On the Use of Starch in Emulsion Polymerizations


1
Department of Chemical and Biological Engineering, Centre for Catalysis Research and Innovation, University of Ottawa, 161 Louis Pasteur Private, Ottawa, ON K1N 6N5, Canada
2
Department of Chemical Engineering, McMaster University, 1280 Main Street West, Hamilton, ON L8S 4L7, Canada
3
Department of Chemical Engineering, Queens University, 19 Division Street, Kingston, ON K7L 3N6, Canada
*
Author to whom correspondence should be addressed.
Processes 2019, 7(3), 140; https://0-doi-org.brum.beds.ac.uk/10.3390/pr7030140
Submission received: 17 January 2019
/
Revised: 14 February 2019
/
Accepted: 28 February 2019
/
Published: 6 March 2019
(This article belongs to the Special Issue Renewable Polymers: Processing and Chemical Modifications)
Abstract
:The substitution of petroleum-based synthetic polymers in latex formulations with sustainable and/or bio-based sources has increasingly been a focus of both academic and industrial research. Emulsion polymerization already provides a more sustainable way to produce polymers for coatings and adhesives, because it is a water-based process. It can be made even more attractive as a green alternative with the addition of starch, a renewable material that has proven to be extremely useful as a filler, stabilizer, property modifier and macromer. This work provides a critical review of attempts to modify and incorporate various types of starch in emulsion polymerizations. This review focusses on the method of initiation, grafting mechanisms, starch feeding strategies and the characterization methods. It provides a needed guide for those looking to modify starch in an emulsion polymerization to achieve a target grafting performance or to incorporate starch in latex formulations for the replacement of synthetic polymers.
1. Introduction
To achieve a sustainable society, we must intensify responsible agriculture and industry and reduce our greenhouse gas (predominantly CO2) emissions and use of non-biodegradable and single-use materials. The polymer industry has a major role to play in this shift in practice [1]. A bio-based economy focusses on extracting useful organic materials from renewable sources such as starch and other saccharides. It is expected that the proportion of plastic products that are bio-based in the market will increase each year and that up to 20% of all chemicals will be bio-based by 2020 [2]. The bioplastics market had an annual growth rate of 40% worldwide and 50% in Europe from 2003 to 2007 and is currently growing at 20–30% per year [3]. Starch-based polymers accounted for 70% of the bio-based market in 2003 (equivalent to 25,000 tons), increasing to 114,000 tons in 2007 and is projected to increase to 810,000 tons in 2020 [4]. In 2009, 75% of the starch-based plastics market was located in Europe [3]. There is a wide range of technologies used for the production of starch-based products, including polymerization reactions, blending and thermoforming and that range from those at the research and development stage to fully commercialized processes. Growth has been slower than that of biofuels, however, due to a lack of government incentives [2].
Emulsion polymerization, as opposed to bulk and solution polymerization, is a method that provides enhanced heat transfer by using water as a dispersion medium, allows for high molecular weight products at simultaneously high reaction rates, enhanced control over polymer morphology and structure, as well as resulting in a final latex with a viscosity independent of molecular weight [5,6]. Paints and coatings are the largest uses of latexes, with 60% being for decorative uses and 10% for industrial applications. Other films are applied for laminates, as construction adhesives or pressure sensitive adhesives. Perhaps most famously, the first large volume commercial product from an emulsion polymerization was styrene butadiene latex rubber as a replacement for natural rubber during World War II [7]. Latexes have also been used to improve the strength and durability of asphalt and concrete, as coatings for textiles, films for packaging, pellets or gels for moulding, as additives for printing inks, as templates for nanostructured optoelectronic materials and as the key ingredient in toner, waxes, polymer alloys, caulks, sealants, engine fluids, pigments and carpet backing [8,9,10,11]. Niche applications include drug delivery, whereby the active molecule is embedded into or immobilized onto the latex polymer particle, calibration material for analytical equipment, as coatings for magnetic recording media and for purification of biological formulations [7,12]
From a green chemistry perspective, it would be ideal to have the entire polymer formulation sustainably sourced and produced. One strategy is to ensure that monomers (feedstock for polymerizations) are synthesized from non-petroleum-based feedstock. Fillers and property modifiers should also be sourced from natural and sustainable feedstock where possible. Biopolymers such as polysaccharides offer naturally occurring and sustainably grown materials to act as fillers or macromers as a substitute for synthetic, petroleum-based monomers in polymer formulations. Polysaccharides have been extensively studied and are among the most abundant, versatile, inexpensive and commercially utilized biopolymers [5,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32].
Starch is a polysaccharide material consisting of amylose and amylopectin arranged in a granular structure with both amorphous and crystalline domains. Starches have found widespread application in many industries for use as food additives, pigment binders in paper production and rheology modifiers for pharmaceutical and textile industries [17,33,34]. Starch can be suspended or, after its granular structure is removed, dissolved in water and its properties are tuneable by physical or chemical modification. When combined with other materials, modified starches impart a wide range of properties to a polymer composite depending on the starch type, its pre-treatment and functionalization [15,28,34,35,36,37,38,39,40,41,42,43]. As a filler or reactive co-polymer, starch can be used to displace reasonable amounts of synthetic material, reducing the products’ dependence on petroleum based feedstock, making the overall process more sustainable. Overcoming certain undesirable properties of starch (i.e., tensile strength, hardness, hygroscopicity), as well as compatibility issues between raw or modified starch and synthetic polymers, is essential for the effective use of starch in industrial latex formulations.
While starch is used extensively as a substrate for graft polymerizations, its use in polymer latexes is an emerging field of interest [44,45]. Because of the many challenges inherent in using starch in water-based formulations in combination with hydrophobic materials, innovative strategies are required. This work focuses on the use of starch as a filler, colloidal stabilizer and reactive polymer in emulsion polymerizations, as well as the feeding strategies that have been employed to incorporate various modified starches into the latex, the effect of reaction variables on grafting performance and final latex properties and finally, on the analysis and characterization methods used to determine grafting performance and other properties. In this review we highlight instances where elevated rates of incorporation of starch and especially of encapsulation, have been reported.
2. Polymer Latex
2.1. Emulsion Polymerization
Emulsion polymerization is a form of heterogeneous free radical chain polymerization, where hydrophobic polymer particles are formed in an aqueous dispersion medium [46]. It satisfies, to a high degree, the 12 principles of green chemistry: as in solution polymerization, the use of small amounts of free radical initiator overcomes the need for excess stoichiometric reagents (principle 9); greater control over molecular weight and final polymer properties at high conversion, relative to solution polymerization, reduces the amount of off-spec and waste materials (principle 1); the use of water as the reaction medium removes the need for solvent use and subsequent evaporation, thus enhancing process safety and reducing energy costs (principles 3, 4 and 5); and lower viscosity profiles lead to enhanced heat transfer and therefore, lower energy requirements (principle 11) [1,12]. The addition of sustainably produced and sourced monomers and fillers (principles 4, 7 and 10) further enhances the “greenness” of the process by displacing typically petroleum-based monomers, giving the overall formulation a smaller environmental footprint.
The mechanism of emulsion polymerization typically begins with the formation of micelles (aggregates of surfactant with their hydrophobic tails oriented inwards) in the aqueous reaction media, which occurs when the surfactant molecules reach their critical micelle concentration (CMC). A significant amount of surfactant remains dissolved in the water phase. When a hydrophobic monomer is added, the monomer partitions into three loci: (1) large monomer droplets that are stabilized by the excess surfactant, (2) micelles and (3) dissolved in the aqueous phase (as a small percentage). Water-soluble initiator is added to the water phase where it slowly dissociates into free radicals during the polymerization. The free radicals react with the monomer dissolved in the aqueous phase to form water-soluble oligomeric radicals. When the oligomeric radicals reach a critical chain length to render them sufficiently hydrophobic, they enter the monomer-swollen micelles, thus nucleating a polymer particle. Due to the large surface area occupied by the micelles compared to the monomer droplets, the nucleation of micelles is the dominant particle nucleation mechanism. The entry of oligomeric radicals into the micelles is called heterogeneous or micellar nucleation and is dominant when surfactant is present above its CMC. Homogeneous particle nucleation can also occur (especially when the surfactant concentration is below its CMC) where the low molecular weight oligomers reach their insolubility limit and precipitate. They are then stabilized by surfactant and become polymer particles. Other ingredients such as buffer, chain transfer agent and crosslinker can be used to modify the reaction kinetics and latex properties. Final latexes can be used as produced or with the addition of post-polymerization additives.
The polymerization can be further described by three reaction intervals (I, II, III) (Figure 1). Interval I comprises nucleation of polymer particles and sees an increase particle number (and consequently the polymerization rate) over time. By the end of this interval (5–15 wt.% monomer conversion), monomer-swollen micelles are no longer present, having been nucleated or the surfactant distributed and absorbed onto the growing polymer particles; the number of particles (and consequently polymerization rate) becomes constant. During interval II, a thermodynamic equilibrium is maintained that keeps the monomer concentration in the polymer particles constant by the continuous transport of monomer from the monomer droplets to the particles. Thus, the polymerization rate remains constant (and no new particles are nucleated), while the polymer particles increase in size and the monomer droplets are depleted. The end of interval II is signalled by the disappearance of the monomer droplets. Interval III begins anywhere between 40–80 wt.% monomer conversion. With the particle number remaining constant and a lack of new monomer added to the system, the polymer concentration in each particle increases and the polymerization rate decreases until full conversion is reached. Final latex polymer particles are typically 50–300 nm in diameter, with a latex viscosity ranging from 30–1000 cp and solids content from 30–70 wt.%. Several particle morphologies are possible such as core-shell, occluded and moon-like. The morphology is dependent on the hydrophobicity and reactivity of the monomers, the glass transition temperature (Tg) of the resultant polymers, the presence of fillers and macromers, the distribution of free radicals within the composite polymer particle and the degree of polymerization between components. Polymerization conditions such as formulation, semi-batch feed strategy, initiation method and temperature are also factors that affect morphology [7,47,48,49,50].
2.2. Film Formation
The mechanism of film formation is generally understood to consist of three stages: drying, deformation and coalescence (Figure 2) [47,51,52,53]. Drying begins once the latex is evenly spread upon a substrate, after which water evaporates at a constant rate until a polymer volume fraction of 0.6–0.75 is reached. During this stage, the particles form a coordinated array, the specific orientation of which depends on ionic strength, viscosity and other factors. Deformation occurs as the evaporation rate slows and particles become closely packed; this occurs above the minimum film formation temperature (MFFT). A thin film may first form at the surface, causing further evaporation to occur by diffusion of water through this polymer “skin.” Particles then contact each other and start to deform to polyhedral structures through air-water, water-polymer and polymer-air interfacial tensions, osmotic forces and surface adhesive forces. Finally, polymer chains diffuse across particle boundaries to reduce surface energy and the spheres coalesce into a continuous film. The type of surfactant used to stabilize the particles can affect their mobility and the subsequent film’s uniformity. The use of additives that modify the density and surface properties of the particles, as well as fillers that alter the zeta-potential with hairy layers or introduce layers of polymer with varying glass transition temperatures, will most likely have an effect not only on the deformation and coalescence of the particles but also on the homogeneity of the final film, as well as its sensitivity to environmental factors.
3. Starch Properties and Modification
3.1. Structure and Properties
Starch is considered a low cost feedstock that offers an economically viable alternative to petroleum sourced materials (e.g., synthetic monomers) [4]. The two largest producers of maize starch worldwide are the United States of America and China. The use of starch as a source of non-food products such as ethanol has increased prices and raised concerns over whether or not to divert food resources to fuels and materials [54].
Two types of polysaccharides make up starch, amylose and amylopectin. Amylose is linear with anhydroglucose repeat units connected by α-1,4-ᴅ-glucoside bonds, while amylopectin is branched and contains α-1,4-ᴅ-glucoside bonded glucose repeat units with short branches of α-1,4-ᴅ-glucopyranose bonded polysaccharide chains at α-1,6 branch points (every ~22 repeat units) (Figure 3) [55].
