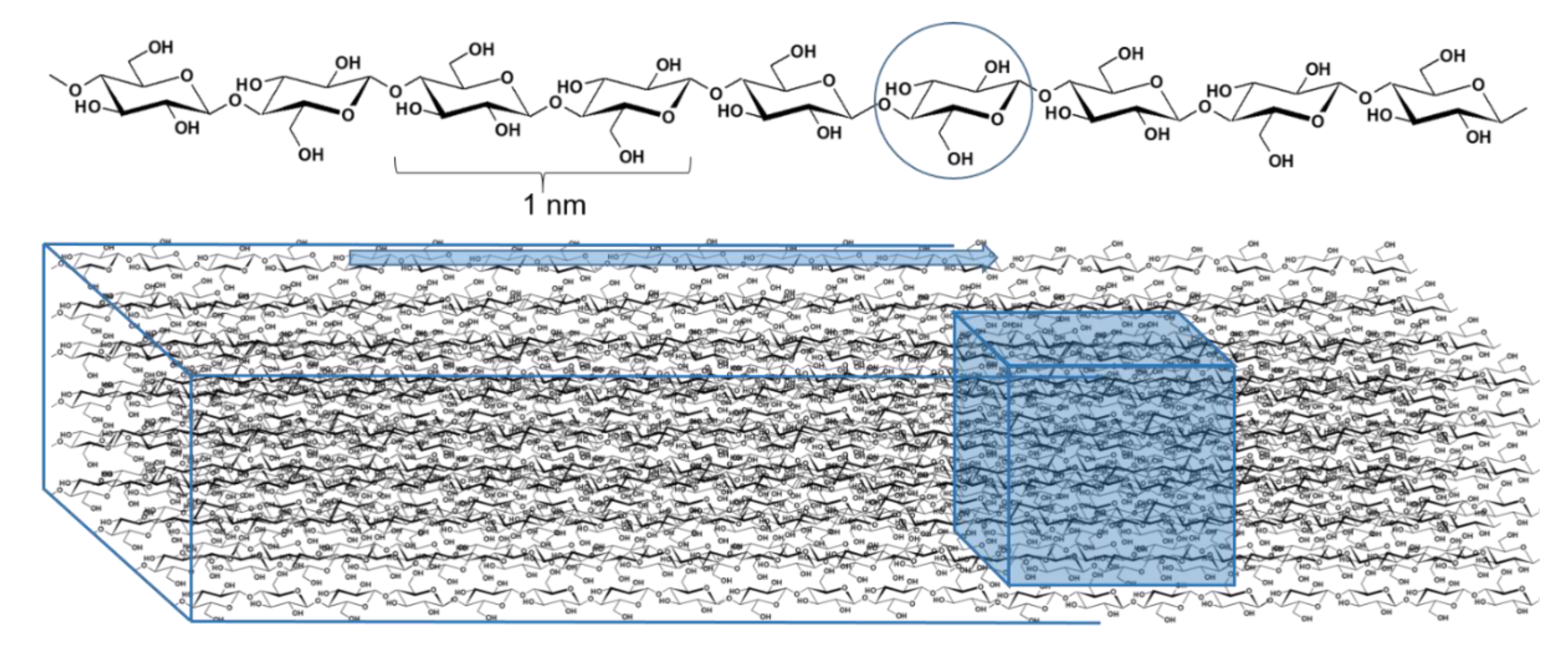
1.1. How Complex?
1.2. The Two- and Three-Dimensional Substituent Distribution
1.3. Analytical Approaches
1.4. Résumé
2. Materials and Methods
2.1. General
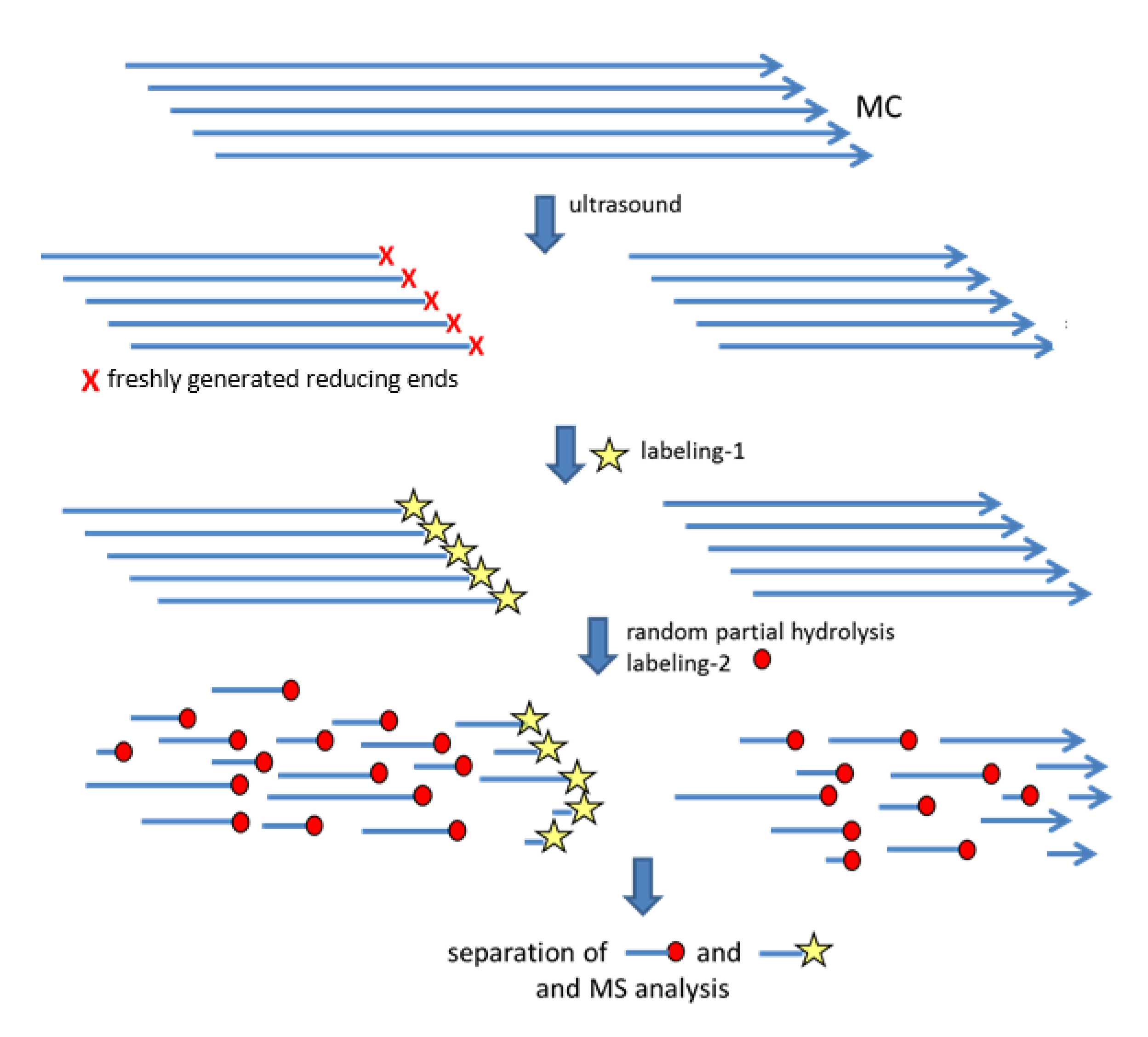
2.2. Preparation of One Labeled Oligosaccharide per Chain
2.2.1. Ultrasonic Treatment
2.2.2. Labeling by Reductive Amination with m-ABA
2.2.3. Partial Hydrolysis