The molecular weights of amylose and amylopectin are dependent on the botanical source but are generally in the range of ~105–106 g/mol and ~107–108 g/mol, respectively [56]. These polymers are naturally occurring and are principally extracted from corn (maize), potato, wheat and rice feedstock. Starch is nontoxic and biocompatible, making it safe for human contact and food applications, while its biodegradable properties make it appealing for sustainable materials [18,57]. The granule and particle size of starch depends on its source (e.g., maize, wheat, potato), number of contaminants (e.g., protein) and morphology (e.g., crystallinity). Starch is first extracted from its source in the form of microscopic granules (2–100 µm) with rings (120–500 nm) of “blocklets” (20–50 nm), each containing alternating layered lamella (9 nm) of crystalline and amorphous regions [5,58,59]. The granules are often colloidally stable in solution, as are other physically modified forms of starch, so long as this stability is not affected by complexing of associable amylose and amylopectin [60]. The crystallinity of extracted starch ranges from 19% in high amylose starch and 39% in waxy maize (low amylose), to 51% in rice starch [59]. Native starch granules have low cold water solubility, are sensitive/responsive to changes in pH, moisture, temperature and mechanical stress [61,62]. Typically, at temperatures above 60 °C, native starch undergoes irreversible gelatinization which begins with swelling of the granules (30 and 100 times by volume for maize and potato, respectively) followed by dissolution into the continuous aqueous phase, forming a viscous solution [17,60]. Once these solutions cool, the linear amylose chains orient themselves into double-helices, while amylopectin crystallizes by attraction of the short branch chains, all of which forms a network that separates from the continuous phase in a process called retrogradation [60,63]. Amylose and amylopectin carry no charge and contain an abundance of hydroxyl functional groups, making them a prime candidate for the addition of customized moieties or for grafting reactions [59].
Starches (particularly native and cooked) suffer from limited solubility, high hydrophilicity, high viscosity, moisture sensitivity and brittleness and thus require some form of physical and chemical modification to make them applicable in polymer composite formulations. At the same time, starch can be viewed as a useful additive to synthetic polymer formulations as a property modifier [26] (e.g., increase oxygen and toluene and decrease water barrier resistance [64]) or to increase the biodegradability [29] and/or sustainability of the final product.
3.2. Physical and Chemical Treatment
Native starch granules (2–100 µm) can be used in polymerizations but are typically first swollen and gelatinized through cooking, which removes the granular structure [5,65,66]. “Native” starch is defined herein as starch that is in the granular form and has not undergone a change in molecular weight, crystallinity, composition or functionality (modification of moieties). Distinction will be made between granules, which are not cooked and gelatinized starch, which has been swollen and become soluble in water. Several mechanical and chemical treatment methods exist that modify the properties of starch but do not introduce functionality to the starch structure. These methods, summarized in Table 1, are often used to extract a specific type or molecular weight of amylose or amylopectin from the granules, to affect granule particle size, degree of crystallinity or overall morphology. Many of these methods are combined for multiple effects. Often treatment is followed by or coupled with, chemical functionalization that introduces reactive or functional moieties, which compatibilizes the starch for a particular polymer formulation or application. A popular and effective strategy for achieving high loadings of starch into polymer latexes at acceptable viscosities has been to severely reduce the amylose and amylopectin molecular weights.
The use of crystalline and amorphous nanoparticles has recently garnered significant attention [5,24,26]. SNCs (Starch nano-crystals) are produced by the removal (usually through hydrolysis) of the amorphous regions of native starch, thus exposing the crystalline platelets (>40% crystallinity). These nano-crystals are typically <100 nm in size, degrade at 251 °C and have a broad distribution of viscosity in solution. SNPs (Starch nano-particles), on the other hand, are more amorphous particles where the crystalline regions have been destroyed (<40% crystallinity), typically by mechanical treatment. Regenerated starch nanoparticles (RSNPs) are near completely amorphous (96–98%) due to treatment via extrusion [5]. These amorphous nano-particles range between 100–500 nm, degrade at 285–294 °C and produce a lower viscosity in solution than native starch.
3.3. Functionalization
Modification of starch has been performed through functionalization by small molecule chemistry (addition of moieties) or by “grafting from” or “grafting to” approaches. The abundance of hydroxyl groups, the reactivity of which is C-6 > C-2 > C-3 [82], provide sites for addition (the likelihood of each hydroxyl to be functionalized also depends on the specific reaction mechanism). However, certain mechanisms allow for the formation of a bond directly to one of the carbons on the starch backbone. Because of its hydrophilicity, starch typically requires hydrophobization or functionalization to become compatible or reactive with hydrophobic synthetic polymers [59]. The literature provides examples of starch modified by small molecule chemistry with a wide variety of moieties (Figure 4, Figure 5 and Figure 6), including anhydrides [13,16,61,83,84,85], esters [16,61,82,83], ethers [16,61,82] and other cationic and anionic functional groups [59,61,82,83,86,87].
4. Measuring Grafting Performance
To determine the extent of grafting and the effect of varying formulations and procedures in achieving the desired particle morphology and composition, there are five types of results that are typically sought (Table 2). After graft polymerization has finished, most analytical methods use solvent extraction to separate homopolymer and/or unreacted starch from the graft polymer prior to analysis. Grafting performance can be expressed by grafting efficiency (Equation (1)), percent grafting through add-on (Equation (2)) or ratio (Equation (3)) or the molecular weight of the grafts (Equation (4)). In cases where the incorporation (embedding) of starch in the synthetic polymer matrix is desired, performance may be reported as the wt.% of starch that cannot be removed from the graft polymer by washing with polar solvent. Incorporation wt.% does not imply grafting but rather the success of the polymerization in removing starch from the continuous water phase and into the synthetic polymer solids. Encapsulation describes a near completely covered starch particle, where a core-shell morphology exists with the starch in the core.
Grafting ratio (GR) = (weight of synthetic polymer / weight of starch) × 100
Add-on (AO) = (weight of synthetic polymer grafted / total weight of grafted polymer) × 100
Grafting efficiency (GE) = weight of grafted polymer / (weight of grafted polymer + weight of homopolymer) × 100
AGU/graft = Mn of grafted polymer × (wt.% of starch in graft copolymer / wt.% of grafted polymer) × (MW of repeat unit)−1
Depending on the desired measure of performance for including starch in an emulsion polymerization, either the unreacted starch or ungrafted synthetic polymer will need to be separated for analysis of the remaining graft polymer. In some instances, both will need to be removed. Either can be achieved by solvent extraction, assuming that an appropriate solvent or non-solvent can be identified. Centrifugation is often used to improve separation and is combined with washing of the dried centrifugation precipitate with the same solvent or with a final separation step with a starch non-solvent such as methanol. A Soxhlet extractor is commonly used to separate a mixture of hydrophobically opposite components. In all procedures used to isolate the grafted polymers, there will always be a portion of the grafts that are removed in the separation stage. Some grafts contain mostly starch and are not sufficiently modified (hydrophobic) to separate out with the bulk of the synthetic polymer. Additionally, the presence of hydrophilic polymer (dependent on the formulation) in the decanted solvent can affect gravimetric calculations. In all cases however, the percentage of starch in the final extracted graft polymer compared to the solvent will be underestimated, meaning final results can be considered as a minimum level of performance.
NMR can be used to determine the presence of synthetic grafts or functionality on starch by identifying either the decrease in starch hydroxyl or backbone hydrogen atoms or the presence of synthetic hydrogen or carbon atoms. Solid state 13C-NMR and 1H-NMR do not typically reveal high resolution spectra and it is recommended using cross polarized magic angle spinning (CP-MAS). The use of IR to determine grafting in an unpurified latex has limitations as homopolymer and unreacted starch will appear on the resulting spectra [88]. In-line IR analysis to track the change in starch hydroxyl or backbone hydrogens in an emulsion polymerization can yield extremely useful information relating to the rate of grafting and reaction kinetics. Gravimetric methods such as centrifugation, precipitation and solvent extraction can be used to separate the starch graft polymer from either the unreacted starch or the synthetic homopolymer, providing quantitative results on grafting performance.
4.1. Characterization of Polymer Products
Once the synthetic homopolymer or unreacted starch is separated from the graft polymer, any of the three samples can be characterized (Table 3).
XPS and elemental analysis are alternative methods to NMR and IR to identify and quantify the concentration of chemical bonds and specific functional groups, respectively. Although strongly affected by the properties of the reaction medium (e.g., turbidity), Raman spectroscopy can be used in-line to monitor the signal of a particular bond, similar to in-line FTIR. The location of particles within an emulsion can be observed in-line by confocal laser scanning microscopy (CLSM). Care must be taken in the preparation of starch based samples for TEM/SEM as the water soluble starches can settle on the latex particles during evaporation and create misleading artefacts [89]. The changes in crystallinity of starch towards that of the synthetic polymer or vice versa, provides strong evidence of grafting [43,90,91,92,93]. The molecular weight of the synthetic polymer grafts can inform us about the effect of reaction conditions and monomer composition on grafting kinetics and can be determined by GPC. The branching of grafted polymer chains decreases the mobility of the grafted polymer, imparts inhibition of the movement of small molecules towards the backbone of the starch and affects the organization of the polymer molecules (e.g., retrogradation). Various methods including 1H-NMR and 13C-NMR [94,95,96], HPLC [97] and GPC [98] techniques have been used to quantify the degree or extent of branching of polymers. Asymmetric flow field-flow fractionation, coupled with a DLS device, can quantifiably detect both synthetic latex particles and more swollen and transparent starch particles by first separating them by size prior to sending them to a detector. Other methods used to determine the particle size of grafted polymers include hydrodynamic chromatography (HDC) using a PSD analyser [99], dynamic image analysis (particles in the micron range) and disc centrifugation (>75 nm). The use of DLS exclusively is not recommended due to the high degree of swelling of some starch particles, giving them a refractive index similar to water. Differential centrifugal sedimentation can be applied to measure the density of nanoparticles when the particle size is known from one of these methods.
4.2. Implicit Proof of Grafting
The characterization methods described previously allow for explicit analysis of grafting performance. To explain the effect of a grafting performance on a particular final property, additional characterization is often undertaken on the latexes or dried films/resin (Table 4 and Table 5). Unless otherwise stated, information in this section is taken from the thesis by Cummings [89].
4.2.1. Latex Properties
Viscosity is typically increased by the presence of starch in the aqueous phase of polymer latex and in some cases can cause coagulation. Even in cases where the starch is not completely encapsulated by the synthetic polymer, latex viscosity (100–1000 cp) can be similar to standard formulations. Molecular weight reduction methods have served to decrease the viscosity of starch dispersions [100,101,102]. Although high viscosity can persist after functionalization and grafting, it is possible to reduce the viscosity by reducing the likelihood of retrogradation, hydrogen bonding and persistent water soluble starches. Crystalline regions of starch contribute to its birefringence properties, which could be weakened or eliminated by deformation and modification [103,104,105]. Starch loaded latex can still exhibit birefringence in the form of blue or green hues upon cooling to room temperature. Due to the abundance of hydroxyl groups on starch molecules, the pH of the water phase can cause or prevent deprotonation, as well as alter the speed at which granules gelatinize or particles disperse. Although modified starches can exhibit strong zeta potential, a much smaller value is seen with starch-synthetic hybrid latexes. Although this should cause instability, the superior steric stabilization properties of starch prevent this [99,106,107]. The homogeneity of films formed from polymer latex containing starch is a key indicator for the presence of water phase compounds and the compatibility of the latex with the substrate. The increased viscosity and hydrogen bonding potential of starch loaded latexes can improve film formation (depending on the substrate), while any exposed starch once the film is dried can alter the composites sensitivity to humidity and temperature. The long term stability of latexes, also known as the shelf life, is affected both by the electrostatic or steric stability of the individual latex particles, as well as the presence of destabilizing or phase separation inducing molecules in the water phase. Starch loaded latex carry the risk of destabilization and phase separation in the cases of retrogradation or persistent non-grafted (hydrophilic) water soluble starch. The presence of grit (large impurities in the final latex) can indicate poor dispersion of the added starch particles/molecules, the burning of the starch due to accumulation on reactor walls or the general coagulation of ungrafted and unreacted starch material.
4.2.2. Film Properties
Even at high levels of incorporation, the presence of small amounts of non-grafted starch on the surface of a polymer film will make it more hydrophilic and increase the contact angle with water droplets. Generally, starch will reduce the adhesive properties of polymer films, especially in dry environments, however shear strength typically increases due to the rigidity, hydrogen bonding and potential crosslinking effect of starch particles. Since the degree of water whitening is dependent on the polarity of the polymer and the diffusivity of water through the matrix, this characteristic can be affected by the presence of starch [108]. The hydrophilicity of starch and the hydrophobicity of the synthetic polymer, combined with crosslinking that may occur during polymerization and intermolecular interactions (hydrogen bonding), all affect the gel and soluble content of the final graft polymer [109]. Starch content increases the polymer film’s sensitivity to humidity and polar plasticizers, all of which reduce the tensile strength. In dry environments, the loading of starch in synthetic polymer formulations tends to increase the tensile strength [41,76,110,111]. Starch is hygroscopic and therefore SNPs do not naturally have good moisture barrier properties. SNCs however, due to their crystalline structure, impart much better moisture and gas barrier properties to polymer composites, as long as deformation of gelatinization of the crystalline regions does not occur. In general, starch has extremely good oxygen barrier properties under low moisture conditions [5,64,76]. The precise functionalization, synthetic graft polymer formulation and final composite particle morphology all have a large effect on final film barrier properties. Starch grafted polymers exhibit a change in Tg towards that of native starch, except in cases where the synthetic polymer can plasticize and disrupt the structural hydrogen bonding of the starch chains [15,88]. In some cases, the Tg remains unaffected by synthetic grafts and in others (where grafting is low) two transitions are observed (synthetic polymer and starch). Typically, starch grafted polymers exhibit thermal stability up to 200–400 °C, with the properties of both starch and the synthetic polymer influencing the decomposition temperature [5,15,75,112,113]. Degradation of a polymer can be photoinduced, thermal, chemical or biological, while usually beginning with a reduction in molecular weight followed by digestion by microorganisms. The presence of biodegradable starch in polymer formulations warrants testing of the final product’s biodegradation potential. Testing typically begins with enzyme and culture experiments, followed by composting conditions in a reactor [29,31]. Starch is readily biodegradable and typically serves to increase the biodegradation of synthetic formulations, so long as there is a mechanism for radiation, water, enzymes and microorganisms to make contact with the starch chains [114,115,116].
5. Grafting Mechanisms
Most polymerizations involving starch have been initiated through radical chain mechanisms. Both native and functionalized starch have been “grafted from” or “grafted to” using polymerization techniques (Figure 7 and Figure 8) [15]. A “grafting from” approach involves generating a radical on the starch backbone or hydroxyl groups through a covalently bound initiating group, which subsequently reacts with a monomer to initiate the growth of a polymer chain. Alternatively, the chain-end of a pre-formed polymeric/oligomeric radical can react with the starch backbone, hydroxyl groups or other added functionality, “grafting to” the starch molecule. Various initiators, monomers and semi-batch feed strategies have been used to include starch into synthetic polymer latexes [117,118].
Attempts to graft or otherwise incorporate starch into synthetic polymers can be categorized based on their mechanism of grafting, which is governed by the initiator system employed. Free radical, controlled/living radical, ring opening and condensation polymerizations have been used to graft synthetic monomer from a starch [45,66,117]. Various "grafting to" formulations are prepared via coupling agents, radical coupling, activated esters or acids, acyl halides and reversible addition-fragmentation chain transfer (RAFT) click chemistry [66].
There are several examples where grafting of starch has been conducted by atom-transfer radical polymerization (ATRP) and RAFT [117]. ATRP has been used to graft starch to MMA, AA, 2-hydroxyethyl acrylate (HEA) and other monomers mostly in heterogeneous polymerizations in organic solvents. In some cases, extremely high graft percentages were reported (up to 1800%, 18x more synthetic mass than starch mass in the graft polymer). RAFT has been used to graft VAc to acid-functionalized, esterified and other starches. Both ATRP and RAFT have the advantages of producing very low amounts of homopolymer while offering control of the molecular weights of the synthetic graft chains [117].
Starch hydroxyl groups can initiate ROP of cyclic esters such as lactones and lactides in the presence of a catalyst (metal, anionic, basic organocatalysis, ionic liquid) [45,66]. Polycondensation polymerizations are stepwise reactions between bifunctional or polyfunctional monomers with liberation of small molecules such as water and have been used to graft starch [66,120,121].
Grafting via free radical polymerization occurs by hydrogen abstraction from starch to an initiator-derived radical, propagation of the starch macroinitiator with vinyl monomers and termination of the graft chains by either an initiator-derived radical, starch coupling or disproportionation. Hydrogen abstraction can either occur with the C-H on the starch backbone or from the hydroxyl groups. Unless initiation is taking place through complexation of starch with the initiating species, it is possible that both types of hydrogen abstraction could occur. Initiator-derived radicals can also directly initiate polymerization, resulting in the production of homopolymer in addition to graft polymer. In the case of persulfates, the grafting efficiency (GE) with starch is often low [66]. Examples of exceptions to this low efficiency are styrene and acrylic acid grafting, where the hydrophilicity of acrylic acid gives higher compatibility with starch and the slower polymerization reaction rate of styrene favours grafting. Ferric initiators gives slightly better grafting efficiencies but is generally lower then cerium ion initiators [66]. Ceric and persulfate initiators are the most widely used for starch grafting and while persulfates typically result in large proportions of homopolymer, ceric initiation produces the best GE, capable of reaching 85–100% with acrylonitrile, acrylamide, methyl methacrylate and styrene [32,45,66]. The molecular weight of graft chains from all methods are found to be in the range of 1–13.5 × 105 g/mol [66]. A common trend with grafting performance (GE and GP) has been a parabolic dependence on polymerization parameters leading to a maximum or optimum condition. An observed decrease in GE beyond a maximum temperature is most likely caused by chain transfer and increased termination of graft chains and with reaction time because of increased local viscosity and polymer concentration around the starch molecules.
The most common types of free radical initiators used to graft synthetic material from starch in emulsion polymerizations can be categorized into how decomposition to produce radicals is induced; thermal decomposition (persulfate, azo), redox (ferrous/peroxide, thiosulfate/persulfate, ceric ion/acid, manganese/acid) or irradiation. The species resulting from the decomposition of persulfate, ferrous and photo initiators typically produce radicals by abstraction of the C2 or C3 hydrogen or a hydroxyl hydrogen, while those deriving from manganese and cerium initiators produce a radical typically by forming a complex with the anhydroglucose unit (Figure 9). A summary of grafting results from starch with each initiator is presented below (Table 6). Preference is given to emulsion polymerization mechanisms, unless studies are limited, in which case applicable cases from solution polymerizations are presented. For emulsion polymerizations, a variety of synthetic surfactants have been employed, including those that are non-ionic, cationic and anionic. Unless otherwise reported, GE is similar between studies when comparing other grafting performance variables.
5.1. Persulfates
Sulphate radicals generated from ammonium persulfate (APS), potassium persulfate (KPS) or a thiosulfate/persulfate redox pair, have been used for the grafting of starch with vinyl monomers according to a “grafting from” mechanism through abstraction of the CH or OH hydrogen of the anhydroglucose unit (Figure 10). The hydrogen is abstracted either by a sulphate or hydroxyl radical and the formed starch radical provides a site for vinyl monomer addition and propagation. Grafting efficiency is limited with persulfate initiation relative to other more selective methods due to the favourability of homopolymerization of the synthetic monomer over radical formation on the starch anhydroglucose repeat unit.
Graft polymerizations of acrylic acid onto hydrolysed starch in solution produced higher PG and GE than identical reactions with AIBN and benzoyl peroxide, presumably due to lower overall reaction rate and thus less homopolymerization [122]. An induction time for grafting was generally observed in grafting polymerizations with KPS, while overall conversion induction time increased with higher starch content. Grafting of VAc onto granular and gelatinized starch exhibited a larger induction time than ceric ammonium nitrate (CAN) due to lack of selectivity of KPS towards hydrogen abstraction from starch. KPS served to add stability when using granular starch due to the addition of charged sulphate groups on the starch surface, since non-gelatinized native starch does not exhibit colloidal stability. A mechanism was proposed for KPS initiation where homopolymerization dominated the beginning of the reaction, at which point chain transfer from polymer radicals initiated graft polymerization on the starch molecules. The VAc polymer would eventually embed itself into the core of the graft polymer while the starch provided stabilization. Increase in starch:monomer ratio served to decrease all grafting performance as well as overall conversion [123,124,136].
Increased homopolymerization, termination of grafted polymeric chains or chain transfer to monomer above 60 °C decreased GE significantly in emulsion polymerizations. An increase in large moieties that cause steric hindrance also decreased GE [125]. Generally GE followed a parabolic trend with time caused by generation and then depletion of starch radicals [126]. A parabolic trend was also seen between GE and initiator concentration [126].
Several methods can be employed to enhance the grafting performance of emulsion polymerization with KPS. Pre-treatment (mixing of starch and KPS in solution prior to polymerization) increased GP and GE due to the increased formation of radicals on the starch molecules as well as a decrease in molecular weight [100,127]. The induction time often seen with persulfate initiators can be reduced with a pre-treatment [102]. A reduction in viscosity was also observed when starch molecular weight was reduced, either via pre-polymerization or in situ. This at times meant the difference between stability and coagulation [101,106]. Alternatively, hydrophobization of starch at any molecular weight increased GE [137]. Gelatinized starch, whether functionalized or not, exhibited stabilization properties and was able to be grafted with several monomers in the absence of surfactant [127,128]. Native and hydrophobized SNPs and SNCs also exhibited excellent stabilization properties in batch emulsion polymerizations [43,129,130,131,132]. In the presence or absence of synthetic surfactant, starch overwhelmingly resided on the surface of the final latex particles, thermodynamically favouring the polar water molecules. It is believed that starch provided stabilization as well as radical grafting sites [135]. Styrene/BA was grafted to gelatinized oxidized cassava starch where it was found that the impact of variables on GE in ascending order was initiator < monomer < temperature < starch, with a maximum efficiency of 42% and on PG was starch < initiator < temperature < monomer, with a maximum value of 301% [88]. More recently, a seeded semi-batch emulsion polymerization was conducted to prepare pressure sensitive adhesives using BA, MMA and AA. SNPs were first crosslinked with sodium trimetaphosphate (STMP), then functionalized with a sugar-based monomer (vinyl group) and finally reacted with butyl vinyl ether (BVE) to add hydrophobization. After in situ modification, the SNP hybrid particles were grafted with the primary monomer feed and KPS initiator, resulting in a core-shell latex particle with the starch completely encapsulated [183].
Enhancement by ultrasound irradiation served to increase the diffusion and collisions between starch, initiator and monomer during polymerization, increasing grafting [102]. Persulfates can also be combined with thiosulfate in a redox pair initiating system to produce generally high GE (>40%) [137,142,143,144,145]. Alternatively, a potassium persulfate/amine redox system or a dual initiator system such as with benzoyl peroxide or acetone sodium bisulphate can be conducted at comparable grafting efficiencies to KPS alone [184,185,186]. Persulfates are commonly used to graft synthetic monomers to starches in emulsion polymerization. While selectivity towards starch continues to limit GE and reaction rate, the above mentioned strategies have worked around this issue to produce useful latexes with starch content.
5.2. Ceric ions
Ceric ions (Ce (IV)) produced from ceric ammonium nitrate and ceric ammonium sulphate can be used to produce starch grafted latexes with high GE relative to other free radical methods [15]. The mechanism of ceric ion initiation is such that the ions form a complex with either the C6 carbon, C6 oxygen, C2-C3 glycol or the C1 hemiacetal at the reducing end of the starch chains (Figure 11) [15]. The most probable grafting site is the C2-C3 glycol [187,188]. The complex then decomposes by abstracting a hydrogen or through the ring opening of the anhydroglucose unit, resulting in a Ce (III) ion and a radical on the starch backbone. Less likely is the chain cleaving mechanism, where the Ce (IV) ion forms a radical on the C1 carbon or the adjacent oxygen from the ether group on a non-reducing end anhydroglucose unit (Figure 12) [32]. Alternatively, a reducing agent other than starch can be used to initiate vinyl monomer polymerization, such as a thiol, glycol, aldehyde or alcohol. Latex formed by the use of heavy metal initiators such as cerium may have a coloured hue.