2.2.4. Separation of Labeled and Non-Labeled O-Me/O-Me-d3-Cellooligosaccharides (COS)
2.3. Instrumentation
Electrospray Ionization-Mass Spectrometry (ESI-MS)
2.4. Fractionation of MCs by Means of Soxhlet-Extraction
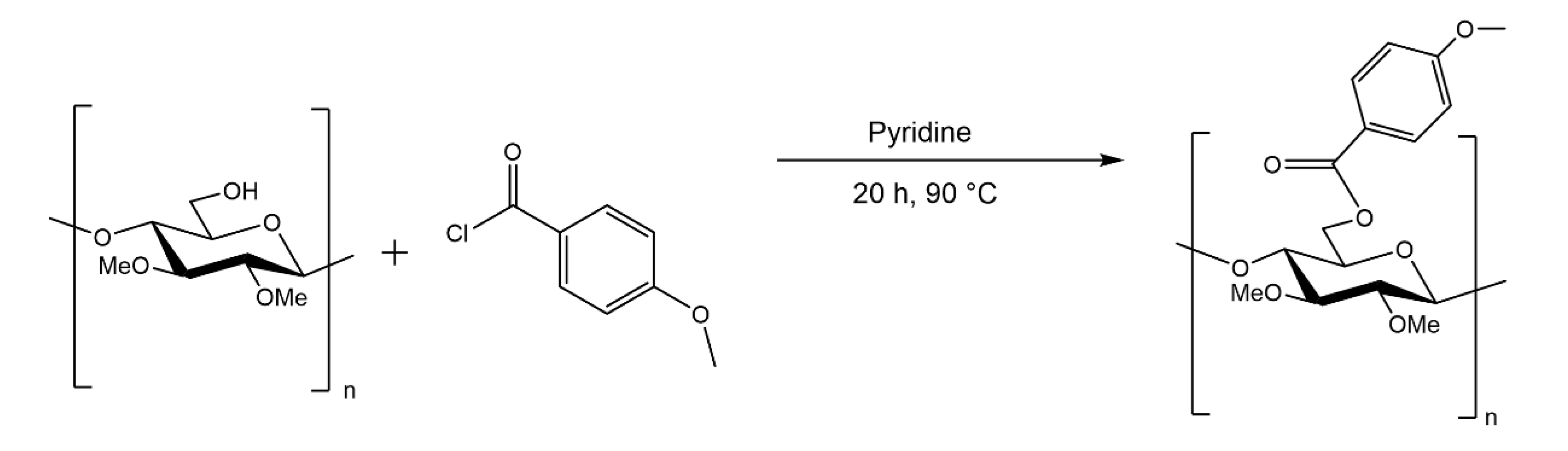
2.5. Fractionation of Esterified MC
2.5.1. Propionylation and Methoxybenzoylation of MC: Impeller Method
2.5.2. Methoxybenzoylation of MC with the Acid Anhydride in Pyridine