The advantage of using Ce (IV) ions is that they do not react directly with vinyl groups on monomers, avoiding or greatly limiting the amount of homopolymer produced and, consequently, achieving high GE. Homopolymer can still be formed, however, for example through chain transfer to monomer or polymer. A disadvantage of this type of initiator in aqueous media is that an acid is often required to avoid hydrolysis of cerium ammonium nitrate to [Ce-O-Ce]6+, which has little to no reactivity [15].
When using ceric initiator to graft AN to granular wheat starch, gelatinization had no effect on grafting performance. This is contrary to the effect of gelatinization in KPS systems, most likely due to the greater need for diffusion of monomer and initiator into the starch matrix for an initiator with low selectivity [147,148]. A “depleted initiator” method was employed with an MMA system where Ce4+ that was not adsorbed to starch was removed. This led to higher grafting efficiencies than the usual continuous method [153]. When AM and N,N’-methylene-bisacrylamide (MBA) were grafted to gelatinized starches of varying amylose content, GE, AO and PG were highest with 50% amylose content and lowest with waxy starch (very low amylose %). It was thought that this occurred due to the more complete gelatinization of starches with higher amylose content compared to those with less, which require a higher cooking temperature. Additionally, the branched nature of the higher molecular weight amylopectin reduced the diffusivity and mobility of the polymer chains, resulting in higher viscosity and resistant chain growth [156].
In emulsion polymerizations, if a monomer is desired that does not react with starch (e.g., isoprene) it can be combined with a monomer that does readily graft in order to incorporate the unreactive monomer into the graft polymer. Gugliemelli et al. used this approach to prepare poly (isoprene-co-acrylonitrile) starch grafts for vulcanized rubber, while studying the effect of monomer ratio and other reaction variables on grafting performance. A disadvantage of this approach is that the unreactive monomer may retard the grafting rate and subsequently the overall polymerization rate (depending on the initiator system employed) [158].
A seeded emulsion polymerization strategy was used with a PMMA latex seed that was subsequently swelled with MMA and semi-batch fed CAN with DTAB surfactant in the presence of oligosaccharides (low molecular weight polysaccharides from enzymatically degraded starch). BA and styrene were sufficiently hydrophobic to make the oligosaccharides amphiphilic with only a few monomer units. Due to this hydrophobicity, there was not a high enough concentration of BA or styrene in the aqueous phase to give a high rate of propagation with the hydrophilic initiator. Coagulum was far lower when MMA was used (1.5%) compared to BA and styrene (20%). The reason given was that the oligosaccharides were grafted with MMA until they became sufficiently surface active to adsorb onto the monomer swollen latex particles, after which the PMMA portion of the graft polymer continued to polymerize inside the particle [99]. Debranched cationic starch was grafted to: (1) pre-synthesized and monomer swollen poly (MMA) particles (GE = 8.5%); (2) MMA monomer in an emulsion polymerization (GE = 24%), and; (3) MMA monomer in the presence of poly (N-isopropylacrylamide). The second feed strategy led to slow particle nucleation and hence a broad particle distribution and low GE [160].
Ozonolyzed amylopectin was grafted with cerium and persulfate/glucose for enhanced activity. Seeded polymer particles were stabilized by the amylopectin and the polymerization continued, where growing latex particles formed within the amylopectin scaffold, which underwent controlled coalescence. The adsorbed amylopectin remained partly exposed, leaving stabilizing chains on the particle surface. A unique use of energy-dispersive spectroscopy (EDS) showed the presence of starch throughout the latex particle, essentially confirming near complete encapsulation [189,190]. Another effective dual initiation system with ceric ions and KPS was used to graft semi-batch fed AM to a DADMAC and gelatinized starch batch charge. GE was 89% while PG was 173%. Higher monomer concentration increased PG to a limit, beyond which radical transfer from starch macroradicals to monomer may have increased, causing a decreased grafting rate. The coupled initiator system was more effective than either initiator alone due to the KPS regenerating depleted Ce3+ ions back into Ce4+ ions that could further initiate radicals on starch [65].
Due to its preference for starch radical formation over monomer initiation, high GE can be achieved with ceric initiators through relatively straightforward strategies [32,149,151,152,153]. Since homopolymerization is not a great concern, focus can be given to optimizing the interaction of starch, monomer and surfactant. Ceric initiators are optimal for attempting to encapsulate starch within latex particles due to the isolation of polymerization to the starch backbone rather than the grafting reaction competing with water phase or micellar polymerization. Starch can be grafted in the aqueous phase until hydrophobic enough to adsorb onto nucleating polymer particles, increasing the chances of incorporation [99].
5.3. Manganic ions
Manganese initiation using manganic (Mn4+) pyrophosphate or potassium permanganate (Mn3+) most likely proceeds either by reaction with an acid (e.g., oxalic, sulphuric) to form a radical species, which abstracts a hydrogen from the starch hydroxyl groups (Figure 13) or by the formation of a complex with the starch similar to that of ceric initiators, followed by hydrogen abstraction and radical formation on the starch backbone (Figure 14) [15,32,187].
The rate determining step of manganic grafting of monomer to starch was found to be the cleaving of the glycol group of the anhydroglucose repeat unit, which was dependent on the pyrophosphate and Mn3+ concentration, acidity and glycol (crosslinker) concentration [21]. An increase in either acid concentration or Mn3+ concentration caused a decrease in molecular weight of the grafts, while AO was parabolically dependent on Mn3+ concentration. The increased number of grafts caused an increase in monomer conversion [21]. When AN was grafted from gelatinized potato starch, a higher agitation rate was necessary compared to granular starch due to the higher viscosity of the gel phase and thus lower GE from diffusion limitations of the monomer and initiator. Monomer conversion (77–84%), AO (48–49%) and the molecular weight of grafts (33,000–430,000) all increased as the starch pre-treatment temperature was increased from 30–85 °C [22]. Grafting efficiencies were generally higher with AN than MMA due to increased rates of termination with MMA [23]. When a methanol:water mixture was used as the solvent, the GE linearly increased to 100% as methanol:water ratio increased to 50:1 (v:v). This behaviour was attributed to the increased solubility of MMA in methanol [163].
5.4. Iron ions
With a ferrous salt and hydrogen peroxide redox system, a hydroxyl radical is first produced by the decomposition of hydrogen peroxide by Fe2+ and then a radical is subsequently formed on the starch backbone by hydrogen abstraction (Figure 15). This initiation system, unlike that of ceric and manganese initiators, is able to polymerize vinyl monomers using hydroxyl free radicals [15,34,91,123,137,191].
AN was grafted more readily to native starch granules than MMA due to its increased hydrophilicity [138]. Grafting efficiency increased as peroxide concentration increased, however degradation of starch began to occur. Oxidized starch was generally more susceptible to grafting than native starch [138]. MAA was grafted to granular starch and it was found that at the termination of the polymerization, the rate of grafting (>85% conversion) was much faster than that of homopolymerization (~10% conversion). Pre-gelatinized starch achieved only half the conversion of granular starch. Explanations for this were given as increased intrinsic reactivity of the starch macroradical, the stability of the propagating free radical species and high local monomer concentration within the starch granule due to association of MAA with the starch hydroxyls [171]. There was a negligible effect of increasing Fe2+ concentration above a minimum, with a brownish hue detected at excess amounts. Longer reaction times are more effective due to added time to homogenize the polymerization as the grafting reaction happens quickly with ferrous initiator pairs and sometimes generates regions of heterogeneity [173].
Semi-batch (monomer and hydrogen peroxide feed) surfactant free emulsion polymerization was used to graft styrene and BA to gelatinized cationic acetylated starch. It was found that conversion increased with H2O2 concentration as well as increased to a maximum with monomer concentration. Grafting efficiency and PG had parabolic dependency on H2O2 concentration and while both variables increased with monomer concentration, GE decreased after a maximum. Grafting efficiency and PG were 35% and 59% respectively [91].
5.5. AIBN
Azobisisobutyronitrile (AIBN) is a hydrophobic initiator that dissociates into two 2-cyanoprop-2-yl radicals capable of initiating polymerization of vinyl monomers (Figure 16). These radicals are capable of abstracting hydrogen from a hydroxyl group on the anhydroglucose units, thus creating a radical on the starch backbone [11,15,122,141,192].
The grafting of AA onto hydrolysed potato starch was conducted using AIBN as initiator in aqueous solution. Monomer conversion approached 100% as expected but GE and PG were limited to 57% and 27% respectively, due to the high amount of homopolymer produced and lack of grafting sites produced on the starch molecules [122]. MMA was grafted to gelatinized starch to achieve PG = 73% and GE = 8% and it was concluded that AIBN radicals were not reactive enough to abstract a hydrogen from starch. Any grafting that occurred came from “grafting to” of polymeric radicals, which were available dependent on the concentration of relatively hydrophobic monomer present, which the radicals were reactive enough to polymerize [140]. Similar observations were made when grafting VAc at PG = 24% and GE = 5% [139]. A starch-based bromide chain transfer agent was grafted with AIBN to vinyl acetate via RAFT polymerization for use in biomedical and other applications. The structure, composition and molecular weight of the grafted polymer was successfully controlled and well defined [193].
Due to the hydrophobicity of AIBN, it is more suited to mini emulsion or suspension polymerization than emulsion polymerization [192,194]. A notable exception was the use of OSA-acetic anhydride (AC) modified SNPs in a seeded emulsion polymerization with styrene. The SNPs were found to first act as Pickering stabilizers of the styrene but then to transition under higher temperature to a core-shell seed with styrene in the core of the starch particle. The polymerization proceeded at the interphase of the swollen SNP seed, with the AIBN hydrophobic initiator initiating creating oligomer radicals that would propagate inward to the core of the particles. The results were an apparent core-shell morphology with most of the starch in the core of the particle [141].
5.6. Other Initiators
Other initiators suitable for grafting from starch include anionic polymerization by alkoxides, metal complexes such as cobalt or chromium (similar to manganese mechanism), copper acetylacetonate-trichloroacetic acid, potassium peroxyvanadate, hydrogen peroxide/iron sulphate/thiourea dioxide, potassium bromate/thiourea dioxide, alkyl hydroperoxide/amino group, vanadium mercaptosuccinic acid, horseradish peroxidase/hydrogen peroxide/2,4 pentane dione, ozone and enzymes [15,32,45,66].
The enzyme horseradish peroxidase (HRP), an oxidoreductase catalyst extracted from the roots of horseradish, has been used on several occasions to perform grafting reactions with starch. BA (GE = 19%) [195], MA (GE = 45%) [196], AM (GE = 66%) [197] and DMDAAC (GE = 99%) [198], have all been grafted from starch in solution polymerization in the presence of hydrogen peroxide as the oxidant and a reductant (e.g., 2,4-pentanedione). Although HRP has been used in emulsion polymerizations, there is no reported work grafting starch using such an approach [199]. This presents a worthwhile avenue to be pursued due to the moderate reaction temperatures and high grafting efficiency associated with HRP, in addition to the fact that it is a bio-catalyst that is renewable.
Benzoyl peroxide was used as the sole initiator for the emulsion polymerization of styrene/maleic anhydride with crosslinked SNPs and hydroxyethyl cellulose as stabilizers. An inverse emulsion was first prepared where SNPs were mixed with water in an organic phase consisting of maleic anhydride, styrene and benzoyl peroxide pre-polymerized to 25% conversion. The SNPs were shown to act as Pickering stabilizers of the water droplets. Subsequently, the inverse emulsion was transferred to an aqueous dispersion of hydroxyethyl cellulose, where the cellulose stabilized a region of the organic media around several smaller SNP stabilized water droplets and the polymerization was continued to completion. Although grafting performance was not reported, FTIR of the purified starch grafted polymer was shown as evidence of esterification between hydroxyl groups and maleic anhydride [107].
5.7. Irradiation
Microwave, UV and high energy (gamma and electron) irradiation have become increasingly popular for grafting synthetic monomer to polysaccharides without the need for additional reagents. High energy ionizing initiation can occur through: (1) a direct method where monomer and polysaccharide are simultaneously initiated; (2) a peroxidation method where hydroperoxide or peroxide functional groups are formed by reaction of the polysaccharide and air, which later form free radical groups under heat; or (3) a pre-irradiation method where the radiation forms free radicals on the polysaccharide in an inert atmosphere, prior to being mixed with monomer [200]. It has been found that high irradiation doses or reaction times can lead to starch degradation [174]. Exposing a photoinitiator (e.g., dimethoxy-2-phenylacetophenone (DMPA), benzoin methyl ether (BME), benzophenone (BP)) to a certain wavelength of radiation causes generation of a radical that can attack vinyl monomer or starch [201]. Irradiation methods can also be used to enhance other initiation methods [202,203]. The precise grafting performance and kinetics of each monomer/initiator pair has to be evaluated separately as their behaviour cannot necessarily be assumed to apply universally [174].
Emulsion polymerization was used to graft styrene, BA and MMA to amylopectin with sodium dodecylbenzene sulfonate as surfactant [177,178]. Using the direct irradiation method, surfactant, water, monomer, starch and initiator were mixed and lightly stirred, purged with N2 and subsequently illuminated. A pre-illumination method was applied where monomer, surfactant and water were added to an addition funnel and purged to form a pre-emulsion, while the starch dispersion was illuminated. Subsequently the pre-emulsion was then added to the reaction vessel with the pre-illuminated starch dispersion. For the direct method, styrene and BA resulted in 50% and 87.8% conversion at ~0% and 11.5% GE respectively. Using pre-illumination, styrene and BA yielded 37.5% and 89.1% conversion and 12.5% and 81.4% GE respectively, a significant improvement over simultaneous illumination. For BA, which has a faster polymerization rate, pre-illumination has a greater effect on GE due to the higher chance of grafting over homopolymerization when the radicals are formed only on the starch backbone.
Irradiative initiation methods present clear benefits over other initiators due to good control over polymerization rate and the potential for radical formation on the starch backbone without the need for addition of potentially harmful chemicals such as initiator, heavy metal salts and acids. So long as degradation is avoided, irradiation can be combined with other initiation methods to enhance GE.
6. Strategies for Starch Incorporation in Emulsion Polymerization
From the numerous studies described above, certain conclusions can be drawn regarding latex formulations and strategies as well as the dependence of grafting performance variables on various reaction parameters. These strategies are summarized below as a guide for using starches in emulsion polymerization.
6.1. Initiator
The appropriate choice of initiator depends on the constraints of a particular system with regard to industrial scalability and final product specifications (mostly GE and PG). Persulfates are the most widely used initiators in industry and there is abundant experience in handling and using these initiators in emulsion polymerizations. If homopolymer formation is not of concern, this is the clear direction to pursue [45]. Ceric initiators can be active at room temperature, do not polymerize vinyl monomers directly and are nearly completely selective towards starch initiation [66]. The need for acid in the polymerization and the presence of heavy metals and colour in the final product may be deterrents for its use [15]. Synergistic effects of multiple initiator systems can be attractive and some creativity in feed strategy can limit the need for a high concentration of heavy metal initiators while achieving improved grafting performance over persulfates alone [65,202,204,205]. The combination of irradiation with persulfate or ceric initiators can more than double the GE [203]. In the case where an initiator can initiate homopolymerization, a pre-treatment step where initiator and starch mix is able to generate starch macroradicals prior to the addition of monomer, as well as reduce the starch molecular weight, which can assist in increasing GE [100,102]. An alternative strategy is to modify the starch through small molecule chemistry to add covalently bonded vinyl groups that can participate in initiation [183,206,207]. These vinyl groups must be incapable of reacting with each other (e.g., maleic anhydrides) or else microgel networks of starch particles will form, while they must readily react with the monomer formulation chosen [89,189].
Although degradation can be caused by prolonged reaction times or irradiation strength, radiation initiation provides a simple method for graft polymerization, provided one has access to the appropriate equipment at scale [174,179]. The degradation of starch by any initiating method can result in lower molecular weight starch radicals, which may improve grafting and lower viscosity. This degradation could also increase the amount of soluble starch that migrates to the water phase, which may derail attempts at achieving a particular final latex particle morphology or stability (such as the degradation of initially well-defined SNPs or SNCs).
The type of initiator used can be expected to have an effect on the properties of the final latex (e.g., viscosity) due to differences in initiation and subsequently, on nucleation mechanisms, amount of homopolymer and water phase polymer, degradation of starch and grafting performance. Generally, the effect of temperature on GE is universally parabolic with the efficiency increasing with temperature due to increased radicals being created on the starch molecules, with subsequent decreases due to higher rates of termination and chain transfer reactions. A similar trend can be expected with reaction time due to consumption of monomer and initiator as well as diffusion limits from longer grafted chains, preventing reaction components from contacting the starch. The temperature of a reaction should be high enough to initiate radical formation but not so high as to produce excess homopolymer formation [125]. Several kinetic models have been prepared for specific systems [32]. One should recall that if the initiator, monomer or starch type change, model parameters will need to be recalculated.
6.2. Monomer
Choice of monomer is made by matching the resulting polymer to the properties of the desired product. The glass transition temperature, mechanical and surface properties and functionality of moieties of the final product are among determining factors when selecting the appropriate monomer (s). In cases of starch grafted polymers, additional attention must be paid to the interaction of the monomer (and resulting polymer) and starch molecules, perhaps the most important compatibility issue when targeting a particular particle morphology or level of grafting performance. Monomer hydrophilicity and reactivity are the primary factors defining this interaction. If a monomer is too hydrophilic it may cause excess water phase homopolymer formation, while extreme hydrophobicity may reduce contact between the monomer and hydrophilic starch. A way around this issue is to functionalize the starch with hydrophobic groups prior to polymerization, although this could add undesired steps to an industrial process and reduce the overall mass of bio-sourced material in the starch pre-cursor material [129,130].
If monomers are required that do not readily graft with starch, they can be coupled with initiating monomers, although this tends to increase the induction time common with certain initiators (such as persulfates), as well as decrease the overall reaction rate [158]. Monomers with sterically hindered vinyl groups can affect reactivity with starch and subsequently the GE [125]. The GE of the same monomers used with persulfates, cerium and manganese initiators vary greatly, suggesting further that such observations cannot be applied universally.
The application of mixed monomer/starch batch seed generation, as well as the use of high shear mixing narrowed the particle size distribution (PSD) and improved the GE [137,160,189,190,208]. Semi-batch feeding of monomer or a pre-emulsion to achieve monomer starved conditions could be favourable for starch incorporation into latex particles, especially in the case where homopolymerization is a concern.
6.3. Surfactant/Stabilizer
Emulsion polymerizations usually use a stabilizer or surfactant to produce well-defined polymer latex particles. These surfactants are low molecular weight synthetic molecules with a hydrophobic tail and charged hydrophilic head. Alternatively, non-charged steric stabilizers can be used, as well as solid spherical particles (Pickering emulsions) or a combination of all three types.
The vast majority of starch types (except uncooked granules) exhibit stabilization properties in emulsion polymerizations. Carbohydrate based surfactants are obviously tailored to be effective stabilizers as direct replacements for typical surfactants such as SDS [209]. SNPs and SNCs are particularly effective at stabilizing latex particles without the need for synthetic surfactants [107,129,130,131,192,210]. Although starch particle sizes are often >1 µm, exceptions exist where they are more appropriately sized at <500 nm [106,131,207,211]. Starch particle stabilizers should be smaller than the latex particles they are stabilizing, which typically range from 50–300 nm. The composition of the continuous phase as well as the hydrophobicity of the monomer and polymer affect the particle size and distribution due to the hydrophilicity and strong hydrogen bonding potential of the starch.
Some strategies for improving stabilization could include crosslinking the starch once it has stabilized the particles, functionalizing the starch with cationic/anionic charged groups and including synthetic surfactants to be cooperating stabilizers or to induce a lower starch CMC [207,212,213]. The electric charge of the latex particles, surfactant, monomer and starch should be considered. Surface tension measurements can shed light on the behaviour of surfactants and stabilizers and their interaction with other molecules. Different forms of starch have been adsorbed onto polymer particles once sufficiently grafted with hydrophobic monomer, after which the particles grow and incorporate the starch into its structure, leaving strands of starch chains on the particle surface for stabilization [99].
The use of synthetic surfactant in starch graft polymerizations can improve GE, reduce viscosity, increase storage stability by reducing retrogradation and prevent amylose aggregation in the water phase [214,215]. Adverse effects of excess surface active molecules can arise from their migration to the surface of the subsequently formed polymer films, thus affecting adhesive and barrier properties [47,215]. Reports of successful grafting in surfactant free systems are regularly reported, while cases of no grafting in such systems also exist [128].
Octenyl succinic anhydride (OSA) functionalized starch has been proven to be an extremely effective stabilizer (low CMC and good particle size control) at particle diameters from 280 nm to 100 µm and concentrations of 1–45 wt.% [199]. Non-functionalized waxy SNPs have stabilized BMA latex particles as small as 135 nm at 10 wt.% concentration, while SNCs have stabilized particles as small as 121 nm at 12 wt.% concentration [129,131]. SNPs have been shown to preferentially stabilize smaller polymer latex particles rather than monomer droplets [129]. The higher molecular weight SNPs are superior at stabilizing oily particles compared to other starches and may have better long term stability that synthetic surfactants [216,217]. In contrast, free amylose derives its stabilization properties from the formation of 3D networks between particles, contributing steric stabilization to resist coagulation [217,218]. The hydroxyl groups of starch have a pKa > 12, indicating a reasonably strong resistance to alkalinity, however one can expect more deprotonation as the pH increases to this point. The presence of salts and buffer can also influence the stability of latex, with or without starch, although latex with low concentrations of water soluble starch are highly salt resistant [38,217,219,220]. Although zeta potential provides an established method to test the stability of latex particles, most starch stabilized latexes exhibit weak zeta potentials (0 to −5 mV) while maintaining good long term stability due to steric hindrance.
6.4. Starch Type
As previously explained, there are several types of starch that can be used to either modify polymer latex properties or for the reduction of synthetic polymer mass. These include granular and gelatinized non-modified starch, as well as non-functionalized and functionalized gelatinized starch, amylose-rich and amylopectin-rich starch, ultra-low molecular weight (ULMW) polysaccharides, SNPs and SNCs. Gelatinization is typically achieved for any starch type by pre-treating in water at 70–95 °C. The temperature of gelatinization influences grafting performance [22,161,162,172].
It is common to degrade starch either by peroxides, acid, persulfates or by enzymatic treatment in order to increase the likelihood of high encapsulation within the latex particles. In fact, apart from vinyl and hydrophobic functionalization, this strategy is the only one to result in significant encapsulation. This may be due to decreased negative effects on viscosity and reaction kinetics, reduced termination of growing graft chains and the speed at which the molecule can become amphiphilic through grafting of the hydrophobic monomer. As with any polar polymer, a decrease in its molecular weight will increase its solubility in non-polar solvents. SNPs with a DS of 0.07 of OSA and 0.158 of AC are sufficient for a very hydrophobic monomer like styrene to swell the core of the SNPs and polymerize, forming varying final morphologies (as observed by SEM). So long as the monomer feed ratio was kept low, the synthetic polymer remained bound to the SNP at full conversion (the synthetic monomer/polymer migrated during the polymerization in all cases) [208].
6.5. Encapsulation
As mentioned earlier, it could be desirable to encapsulate all or most of the starch material into the latex particles, to maintain the properties of the synthetic standard. Apart from degradation or hydrophobization, vinyl functionalization can transform the starch into a reactive polymer (or macromer) capable of participating in free radical polymerization. This strategy has been pursued with bifunctional groups as well as with glucose surfmer (reactive surfactant) functionalized SNPs [183,206,207,224].
Enzymatically degraded starch was reported to be encapsulated within pMMA latex particles either by early adsorption of the amphiphilic graft polymers onto the nucleated latex particles and subsequent particle growth or by the crosslinking of grafted starches and subsequent monomer swelling and polymerization [160]. The authors demonstrated this by analysing the starch and graft polymer by newly developed and apparent error-free solution 1H-NMR and solid 13C CP-MAS quantification methods, as well as TEM.