2.5.3. Fractionation of Esterified MC by Solid Phase Extraction
2.5.4. Fractionation of MC Methoxybenzoate by LAC-HPLC
2.5.5. Constituent Analysis of Esterified MC Fractions
3. Results and Discussion
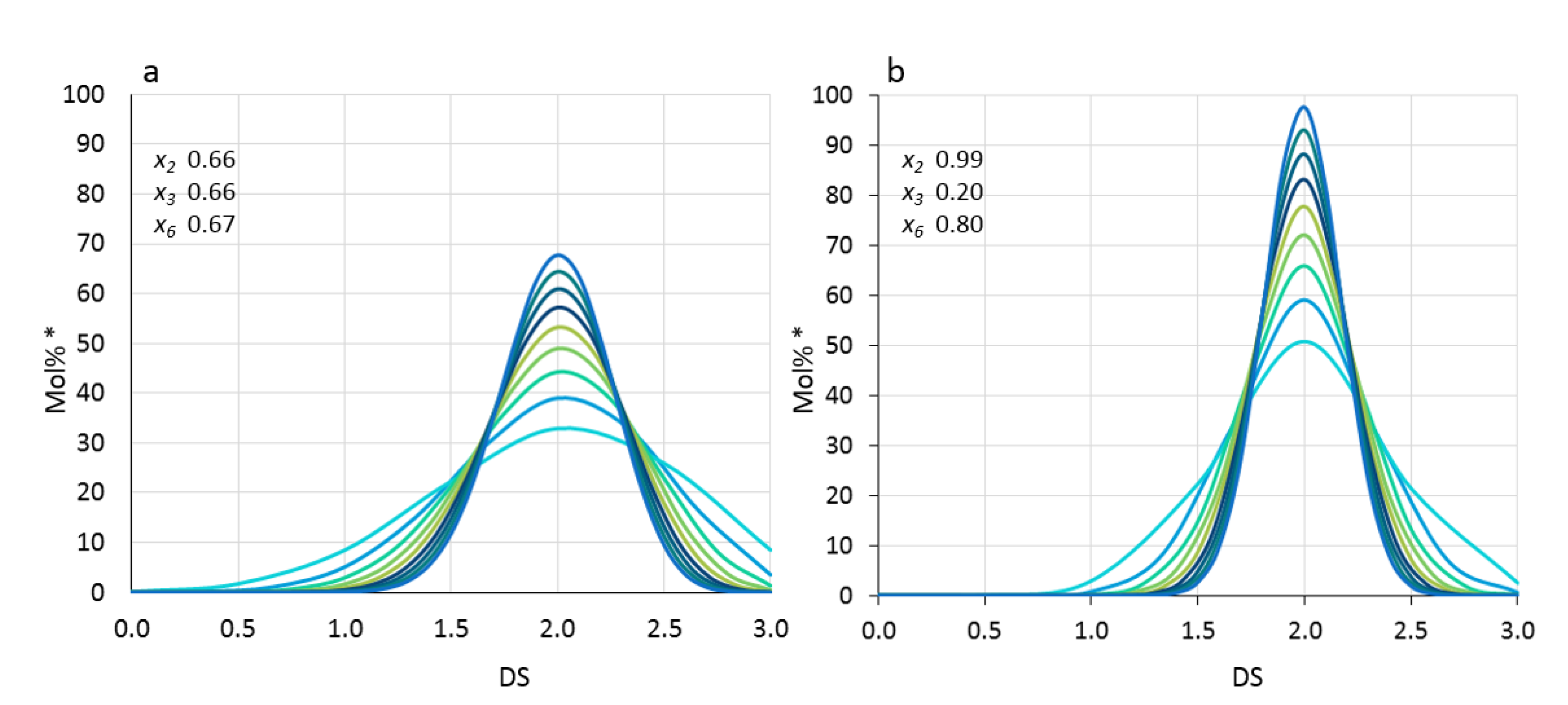
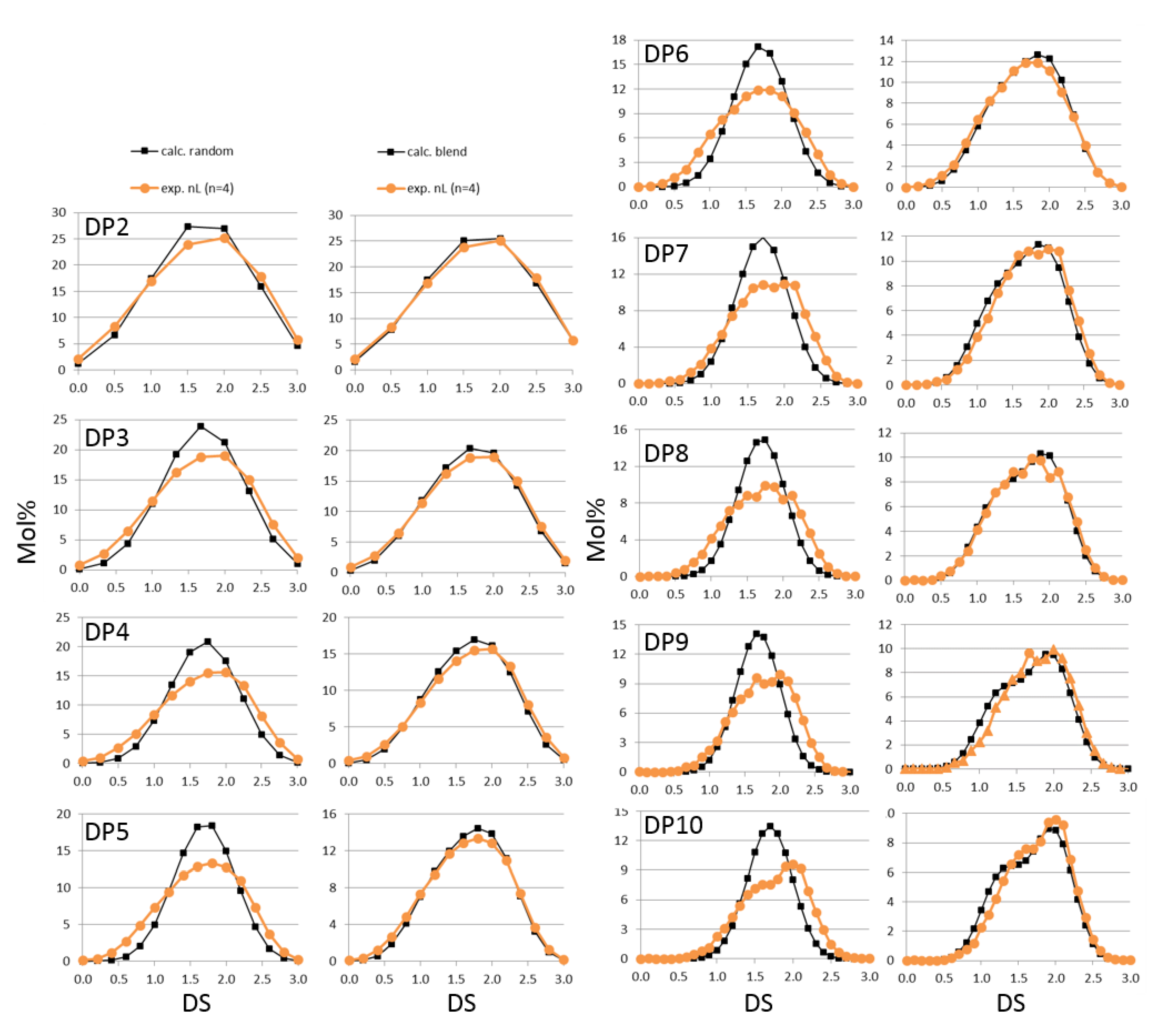
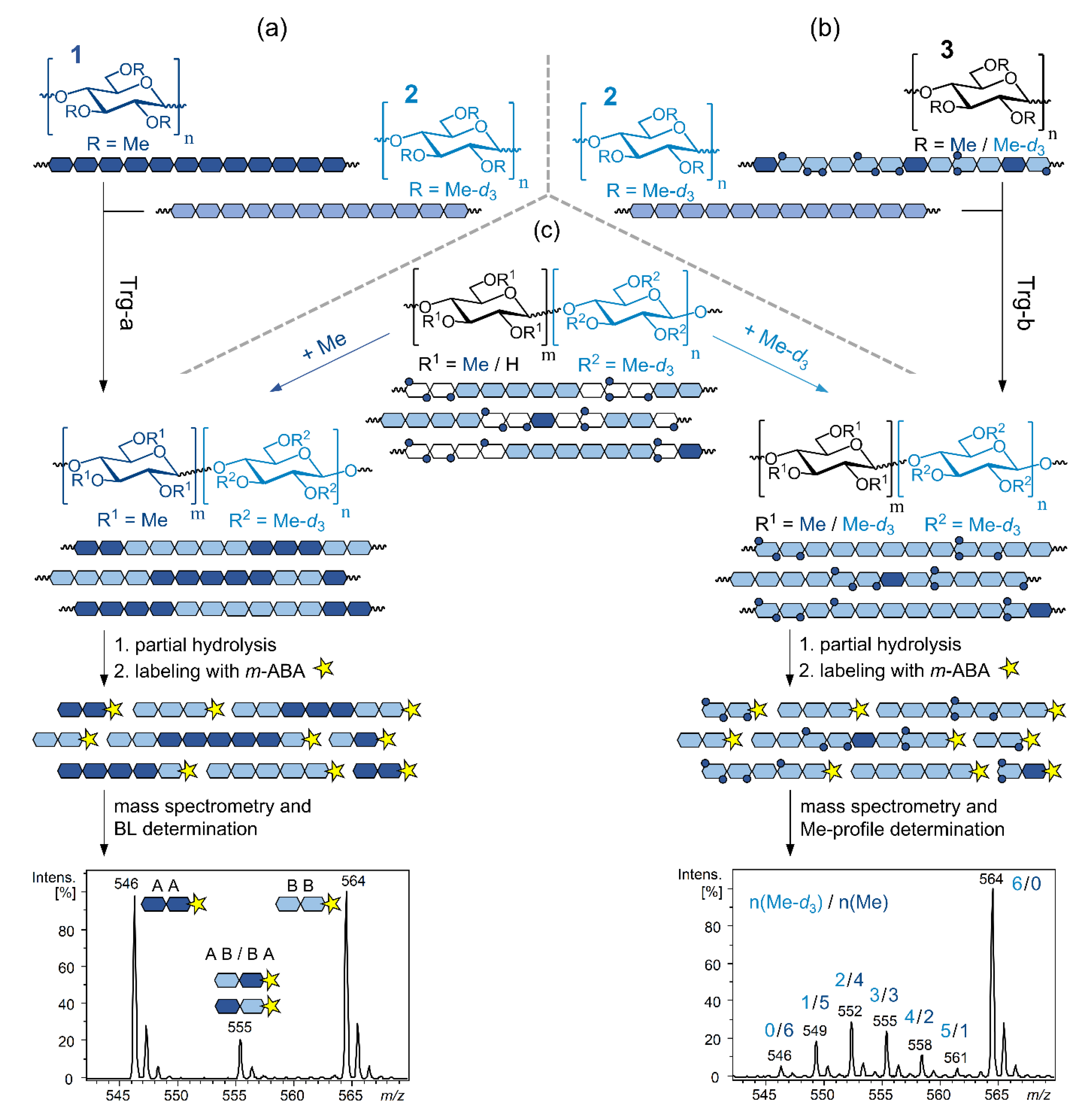
3.1. Is There Any Difference between the Heterogeneities of First and Second Order? A Practical Method Resulting in a Gedankenexperiment