OSA-AC modified SNPs were found to first form Pickering emulsions with styrene but then transitioned under higher temperature to a core-shell emulsion with styrene in the core of the starch particle. Upon addition of hydrophobic initiator an apparent core-shell latex particle morphology was formed containing the majority of the starch in the core of the particle, with small amounts of starch chains extending into the shell [141].
Another approach reported by Cummings et al., consisted of RSNPs functionalized with vinyl functionalized glucose-based surfmer. When used in an emulsion polymerization of BA/MMA, these functionalized RSNPs produced 10–40 wt.% incorporation of starch in the polymer latex [206,207]. Subsequently, this functionalization was combined with an additional internal crosslinking of the RSNP with sodium meta triphosphate (SMTP) to reduce the amount of water soluble starch and increase the crosslink density of the RSNPs prior to polymerization. This modification improved incorporation greatly, where the starch was reported to be fully encapsulated in the latex particle [183]. Furthermore, the use of a tie-layer monomer (e.g., butyl vinyl ether) with low reactivity to preferentially graft to the starch vinyl groups, served to hydrophobize the RSNPs prior to the primary monomer feed. The result was successful encapsulation and core-shell morphology of the SNPs within the final latex particles. Although the maximum starch content reported to date was 17 wt.%, this work is significant in that it follows a relatively simple emulsion polymerization procedure with a very common initiator (KPS) to produce completely encapsulated particles [183].
Latexes with low encapsulation can still appear white at low conversion, while those with high encapsulation can still undergo significant increases in viscosity during the polymerization (even when only small amounts of water phase starch is present).
6.6. Starch Loading and Solids Content
Challenges persist in regard to the preparation of latexes with high starch loading and solids content and manageable viscosity and grafting (ideally encapsulation). Achievement of such an industrially relevant formulation that could impart some beneficial property modification while displacing a large amount of synthetic (petroleum sourced) material represents a large step forward in applying the 12 principles of green chemistry to the production of synthetic-starch graft polymers.
Solids content limitations typically arise from an increase in viscosity caused by persistent water phase starches. These starches additionally interfere with the diffusion of initiator and monomer to the polymer particles (causing a decrease in reaction rate, potential monomer droplet initiation and subsequent coagulation) and reduce the stability of the latex particles at high or complete conversion [183,206,207].
Starch loading is limited by similar effects but mostly by the decrease in polymerization rate of initiator-starch initiation compared to that of the initiator-monomer, the persistence of water phase starches affecting viscosity and poor GE leading to a product with limited usefulness [183,206,207]. The slower initiator-starch initiation is caused by the higher dissociation energy of the starch hydroxyl hydrogen compared to the π-bond of the monomer's vinyl group, resulting in the faster homopolymerization reaction being preferred when both monomer and starch are present. Achieving a particular loading is usually not of concern in cases where starch is being used as a surfactant/stabilizer, in contrast to when it is used as a filler or synthetic polymer replacement.
Surveying the achieved solids contents and starch loadings in the literature, the highest solids content achieved was ~60 wt.% when using VAc to graft onto waxy maize dextrin (ULMW starch) at a starch loading of 25 wt.% [137]. Also using dextrin, the highest loading of 93 wt.% was achieved at 5 wt.% solids [225]. A degraded amylose was loaded at 70 wt.% with VAc at 45 wt.% solids [214]. Beyond low molecular weight starches, 50 wt.% loading and solids was achieved with native starch being used as a filler and stabilizer with styrene and BA with KPS [226]. For modified starches, high solids and loadings are rarer, with cationic acetylated starch being loaded at 20 wt.% with styrene and BA at 52 wt.% solids with ferrous initiator [91] and SNCs being loaded up to 50 wt.% with styrene at low 6 wt.% solids with KPS [92]. OSA-AC modified SNPs that achieved excellent encapsulation were performed at only 9 wt.% loading at 6 wt.% solids with AIBN [141]. Further, other OSA modified SNPs were used at 2 wt% loading and 53 wt.% solids with KPS [130]. It is increasingly difficult to apply efficient encapsulation strategies at high loadings and solids contents. Brune and Eben-Worlée achieved 30–50 wt.% loading at 30–70 wt.% solids and although copolymerization was assumed, no grafting performance was reported [224]. Our group has achieved partial incorporation of waxy (20 wt.%) and dent (40 wt.%) RSNPs modified with a vinyl functional glucose surfmer. Waxy RSNPs were included at 40 wt.% (20 wt% incorporation) and 50 wt.% (10 wt.% incorporation) loadings and 40 wt.% solids, while dent RSNPs had a limit of 25 wt.% loading at 40 wt.% solids (40 wt.% incorporation) [206,207]. A positive achievement of these latexes is their low viscosity (<250 cp), reasonable particle size (<300 nm), good film formation and good long-term stability.
7. Starch-Based Latex Applications
7.1. Starch
With its abundance, biodegradability and potential for modification and functionalization, starch has been widely used in industry and for academic study. In fact, a third of the biodegradable polymers being commercially produced are starch based [114]. Despite this, starch is readily plasticized by water, has poor barrier resistance to water vapor and is overly rigid, making it difficult for use in many coating and film applications. Researchers have used starch in composites to impart unique properties and have shown it is applicable for the food, cosmetics, pharmaceuticals, paper making and tire industries [76,227].
Two of the most popular commercialized starch products are Mater-BiTM (carbon black replacement for tires) and Eco-SphereTM (bio-based latex for coatings and paper binder) [59]. Due to its stabilizing properties, starch can be used in emulsions for cosmetics, pharmaceuticals, paper products, textiles, packaging and industrial latexes for paints and coatings [228,229]. While the most common use of starch products are for packaging applications, it holds promise for drug delivery and other niche applications [3]. In the food industry, starch can replace fats while maintaining similar rheological properties [14,227,230]. SNPs have been used to reinforce packaging for foodstuffs. In the pharmaceutical industry, starch has been extensively investigated as a drug carrier, although further work must be done to determine the dangers of starch based particles in the blood stream [15,227,231]. These particles are a low cost alternative as an adsorbent for wastewater treatment. In comparison to native starch, SNPs have exhibited improved properties as a paper binder [14,227,230]. Metal implants can conceivably be replaced by starch based biodegradable material. SNC have good reinforcing properties for composites and latexes such as natural rubber and various synthetics [26].
Commercialized starch blends include Bioplast®, NovonTM, Biopar®, Baialene®, Solanyl®, Ecobras, Biolice, Cornpole, Pentafood, Pentaform, Plantic and products Trellis Earth among others [110,232]. Due to the requirements for vegetable packaging, this provides a new area to be explored. Current starch based products include thermoformed trays and cups, injection moulded cutlery, plates and bottles, as fibres for agricultural products and clothing, as thickeners and as additives in adhesives [4,233]. Some commercial leaders in the field include Novamont, Rodenburg, Biotec, Biop, Limagrain, Livan, EcoSynthetix, Cereplast and Plantic, with the bulk of the industry existing in Europe [114]. The future of commercialized starch products is sure to include a high growth rate in innovation and commercialization, especially in the field of starch graft polymer composites for a wide array of previously unexplored applications, including for uses as consumer plastic bags, straws, tapes, films, wraps and paints [114].
7.2. Starch-g-synthetic Latex
As mentioned previously, although starch is a useful material in and of itself, its performance can be greatly enhanced by customizing its functional moieties and adding grafted polymer chains. This will allow previously unexplored applications to be tested, as well as open the possibility of replacing purely synthetic formulations with increasing amounts of renewable starch material. Starch material modified with synthetic polymers have shown potential to be used in biomedical and pharmaceutical fields, as an adsorbent for wastewater treatment and in polymer composites as a property modifier.
Other applications for starch graft polymer latexes are as flocculants [15,66], resins in ion exchange filtration [15], superabsorbent/hydrogel for aqueous solutions (water treatment) [15,32], sizing agents for textiles [32,66,117] and in various papermaking applications [15,32,45,66]. Biomedical applications include drug delivery, enzyme immobilization, biosensing and catalysis [15,45,66,117,234]. Biodegradable plastics (including for tissue engineering and implants) can contain a significant amount of starch and continue to have desired properties [15,45,234]. Additionally, there are several oilfield applications where starch graft polymers have been used [15].
8. Conclusions and Future Perspectives
The grafting of vinyl monomers from granular, native and functionalized amylose, amylopectin, SNCs and SNPs has been extensively explored in the literature. Initiation methods investigated include persulfates, irradiation, azo and ferrous, ceric and manganese redox initiators. Undesired homopolymer formation during the grafting polymerization is often a challenge, as is proper encapsulation of the starch material. Controlled radical polymerization strategies such as ATRP, RAFT and more recently nitroxide-mediated radical polymerization (NMP), have been investigated for polysaccharide modification and grafting and have the advantage of avoiding homopolymer formation. The bio-catalyst HRP has also garnered recent attention for the grafting of starch and shows promise as a “green” pathway for the production of starch grafted polymers. The use of emulsion polymerizations to produce starch incorporated latexes for coatings and other applications of polymer films shows promise for the enhancement of properties, the reduction of synthetic (petroleum-based) monomers and the further improvement of overall process sustainability. Challenges persist due to the difficulty in maintaining latex stability in the presence of high molecular weight starches and the persistence of water phase starches in the final latex. Although some strategies exist in the literature as proofs of concepts, more work must be done to develop procedures for full encapsulation of starch within the latex particles (thermodynamically unfavourable) for a range of monomers. The resulting core-shell particles would be expected to exhibit the properties of the vinyl monomer rather than that of the encapsulated starch. This may also increase the starch loading limit before undesired effects begin to exhibit themselves during the polymerization. Some of these strategies have included hydrophobization or vinyl functionalization of starches (of mainly SNPs and SNCs) to make them more compatible with vinyl monomer and initiator or the use of sequential polymerization stages to first create an amphiphilic grafted starch seed that can then be encapsulated with semi-batch fed monomer or a pre-emulsion. Another recent approach has been to include carbohydrate-based monomer in polymerizations to increase the bio-content of the final product [235]. Starch is generally cheaper than petroleum by-products and has the potential to be sustainably sourced and biodegradable, making it extremely attractive in an industry looking to reduce its environmental footprint. Even so, the commercialization of starch loaded latexes must also accelerate to increase public acceptance and support of starch-based products, government funding of academic research and industrial start-ups and more comprehensive analysis of the effectiveness of these products and the environmental impact of their full life cycle. We have presented in this work a survey of the attempts to include starch in polymerizations, specifically those carried out through the more sustainable emulsion polymerization process, including those based on granular, native, functionalized and nano-sized starch materials. Strategies for future works were presented and a comprehensive analysis of characterization methods and applications of starch grafted latexes and films was provided.
Author Contributions
Conceptualization, S.C., M.D. and N.S.; methodology, S.C., M.D. and N.S.; software, S.C.; validation, M.D., N.S., M.C. and Y.Z.; formal analysis, S.C.; investigation, S.C.; resources, M.D. and S.C.; data curation, S.C.; writing—original draft preparation, S.C.; writing—review and editing, S.C., M.D., N.S. and M.C.; visualization, S.C., M.D., M.C., N.S. and Y.Z.; supervision, M.D., M.C. and N.S.; project administration, M.D.; funding acquisition, M.D. and M.C.
Funding
This research was funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada, grant number CRDPJ 476580-14.