3.2. Analysis of Two Mixed MCs-Blend or Block Structure?
3.3. Detection of Transition Domains
3.4. Fractionation According to Heterogeneity of First Order
3.4.1. Fractionation of MC Esters on NP and RP Phases
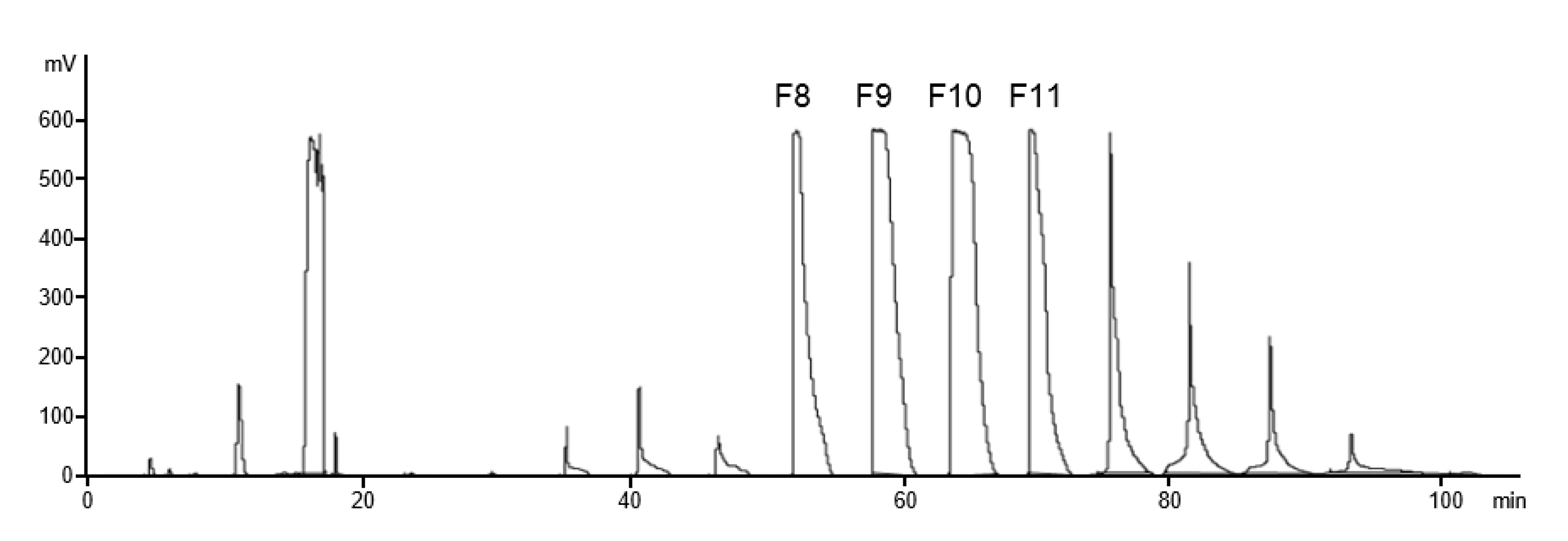
3.4.2. Fractionation of MC Methoxybenzoates by Gradient HPLC
- Complete acylation of MCs and purification is possible without selective losses of material (except hemicelluloses).
- Solutions of fully protected MC-MeOBz can be micro-filtrated without losses.
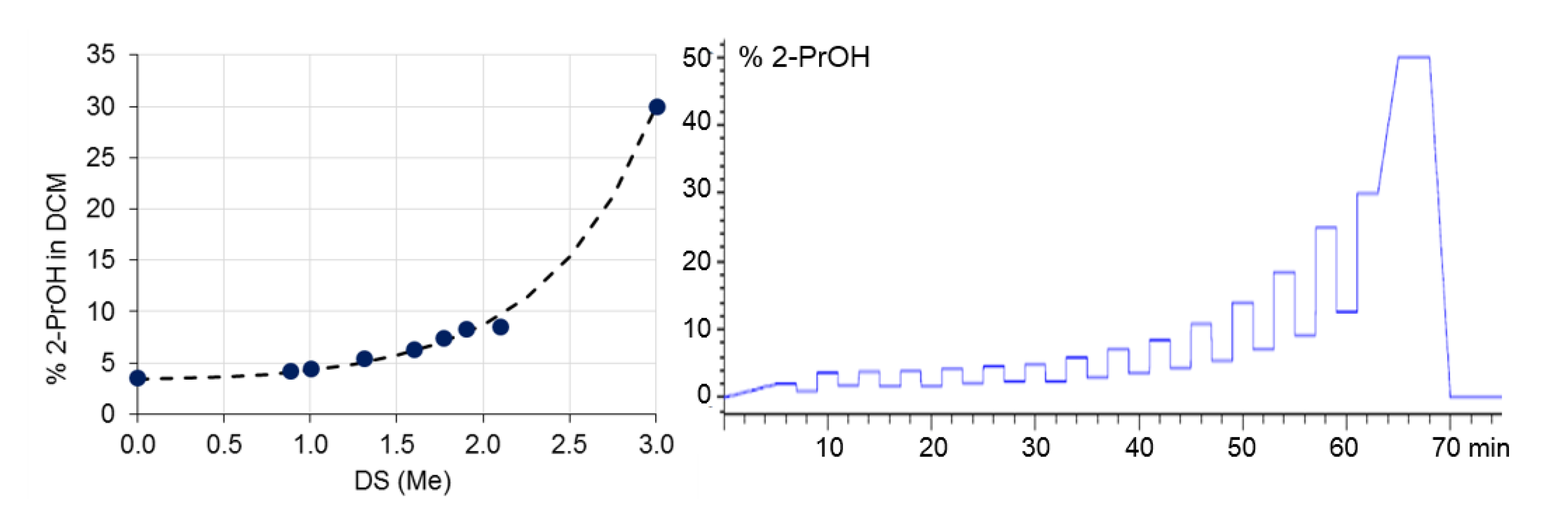
- MC-MeOBz are completely recovered from the silica column with the DCM/2-PrOH eluent system. This was proven by measuring the signal intensities of the UV detector after direct injection (without column) of Me-MeBz solutions, and injection with column. Both signals were equal.