Acknowledgments
EcoSynthetix, Inc. (Burlington, ON) is acknowledged for their financial support of the research.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 1H-NMR | proton nuclear magnetic resonance |
| 13C-NMR | carbon 13 nuclear magnetic resonance |
| AA | acrylic acid |
| AC | acetic anhydride |
| AIBN | azobisisobutyronitrile |
| AGU | anhydroglucose units |
| AM | acrylamide |
| AN | acrylonitrile |
| AO | add-on |
| APS | ammonium persulfate |
| ATRP | atom-transfer radical polymerization |
| BA | butyl acrylate |
| BAM | N-tert-butyl acrylamide |
| BMA | butyl methacrylate |
| BME | benzoin methyl ether |
| BP | benzophenone |
| CAN | ceric ammonium nitrate |
| CLSM | confocal laser scanning microscopy |
| CMC | critical micelle concentration |
| CP-MAS | cross polarized magic angle spinning |
| DADMAC/DMDAAC | diallyl dimethyl ammonium chloride |
| DAAM | diacetone acrylamide |
| DLS | dynamic light scattering |
| DMA | dynamic mechanical analysis |
| DMAEMA | 2-(dimethylamino)ethyl methacrylate |
| DMPA | dimethoxy-2-phenylacetophenone |
| DTAB | dodecyltrimethylammonium bromide |
| EA | ethyl acrylate |
| EMA | ethyl methacrylate |
| FTIR | Fourier-transform infrared |
| GE | grafting efficiency |
| GMA | glycidyl methacrylate |
| GPC | gel permeation chromatography |
| GR | grafting ratio |
| HDC | hydrodynamic chromatography |
| HPLC | high performance liquid chromatography |
| IA | itaconic acid |
| KPS | potassium persulfate |
| MA | methyl acrylate |
| MAA | methacrylic acid |
| MBA | N,N’-methylene-bisacrylamide |
| MFFT | minimum film formation temperature |
| MMA | methyl methacrylate |
| MW | molecular weight |
| NMA | N-methylol acrylamide |
| OSA | octenyl succinic anhydride |
| PG | percent grafting |
| PSD | particle size distribution |
| RAFT | reversible addition fragmentation chain transfer |
| RSNP | regenerated starch nano-particle |
| SEM | scanning electron microscopy |
| SMTP | sodium meta triphosphate |
| SNC | starch nano-crystal |
| SNP | starch nano-particle |
| St | styrene |
| TEM | transmission electron microscopy |
| TGA | thermogravimetric analysis |
| VAc | vinyl acetate |
| ULMW | ultra-low molecular weight |
| XRD | x-ray diffraction |
References
- Zhang, Y.; Dubé, M.A. Green Emulsion Polymerization Technology. Adv. Polym. Sci. 2017, 1–36. [Google Scholar]
- Marques, S.; Moreno, A.D.; Ballesteros, M.; Gírio, F. Biomass and Green Chemistry, 1st ed.; Vaz, S., Jr., Ed.; Springer: Cham, Switzerland, 2018; pp. 69–94. [Google Scholar]
- Laycock, B.G.; Halley, P.J. Starch Polymers, 1st ed.; Halley, P.J., Averous, L., Eds.; Elsevier: New York, NY, USA, 2014; pp. 381–419. [Google Scholar]
- Tadini, C.C. Starch-Based Materials in Food Packaging: Processing, Characterization and Applications, 1st ed.; Barbosa, S.E., Garcia, M.A., Castillo, L., Lopez, O.V., Vilar, M., Eds.; Academic Press: London, UK, 2017; pp. 19–36. [Google Scholar]
- Le Corre, D.; Dufresne, A. Preparation of Starch Nanoparticles. Biopolym. Nanocomposites Process. Prop. Appl. 2013, 153–180. [Google Scholar]
- Belgacem, M.N.; Gandini, A.; Silvestre, A.; Galbis, J.A.; García-Martín, M.G.; Benvegnu, T.; Plusquellec, D.; Lemiègre, L.; Pizzi, A.; Gellerstedt, G.; et al. Monomers, Polymers and Composites From Renewable Resources, 1st ed.; Belgacem, M.N., Gandini, A., Eds.; Elsevier: New York, NY, USA, 2008. [Google Scholar]
- Hansen, F.K.; van Herk, A.M.; Gilbert, R.G.; Leiza, J.R.; Meuldijk, J.; Charleux, B.; Monteiro, M.J.; Heuts, H.; Mercado, Y.R.; Akhmastkaya, E.; et al. Chemistry and Technology of Emulsion, 2nd ed.; van Herk, A.M., Ed.; John Wiley & Sons: West Sussex, UK, 2013. [Google Scholar]
- Anderson, C.D.; Daniels, E.S. Rapra Rev and Latex Applications. Rapra Rev. Rep. 2003, 14, 1–146. [Google Scholar]
- Guyot, A.; Chu, F.; Schneider, M.; Graillat, C.; McKenna, T.F. High Solids Content Latexes. Prog. Polym. Sci. 2002, 27, 1573–1615. [Google Scholar] [CrossRef]
- Rudin, A.; Choi, P. The Elements of Polymer Science & Engineering, 3rd ed.; Elsevier: New York, NY, USA, 2013; pp. 427–447. [Google Scholar]
- Odian, G. Principles of Polymerization, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2004; pp. 198–349. [Google Scholar]
- Dubé, M.A.; Salehpour, S. Applying the Principles of Green Chemistry to Polymer Production Technology. Macromol. React. Eng. 2014, 8, 7–28. [Google Scholar] [CrossRef]
- Ačkar, Đ.; Babić, J.; Jozinović, A.; Miličević, B.; Jokić, S.; Miličević, R.; Rajič, M.; Šubarić, D. Starch Modification by Organic Acids and Their Derivatives: A Review. Molecules 2015, 20, 19554–19570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.Y.; Park, S.S.; Lim, S.T. Preparation, Characterization and Utilization of Starch Nanoparticles. Colloids Surf. B Biointerfaces 2015, 126, 607–620. [Google Scholar] [CrossRef] [PubMed]
- Jyothi, A.N.; Carvalho, A.J.F. Polysaccharide Based Graft Copolymers, 1st ed.; Kalia, S., Sabaa, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 59–110. [Google Scholar]
- Mollaahmad, M.A. Sustainable Fillers for Paper. Masters’s Thesis, Luleå University of Technology, Luleå, Sweden, 2008. [Google Scholar]
- Le Corre, D. Starch Nanocrystals: Preparation and Application to Bio-Based Flexible Packaging. Ph.D. Thesis, Université De Grenoble, Grenoble, France, 2011. [Google Scholar]
- Dudhani, A.R. Starch-Based Polymeric Materials and Nanocomposites, 1st ed.; Ahmed, J., Tiwari, B.K., Imam, S.H., Rao, M.A., Eds.; CRC Press: Boca Raton, FL, USA, 2012; pp. 361–372. [Google Scholar]
- Angellier, H.; Molina-Boisseau, S.; Dufresne, A. Mechanical Properties of Waxy Maize Starch Nanocrystal Reinforced Natural Rubber. Macromolecules 2005, 38, 9161–9170. [Google Scholar] [CrossRef]
- Dennenberg, R.J.; Abbott, T.P. Rapid Analysis of Starch Graft Copolymers. J. Polym. Sci. Polym. Lett. Ed. 1976, 14, 693–696. [Google Scholar] [CrossRef]
- Mehrotra, R.; Ranby, B. Graft Copolymerization Onto Starch. II. Grafting of Acrylonitrile to Granular Native Potato Starch by Manganic Pyrophosphate Initiation. Effect of Reaction Conditions on Grafting Parameters. J. Appl. Polym. Sci. 1977, 21, 3407–3415. [Google Scholar] [CrossRef]
- Mehrotra, R.; Ranby, B. Graft Copolymerization Onto Starch. III. Grafting of Acrylonitrile to Gelatinized Potato Starch by Manganic Pyrophosphate Initiation. J. Appl. Polym. Sci. 1978, 22, 2991–3001. [Google Scholar] [CrossRef]
- Mehrotra, R.; Ranby, B. Graft Copolymerization Onto Starch. IV. Grafting of Methyl Methacrylate to Granular Native Potato Starch by Manganic Pyrophosphate Initiation. J. Appl. Polym. Sci. 1978, 22, 3003–3010. [Google Scholar] [CrossRef]
- Dufresne, A. Polysaccharides: Bioactivity and Biotechnology, 1st ed.; Ramawat, K.G., Merillon, J.-M., Eds.; Springer International Publishing: Basel, Switzerland, 2015; pp. 417–449. [Google Scholar]
- Heinze, T.; Koschella, A. Carboxymethyl Ethers of Cellulose and Starch—A Review. Macromol. Symp. 2005, 223, 13–39. [Google Scholar] [CrossRef]
- Le Corre, D.; Angellier-Coussy, H. Preparation and Application of Starch Nanoparticles for Nanocomposites: A Review. React. Funct. Polym. 2014, 85, 97–120. [Google Scholar] [CrossRef]
- Dufresne, A.; Castaño, J. Polysaccharide Nanomaterial Reinforced Starch Nanocomposites: A Review. Starch/Stärke 2016, 68, 1–19. [Google Scholar] [CrossRef]
- Carvalho, O.; Avérous, L.; Tadini, C. Mechanical Properties of Cassava Starch-Based Nano-Biocomposites. In Proceedings of the 11th International Confress of Engineering and Food, Athens, Greece, 22–26 May; Saravacos, G., Ed.; Elsevier Procedia: New York, NY, USA, 2011; pp. 111–112. [Google Scholar]
- Leja, K.; Lewandowicz, G. Polymer Biodegradation and Biodegradable Polymers—A Review. Polish J. Environ. Stud. 2010, 19, 255–266. [Google Scholar]
- Mose, B.R.; Maranga, S.M. A Review on Starch Based Nanocomposites for Bioplastic Materials. J. Mater. Sci. Eng. B 2011, 1, 239–245. [Google Scholar]
- Lu, D.R.; Xiao, C.M.; Xu, S.J. Starch-Based Completely Biodegradable Polymer Materials. Express Polym. Lett. 2009, 3, 366–375. [Google Scholar] [CrossRef]
- Athawale, V.D.; Rathi, S.C. Graft Polymerization: Starch as A Model Substrate. J. Macromol. Sci. Part C Polym. Rev. 1999, 39, 445–480. [Google Scholar] [CrossRef]
- Santana, Á.L.; Angela, M.; Meireles, A. New Starches Are the Trend for Industry Applications: A Review. Food Public Heal. 2014, 4, 229–241. [Google Scholar] [CrossRef]
- Meshram, M.W.; Patil, V.V.; Mhaske, S.T.; Thorat, B.N. Graft Copolymers of Starch and Its Application In Textiles. Carbohydr. Polym. 2009, 75, 71–78. [Google Scholar] [CrossRef]
- Li, M.C.; Ge, X.; Cho, U.R. Emulsion Grafting Vinyl Monomers Onto Starch for Reinforcement of Styrene-Butadiene Rubber. Macromol. Res. 2013, 21, 519–528. [Google Scholar] [CrossRef]
- van den Oever, M.; Molenveld, K. Replacing Fossil Based Plastic Performance Products by Bio-Based Plastic Products-Technical Feasibility. New Biotechnol. 2017, 37, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Glavas, L. Starch and Protein Based Wood Adhesives. Masters’s Thesis, Kungluga Tekniska Högskolan, Stockholm, Sweden, 2011. [Google Scholar]
- Eutamene, M.; Benbakhti, A.; Khodja, M.; Jada, A. Preparation and Aqueous Properties of Starch-Grafted Polyacrylamide Copolymers. Starch/Staerke 2009, 61, 81–91. [Google Scholar] [CrossRef]
- Shin, J.Y.; Jones, N.; Lee, D.I.; Fleming, P.D.; Joyce, M.K.; Dejong, R.; Bloembergen, S. Rheological Properties of Starch Latex Dispersions and Starch Latex-Containing Coating Colors. In Proceedings of the“Growing the Future”, Proceedings of the Paper Conference and Trade Show, TAPPI Press, Peachtree Corners, GA, USA, 22–25 April 2012; pp. 1–25. [Google Scholar]
- Misman, M.A.; Azura, A.R.; Hamid, Z.A.A. The Physical and Degradation Properties of Starch-Graft-Acrylonitrile/Carboxylated Nitrile Butadiene Rubber Latex Films. Carbohydr. Polym. 2015, 128, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; Sun, X. Mechanical Properties of Poly(Lactic Acid)/Starch Composites Compatibilized by Maleic Anhydride. Biomacromolecules 2004, 5, 1446–1451. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.M.; Tan, J.; Xue, G.N. Synthesis and Properties of Starch-g-Poly(Maleic Anhydride-Co-Vinyl Acetate). Express Polym. Lett. 2010, 4, 9–16. [Google Scholar] [CrossRef]