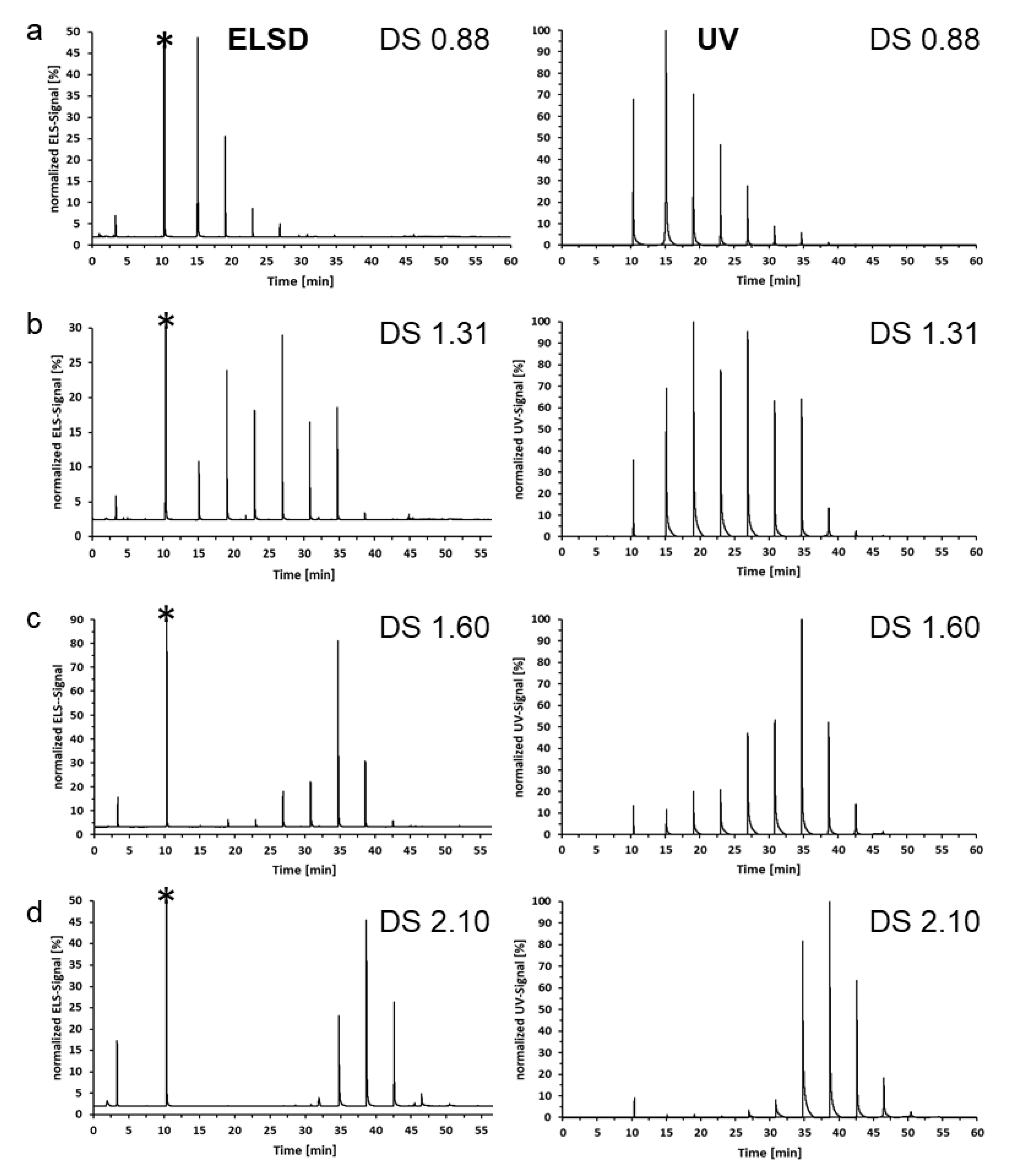
- MC-MeOBz can be separated according to polarity, mainly defined by the ratio of DSMe/DSMeOBz, by liquid adsorption chromatography on normal (DCM/ROH) or reversed phases (ACN/H2O).
- Portions of MC-MeOBz can be eluted from cartridges or HPLC columns by stepwise enhancing the volume of the displacer and intermittent trapping of the residual material by reducing the stronger eluent again.
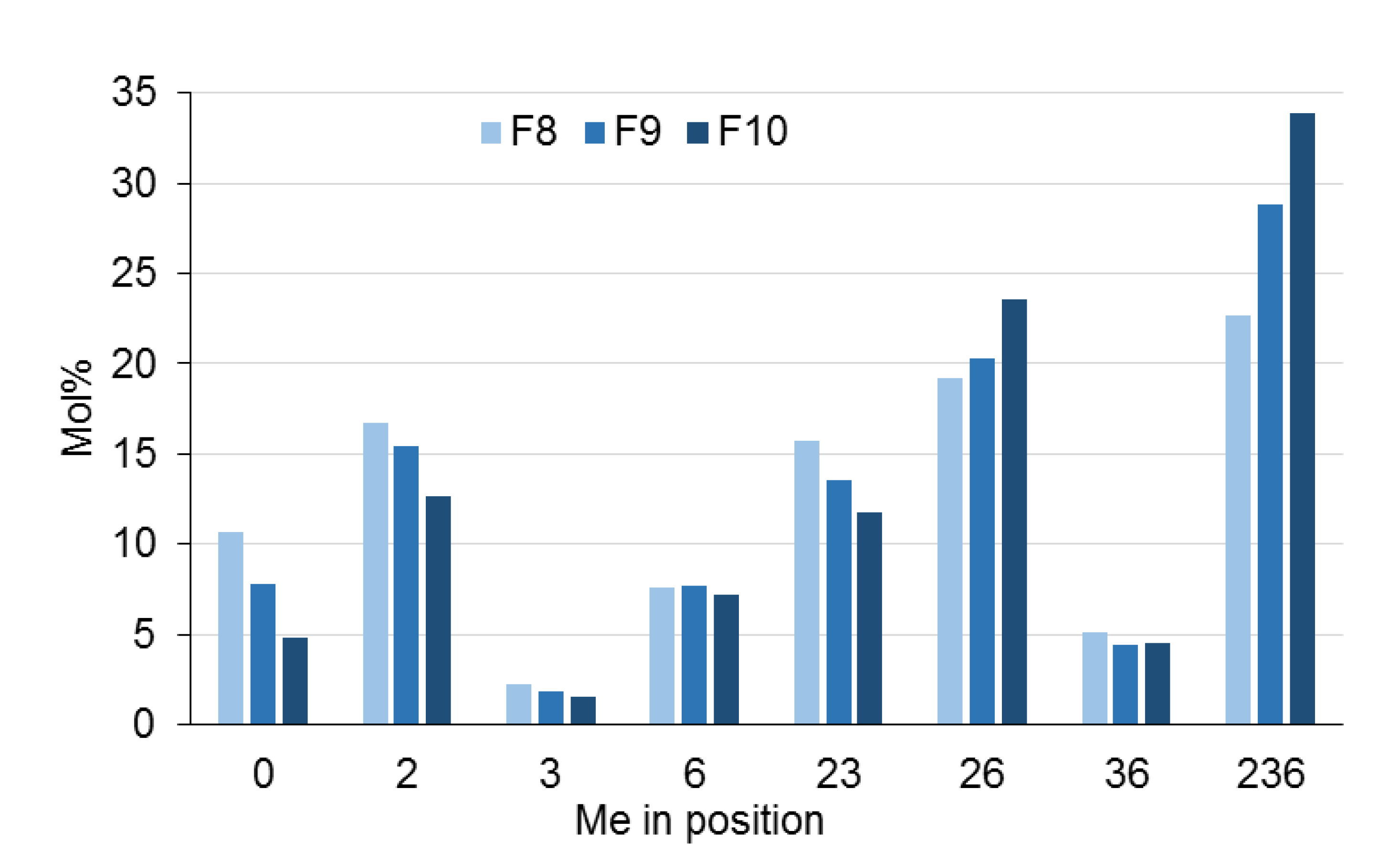
- The fractions obtained with NP showed increasing DSMe with the fraction number, and the opposite is true for RP.
- Sufficient material for further methyl pattern analysis can be obtained by semi-preparative HPLC. MC-MeOBz have been submitted to the alditol acetate method after deprotection with sodium methanolate. The average DS of subsequent main fractions increased by 7–8%.
- Re-analysis of the preparatively obtained fractions still show a wide DS range, which can probably be narrowed by accumulating fractions from several runs with less material per injection. Resolution should be improved, and quantitative reconstruction of the sample needs to be performed.
- If more material is collected, it can also be used for O-Me/O-Me-d3-COS preparation and MS analysis to determine the methyl distribution along the polysaccharide chain.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fannon, J.E.; Gray, J.A.; Gunawan, N.; Huber, K.C.; BeMiller, J.N. Heterogeneity of starch granules and the effect of granule channelization on starch modification. Cellulose 2004, 11, 247–254. [Google Scholar] [CrossRef]
- Tomasik, P.; Schilling, C.H. Chemical modification of starch. Adv. Carbohydr. Chem. Biochem. 2004, 59, 175–403. [Google Scholar] [PubMed]
- Whistler, R.L.; Madson, M.A.; Zhao, J.; Daniel, J.R. Surface derivatization of corn starch granules. Cereal Chem. 1998, 75, 72–74. [Google Scholar] [CrossRef]
- Steeneken, P.A.M.; Woortman, A.J.J. Surface effects in the acetylation of granular potato starch. Carbohydr. Res. 2008, 343, 2278–2284. [Google Scholar] [CrossRef] [PubMed]
- Steeneken, P.A.M.; Smith, E. Topochemical effects in the methylation of starch. Carbohydr. Res. 1991, 209, 239–249. [Google Scholar] [CrossRef]
- Muetgeert, J. Fractionation of starch. Adv. Carbohydr. Chem. 1961, 16, 299–333. [Google Scholar] [PubMed]
- Medronho, B.; Romano, A.; Miguel, M.G.; Stigsson, L.; Lindman, B. Rationalizing cellulose (in)solubility: Reviewing basic physicochemical aspects and role of hydrophobic interactions. Cellulose 2012, 19, 581–587. [Google Scholar] [CrossRef]
- Glasser, W.G.; Atalla, R.H.; Blackwell, J.; Brown, R.M., Jr.; Burchard, W.; French, A.D.; Klemm, D.O.; Nishiyama, Y. About the structure of cellulose: Debating the Lindman hypothesis. Cellulose 2012, 19, 589–598. [Google Scholar] [CrossRef]
- Klemm, D.; Heublein, B.; Fink, H.-P.; Bohn, A. Cellulose: Fascinating Biopolymer and Sustainable Raw Material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef] [PubMed]
- Klemm, D.; Philipp, B.; Heinze, T.; Heinze, U.; Wagenknecht, W. Comprehensive Cellulose Chemistry: Fundamentals and Analytical Methods, Volume 1, and Functionalization of Cellulose; Wiley-VCH: Weinheim, Germany, 1998; Volume 2. [Google Scholar]
- Mann, G.; Kunze, J.; Loth, F.; Fink, H.-P. Cellulose ethers with a block-like distribution of the substituents by structure-selective derivatization of cellulose. Polymer 1998, 39, 3155–3165. [Google Scholar] [CrossRef]
- Wüstenberg, T. Cellulose and Cellulose Derivatives in the Food Industry; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Ott, E.; Spurlin, H.M.; Grafflin, M.G. Cellulose and Cellulose Derivatives, High Polymers V, 1-3, 2nd ed.; Interscience Publishers: New York, NY, USA, 1954. [Google Scholar]
- Spurlin, H.M. Arrangement of substituents in cellulose derivatives. J. Am. Chem. Soc. 1939, 61, 2222–2227. [Google Scholar] [CrossRef]
- Heinze, T.; Liebert, T.; Koschella, A. Esterification of Polysaccharides; Springer: Berlin/Heidelberg, Germany, 2006; p. 232. [Google Scholar]
- Koschella, A.; Heinze, T.; Klemm, D. First synthesis of 3-O-functionalized cellulose ethers via 2,6-di-O-protected silyl cellulose. Macromol. Biosci. 2001, 1, 49–54. [Google Scholar] [CrossRef]
- Kamitakahara, H.; Koschella, A.; Mikawa, Y.; Nakatsubo, F.; Heinze, T.; Klemm, D. Synthesis and comparison of 2,6-di-O-methyl-celluloses from natural and synthetic cellulloses. Macromol. Biosci. 2006, 8, 690–700. [Google Scholar] [CrossRef]
- Köhler, S.; Liebert, T.; Heinze, T.; Vollmer, A.; Mischnick, P.; Möllmann, E.; Becker, W. Interactions of Ionic Liquids with Polysaccharides, 9. Hydroxyalkylation of Cellulose without Additional Inorganic Bases. Cellulose 2010, 17, 437–448. [Google Scholar] [CrossRef]
- Reuben, J. Description and Analysis of Hydroxyethylcelluloses. Macromolecules 1984, 17, 156–161. [Google Scholar] [CrossRef]
- Huebner, B.; Knarr, M.; Adden, R.; Sammler, R.; Adden, A. Methods and Compositioins for Inducing Satiety. U.S. 9295712B2, 16 March 2016. [Google Scholar]
- Kamitakahara, F.; Nakatsubo, F.; Klemm, D. Block co-oligoers of tri-O-methylated and unmodfied cello-oligosaccharides as model compounds for methylcellulose and its dissolution/gelation behavior. Cellulose 2005, 13, 375–392. [Google Scholar] [CrossRef]
- Kamitakahara, H.; Funakoshi, T.; Takanao, T.; Nakatsubo, F. Synthesis of 2,6-O-alkyl celluloses: Influence of methyl and ethyl groups regioselectively introduced at O-2 and O-6 positions on their solubility. Cellulose 2009, 16, 1167–1178. [Google Scholar] [CrossRef]
- Arisz, P.W.; Kauw, H.J.J.; Boon, J.J. Substituent distribution along the cellulose backbone in O-methylcelluloses using GC and FAB MS for monomer and oligomer analysis. Carbohydr. Res. 1995, 271, 1–14. [Google Scholar] [CrossRef]
- Mischnick, P.; Kühn, G. Correlation between reaction conditions and primary structure: Model studies on methyl amylose. Carbohydr. Res. 1996, 290, 199–207. [Google Scholar] [CrossRef]
- Mischnick, P.; Adden, R. Fractionation of Polysaccharide Derivatives and Subsequent Analysis to Differentiate Heterogeneities on Various Hierarchical Levels. Macromol. Symp. 2008, 262, 1–7. [Google Scholar] [CrossRef]
- Adden, R.; Mischnick, P. A novel method for the analysis of the substitution pattern of O-methyl-α- and β-1,4-glucans by means of electrospray-ionization mass spectrometry/collision induced dissociation. Int. J. Mass Spectrom. 2005, 242, 63–73. [Google Scholar] [CrossRef]
- Mischnick, P. Analysis of the Substituent Distribution in Cellulose Ethers—Recent Contributions. In Cellulose Science and Technology, 1st ed.; Rosenau, T., Potthast, A., Hell, J., Eds.; Wiley: Weinheim, Germany, 2019; pp. 143–173. [Google Scholar]
- Kondo, T. Preparation of 6-O-alkyl-celluloses. Carbohydr. Res. 1993, 238, 231–240. [Google Scholar] [CrossRef]
- Xu, D.; Voiges, K.; Elder, T.; Mischnick, P.; Edgar, K. Regioselective Synthesis of Cellulose Ester Homopolymers. Biomacromolecules 2012, 13, 2195–2201. [Google Scholar] [CrossRef]
- Adden, R.; Niedner, W.; Müller, R.; Mischnick, P. Comprehensive analysis of the substituent distribution in the glucosyl units and along the polymer chain of hydroxyethylmethylcelluloses and statistical evaluation. Anal. Chem. 2006, 78, 1146–1157. [Google Scholar] [CrossRef]
- Mischnick, P.; Momcilovic, D. Chemical Structure Analysis of Starch and Cellulose Derivatives. Adv. Carbohydr. Chem. Biochem. 2010, 64, 117–210. [Google Scholar] [CrossRef] [PubMed]
- Steeneken, P.A.M. Reactivity of amylose and amylopectin in potato starch. Starch-Stärke 1984, 36, 13–18. [Google Scholar] [CrossRef]
- Steeneken, P.A.M.; Woortman, A.J.J. Substitution pattern in methylated starch as studied by enzymic degradation. Carbohydr. Res. 1994, 258, 207–221. [Google Scholar]
- Steeneken, P.A.M.; Tas, A.C.; Woortman, A.J.J.; Sanders, P.; Mijland, P.J.H.C.; de Weijs, L.G.R. Substitution patterns in methylated starch as revealed from the structure and composition of fragments in enzymatic digests. Carbohydr. Res. 2008, 343, 2411–2416. [Google Scholar] [CrossRef] [PubMed]
- Nojiri, M.; Kondo, T. Application of regioselectively substituted methylcelluloses to characterize the reaction mechanism of cellulase. Macromolecules 1996, 29, 2392–2395. [Google Scholar] [CrossRef]
- Horner, S.; Puls, J.; Saake, B.; Klohr, E.-A.; Thielking, H. Enzyme-aided characterisation of carboxymethylcellulose. Carbohyd. Polym. 1999, 40, 1–7. [Google Scholar] [CrossRef]
- Saake, B.; Puls, J.; Wagenknecht, W. Endoglucanase fragmentation of cellulose sulfates derived from different synthesis concepts. Carbohydr. Polym. 2002, 48, 7–14. [Google Scholar] [CrossRef]
- Melander, C.; Adden, R.; Brinkmalm, G.; Gorton, L.; Mischnick, P. New approaches to the analysis of enzymatically hydrolyzed methyl cellulose. Part 2. comparison of various enzyme preparations. Biomacromolecules 2006, 7, 1410–1421. [Google Scholar] [CrossRef] [PubMed]
- Schagerlöf, U.; Schagerlöf, H.; Momcilovic, D.; Brinkmalm, G.; Tjerneld, F. Endoglucanase sensitivity for substituents in methyl cellulose hydrolysis studied using MALDI-TOFMS for oligosaccharide analysis and structural analysis of enzyme active sites. Biomacromolecules 2007, 8, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Enebro, J.; Momcilovic, D.; Siika-aho, M.; Karlsson, S. Liquid chromatography combined with mass spectrometry for investigation of endoglucanase selectivity on carboxymethyl cellulose. Carbohydr. Res. 2009, 344, 2173–2181. [Google Scholar] [CrossRef] [PubMed]
- Cuers, J.; Unterieser, I.; Burchard, W.; Adden, R.; Rinken, M.; Mischnick, P. Simultaneous determination of substituent patterns in partially acid hydrolyzed O-Me/O-Me-d3-cellulose and quantification of the obtained oligomers by HPLC-ESI-MS. Carbohydr. Res. 2012, 348, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Mischnick, P.; Unterieser, I.; Voiges, K.; Cuers, J.; Rinken, M.; Adden, R. A new method for the analysis of the substitution pattern of hydroxyethyl(methyl) celluloses over the polysaccharide chain. Macromol. Chem. Phys. 2013, 214, 1363–1374. [Google Scholar] [CrossRef]
- Gohdes, M.; Mischnick, P. Determination of the substitution pattern in the polymer chain of cellulose sulfates. Carbohydr. Res. 1998, 309, 109–115. [Google Scholar] [CrossRef]
- Brun, Y. The mechanism of copolymer retention in interactive polymer chromatography. I. Critical point of adsorption for statistical copolymers. J. Liquid Chromatogr. Rel. Techn. 1999, 22, 3027–3065. [Google Scholar] [CrossRef]
- Pasch, H.; Thrathnigg, B. Multidimensional HPLC of Polymers. In Springer Laboratory Manuals in Polymer Science; Alig, I., Pasch, H., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Radke, W. Polymer separations by liquid interaction chromatography: Principles—prospects—limitations. J. Chromatogr. A 2014, 1335, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Shakun, M.; Heinze, T.; Radke, W. Determination of the DS distribution of non-degraded sodium carboxymethyl cellulose by gradient chromatography. Carbohydr. Polym. 2013, 98, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Shakun, M.; Heinze, T.; Radke, W. Characterization of sodium carboxymethyl cellulose by comprehensive two-dimensional liquid chromatography. Carbohydr. Polym. 2015, 130, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Ghareeb, H.O.; Radke, W. Separation of cellulose acetates by degree of substitution. Polymer 2013, 54, 2632–2638. [Google Scholar] [CrossRef]
- Ghareeb, H.O.; Radke, W. Characterization of cellulose acetates according to DS and molar mass using two-dimensional chromatography. Carbohydr. Polym. 2013, 98, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Krasselt, K.; Schmidt, I.; Weightman, D. Distribution of substituents along the cellulose chain on cellulose xanthate and carboxymethyl cellulose. Macromol. Symp. 2005, 223, 109–120. [Google Scholar] [CrossRef]
- Fischer, S.; Thümmler, K.; Volkert, B.; Hettrich, K.; Schmidt, I.; Fischer, K. Properties and applications of cellulose acetate. Macromol. Symp. 2008, 262, 89–96. [Google Scholar] [CrossRef]
- Fitzpatrick, F.; Schagerlöf, H.; Andersson, T.; Richardson, S.; Tjerneld, F.; Wahlung, K.-G.; Wittgren, B. NMR, cloud-point measurements and enzymatic depolymerization: Complementary tools to investigate substituent patterns in modified celluloses. Biomacromolecules 2006, 7, 2909–2917. [Google Scholar] [CrossRef] [PubMed]
- Oudhoff, K.A.; Buijtenhuijs, F.A.; Winjen, P.H.; Schoenmakers, P.J.; Kok, W.T. Determination of the degree of substitution and its distribution of carboxymethylcelluloses by capillary zone electrophoresis. Carbohydr. Res. 2004, 339, 1917–1924. [Google Scholar] [CrossRef] [PubMed]
- Bol, M.; Dobos, M.A.; Lebioda, S.; Saake, B.; Mischnick, P. Methanolysis of Carboxymethyl Cellulose—A Comprehensive Study. Cellulose 2019, 26, 383–397. [Google Scholar] [CrossRef]
- Adden, R.; Müller, R.; Mischnick, P. Fractionation of methyl cellulose according to polarity—A tool to differentiate first and second order heterogeneity of the substituent distribution. Macromol. Chem. Phys. 2006, 207, 954–965. [Google Scholar] [CrossRef]
- Greiderer, A.; Steeneken, L.; Aalbers, T.; Vivó-Truyois, G.; Schoenmakers, P. Characterizaton of hydroxypropylmethylcellulose (HPMC) using comprehensive two-dimensional liquid chromatography. J. Chromatogr. A 2011, 1218, 5787–5793. [Google Scholar] [CrossRef] [PubMed]
- Viridén, A.; Wittgren, B.; Andersson, T.; Arahmsén-Alami, S.; Larsson, A. Influence of substitution pattern on solution behavior of hydroxypropyl methylcellulose. Biomacromolecules 2009, 10, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Wirick, M.G.; Waldman, M.H. Some solution properties of fractionated water-soluble hydroxypropylcelluloses. J. Appl. Polm. Sci. 1970, 14, 579–597. [Google Scholar] [CrossRef]
- Spurlin, H.M. Homogeneity and Properties of Nitrocellulose. Industr. Eng. Chem. 1938, 30, 538–542. [Google Scholar] [CrossRef]
- Cuers, J.; Rinken, M.; Adden, R.; Mischnick, P. Critical investigation of the substituent distribution in the polymer chains of hydroxypropyl methylcelluloses by LC-ESI-MS. Anal. Bioanal. Chem. 2013, 405, 9021–9032. [Google Scholar] [CrossRef] [PubMed]
- Voiges, K.; Adden, R.; Rinken, M.; Mischnick, P. Critical Re-Investigation of the Alditol Acetate Method for Analysis of Substituent Distribution in Methyl Cellulose. Cellulose 2012, 19, 993–1004. [Google Scholar] [CrossRef]
- Basedow, A.M.; Ebert, K. Ultrasonic degradation of polymers in solution. In Advances in Polymer Science—Physical Chemistry; Springer: Berlin/Heidelberg, Germany, 1977; pp. 83–148. [Google Scholar]
- Schittenhelm, N.; Kulicke, W.M. Producing homologous series of molar masses for establishing structure-property relationships with the aid of ultrasonic degradation. Macromol. Chem. Phys. 2000, 201, 1976–1984. [Google Scholar] [CrossRef]
- Grönroos, A.; Pirkonen, P.; Ruppert, O. Ultrasonic depolymerization of aqueous carboxymethylcellulose. Ultrasonics Sonochem. 2004, 11, 9–12. [Google Scholar] [CrossRef]
- Stefanovic, B.; Rosenau, T.; Potthast, A. Effect of sonochemical treatments on the integrity and oxidation state of cellulose. Carbohydr. Polym. 2013, 92, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Rother, M.; Radke, W.; Mischnick, P. Block-structured 1,4-glucans by transglycosidation of cellulose ethers. Macromol. Chem. Phys. 2016, 217, 889–900. [Google Scholar] [CrossRef]
- Hashemi, P.; Mischnick, P. 1,4-D-Glucan Block Copolymers—Synthesis and Comprehensive Structural Characterization. Anal. Bioanal. Chem. 2020, 412, 1597–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, L.; Seger, B.; Burchard, W. Structures of cellulose in solution. Macromol. Chem. Phys. 2000, 201, 2008–2022. [Google Scholar] [CrossRef]
- Morooka, T.; Norimoto, M.; Yamada, T.; Shiraishi, N. Dielectric properties of cellulose acylates. J. Appl. Polym. Sci. 1984, 29, 3981–3990. [Google Scholar] [CrossRef]
- Penduff, P. Analytical to Preparative HPLC Method Transfer. An Easy Way to Scale up from UHPLC to Preparative HPLC Using Focused Gradient; Agilent Technology: Santa Clara, CA, USA, 2013; Publication Number 5991-2013EN. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mischnick, P.; Voiges, K.; Cuers-Dammann, J.; Unterieser, I.; Sudwischer, P.; Wubben, A.; Hashemi, P. Analysis of the Heterogeneities of First and Second Order of Cellulose Derivatives: A Complex Challenge. Polysaccharides 2021, 2, 843-865. https://0-doi-org.brum.beds.ac.uk/10.3390/polysaccharides2040051
Mischnick P, Voiges K, Cuers-Dammann J, Unterieser I, Sudwischer P, Wubben A, Hashemi P. Analysis of the Heterogeneities of First and Second Order of Cellulose Derivatives: A Complex Challenge. Polysaccharides. 2021; 2(4):843-865. https://0-doi-org.brum.beds.ac.uk/10.3390/polysaccharides2040051
Chicago/Turabian StyleMischnick, Petra, Kristin Voiges, Julia Cuers-Dammann, Inga Unterieser, Patrick Sudwischer, Anika Wubben, and Payam Hashemi. 2021. "Analysis of the Heterogeneities of First and Second Order of Cellulose Derivatives: A Complex Challenge" Polysaccharides 2, no. 4: 843-865. https://0-doi-org.brum.beds.ac.uk/10.3390/polysaccharides2040051