- Wang, C.; Pan, Z.; Wu, M.; Zhao, P. Effect of Reaction Conditions on Grafting Ratio and Properties of Starch Nanocrystals-g-Polystyrene. J. Appl. Polym. Sci. 2014, 131, 1–7. [Google Scholar] [CrossRef]
- Noordergraaf, I.-W.; Fourie, T.K.; Raffa, P. Free-Radical Graft Polymerization Onto Starch as a Tool to Tune Properties in Relation to Potential Applications. A Review. Processes 2018, 6, 31. [Google Scholar] [CrossRef]
- Smeets, N.M.B.; Imbrogno, S.; Bloembergen, S. Carbohydrate Functionalized Hybrid Latex Particles. Carbohydr. Polym. 2017, 173, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Asua, J.M. Emulsion Polymerization: From Fundamental Mechanisms to Process Developments. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 1025–1041. [Google Scholar] [CrossRef]
- Steward, P.A.; Hearn, J.; Wilkinson, M.C. Overview of Polymer Latex Film for mation and Properties. Adv. Colloid Interface Sci. 2000, 86, 195–267. [Google Scholar] [CrossRef]
- Okubo, M.; Yamada, A.; Matsumoto, T. Estimation of Morphology of Composite Polymer Emulsion Particles by the Soap Titration Method. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 3219–3228. [Google Scholar] [CrossRef]
- Okubo, M.; Katsuta, Y.; Matsumoto, T. Studies on Suspension and Emulsion. LI. Peculiar Morphology of Composite Polymer Particles Produced by Seeded Emulsion Polymerization. J. Polym. Sci. Polym. Lett. Ed. 1982, 20, 45–51. [Google Scholar] [CrossRef]
- Sundberg, D.C.; Durant, Y.G. Latex Particle Morphology, Fundamental Aspects: A Review. Polym. React. Eng. 2003, 11, 379–432. [Google Scholar] [CrossRef]
- Singh, K.B. Understanding Film for mation Mechanism In Latex Dispersions. Ph.D. Thesis, Indian Institute of Technology Bombay, Mumbai, India, 2008. [Google Scholar]
- Dalnoki-Veress, K.; Dutcher, J.R.; for rest, J.A. Dynamics and Pattern for mation in Thin Polymer Films. Phys. Can. 2003, 59, 75–84. [Google Scholar]
- Keddie, J.L. Film for mation of Latex. Mater. Sci. Eng. 1997, 21, 101–170. [Google Scholar] [CrossRef]
- Ranum, P.; Peña-Rosas, J.P.; Garcia-Casal, M.N. Global Maize Production, Utilization, and Consumption. Ann. N. Y. Acad. Sci. 2014, 1312, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Rudin, A.; Choi, P. The Elements of Polymer Science & Engineering, 3rd ed.; Elsevier: New York, NY, USA, 2013; pp. 521–535. [Google Scholar]
- Olsson, E. Effects of Citric Acid on Starch-Based Barrier Coatings. Ph.D. Thesis, Karlstad University, Karlstad, Sweden, 2013. [Google Scholar]
- Li, M.; Witt, T.; Xie, F.; Warren, F.J.; Halley, P.J.; Gilbert, R.G. Biodegradation of Starch Films: The Roles of Molecular and Crystalline Structure. Carbohydr. Polym. 2015, 122, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Gallant, D.J.; Bouchet, B.; Baldwin, P.M. Microscopy of Starch: Evidence of A New Level of Granule Organization. Carbohydr. Polym. 1997, 32, 177–191. [Google Scholar] [CrossRef]
- Le Corre, D.; Bras, J.; Dufresne, A. Starch Nanoparticles: A Review. Biomacromolecules 2010, 11, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Singh, J.; Kaur, L.; Sodhi, N.S.; Gill, B.S. Morphological, Thermal and Rheological Properties of Starches From Different Botanical Sources. Food Chem. 2003, 81, 219–231. [Google Scholar] [CrossRef]
- Namazi, H.; Fathi, F.; Heydari, A. The Delivery of Nanoparticles, 1st ed.; Hashim, A.A., Ed.; Intech: London, UK, 2012; pp. 149–184. [Google Scholar]
- Daniel-da-Silva, A.L.; Trindade, T. Advances in Nanocomposite Technology, 1st ed.; Hashim, A.A., Ed.; Intech: London, UK, 2011; pp. 275–291. [Google Scholar]
- Davidson, R.L. Handbook of Water-Soluble Gums and Resins, 1st ed.; Davidson, R.L., Ed.; McGraw-Hill: New York, NY, USA, 1980. [Google Scholar]
- Rindlav-Westling, A.; Stading, M.; Hermansson, A.-M.; Gatenholm, P. Structure, Mechanical and Barrier Properties of Amylose and Amylopectin Films. Carbohydr. Polym. 1998, 36, 217–224. [Google Scholar] [CrossRef]
- Lu, S.; Lin, S.; Yao, K. Study on The Synthesis and Application of Starch-Graft-Poly(AM-Co-DADMAC) by Using A Complex Initiation System of CS-KPS. Starch/Stärke 2004, 56, 138–143. [Google Scholar] [CrossRef]
- Meimoun, J.; Wiatz, V.; Saint-Loup, R.; Parcq, J.; Favrelle, A.; Bonnet, F.; Zinck, P. Modification of Starch by Graft Copolymerization. Starch/Stärke 2018, 70, 1–23. [Google Scholar] [CrossRef]
- Liu, W.C.; Halley, P.J.; Gilbert, R.G. Mechanism of Degradation of Starch, a Highly Branched Polymer, during Extrusion. Macromolecules 2010, 43, 2855–2864. [Google Scholar] [CrossRef]
- Liu, D.; Wu, Q.; Chen, H.; Chang, P.R. Transitional Properties of Starch Colloid with Particle Size Reduction from Micro- to Nanometer. J. Colloid Interface Sci. 2009, 339, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.; Naqash, F.; Gani, A.; Masoodi, F.A. Art and Science behind Modified Starch Edible Films and Coatings: A Review. Compr. Rev. Food Sci. Food Saf. 2016, 15, 568–580. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F. Composition, Structure, Physicochemical Properties, and Modifications of Cassava Starch. Carbohydr. Polym. 2015, 122, 456–480. [Google Scholar] [CrossRef] [PubMed]
- Shi, A.M.; Li, D.; Wang, L.J.; Li, B.Z.; Adhikari, B. Preparation of Starch-Based Nanoparticles through High-Pressure Homogenization and Miniemulsion Cross-Linking: Influence of Various Process Parameters on Particle Size and Stability. Carbohydr. Polym. 2011, 83, 1604–1610. [Google Scholar] [CrossRef]
- Li, W.; Cao, F.; Fan, J.; Ouyang, S.; Luo, Q.; Zheng, J.; Zhang, G. Physically Modified Common Buckweat Starch and Their Physicochemical and Structural Properties. Food Hydrocoll. 2014, 40. [Google Scholar] [CrossRef]
- Bel Haaj, S.; Thielemans, W.; Magnin, A.; Boufi, S. Starch Nanocrystals and Starch Nanoparticles from Waxy Maize as Nanoreinforcement: A Comparative Study. Carbohydr. Polym. 2016, 143, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Zondag, M.D. Effect of Microwave Heat-Moisture and Annealing Treatments on Buckwheat Starch Characteristics. Masters’s Thesis, University of Wisconsin, Madison, WI, USA, 2003. [Google Scholar]
- Liu, X.; Wang, Y.; Yu, L.; Tong, Z.; Chen, L.; Liu, H.; Li, X. Thermal Degradation and Stability of Starch under Different Processing Conditions. Starch/Staerke 2013, 65, 48–60. [Google Scholar] [CrossRef]
- Šárka, E.; Dvořáček, V. New Processing and Applications of Waxy Starch (A Review). J. Food Eng. 2017, 206, 77–87. [Google Scholar] [CrossRef]
- Kim, H.Y.; Lee, J.H.; Kim, J.Y.; Lim, W.J.; Lim, S.T. Characterization of Nanoparticles Prepared by Acid Hydrolysis of Various Starches. Starch/Stärke 2012, 64, 367–373. [Google Scholar] [CrossRef]
- Kim, J.Y.; Yoon, J.W.; Lim, S.T. Formation and Isolation of Nanocrystal Complexes between Dextrins and N-Butanol. Carbohydr. Polym. 2009, 78, 626–632. [Google Scholar] [CrossRef]
- Saari, H.; Fuentes, C.; Sjöö, M.; Rayner, M.; Wahlgren, M. Production of Starch Nanoparticles by Dissolution and Non-Solvent Precipitation for Use in Food-Grade Pickering Emulsions. Carbohydr. Polym. 2017, 157, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Thio, Y.S.; Deng, Y. Starch Nanoparticle for mation Via Reactive Extrusion and Related Mechanism Study. Carbohydr. Polym. 2011, 85, 208–214. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, D.J.; Lim, S.T. Fragmentation of Waxy Rice Starch Granules by Enzymatic Hydrolysis. Cereal Chem. 2008, 85, 182–187. [Google Scholar] [CrossRef]
- Song, D. Starch Crosslinking for Cellulose Fiber Modification and Starch Nanoparticle for Mation. Ph.D. Thesis, Georgia Institute of Technology, Atlanta, GA, USA, 2011. [Google Scholar]
- Lin, N.; Huang, J.; Chang, P.R.; Anderson, D.P.; Yu, J. Preparation, Modification, and Application of Starch Nanocrystals In Nanomaterials: A Review. J. Nanomater. 2011, 2011, 1–13. [Google Scholar] [CrossRef]
- Hui, R.; Qi-he, C.; Ming-liang, F.; Qiong, X.; Guo-qing, H. Preparation and Properties of Octenyl Succinic Anhydride Modified Potato Starch. Food Chem. 2009, 114, 81–86. [Google Scholar] [CrossRef]
- Rzayev, Z.M.O. Graft Copolymers of Maleic Anhydride and Its Isostructural Analogues: High Performance Engineering Materials. Int. Rev. Chem. Eng. 2011, 3, 1689–1699. [Google Scholar]
- Xu, Y.; Ding, W.; Liu, J.; Li, Y.; Kennedy, J.F.; Gu, Q.; Shao, S. Preparation and Characterization of Organic-Soluble Acetylated Starch Nanocrystals. Carbohydr. Polym. 2010, 80, 1078–1084. [Google Scholar] [CrossRef]
- Chakraborty, S.; Sahoo, B.; Teraoka, I.; Miller, L.M.; Gross, R.A. Enzyme-Catalyzed Regioselective Modification of Starch Nanoparticles. Macromolecules 2005, 38, 61–68. [Google Scholar] [CrossRef]
- Cheng, S.; Zhao, W.; Wu, Y. Optimization of Synthesis and Characterization of Oxidized Starch-Graft-Poly(Styrene-Butyl Acrylate) Latex for Paper Coating. Starch/Stärke 2015, 67, 493–501. [Google Scholar] [CrossRef]
- Cummings, S. The Incorporation of Vinyl Modified Regenerated Starch Nanoparticles in Emulsion Polymerizations. Ph.D. Thesis, University of Ottawa, Ottawa, ON, Canada, 2017. [Google Scholar]
- Li, M.; Mun, Y.; Cho, U.R. Synthesis of Environmental-Friendly Starch-Acrylic Coating Sols by Emulsion Polymerization. Elastom. Compos. 2010, 45, 272–279. [Google Scholar]
- Mou, J.; Li, X.; Wang, H.; Fei, G.; Liu, Q. Preparation, Characterization, and Water Resistance of Cationic Acetylated Starch-g-Poly(Styrene-Butyl Acrylate) Surfactant-Free Emulsion. Starch/Stärke 2012, 64, 826–834. [Google Scholar] [CrossRef]
- Song, S.; Wang, C.; Pan, Z.; Wang, X. Preparation and Characterization of Amphiphilic Starch Nanocrystals. J. Appl. Polym. Sci. 2008, 107, 418–422. [Google Scholar] [CrossRef]
- Gao, J.-P.; Tian, R.-C.; Yu, J.-G.; Duan, M.-L. Graft Copolymers of Methy Methacrylate onto Canna Starch Using Manganic Pyrophosphate as an Initiator. J. Appl. Polym. Sci. 1994, 53, 1091–1102. [Google Scholar] [CrossRef]
- Wyatt, V.T.; Strahan, G.D. Degree of Branching in Hyperbranched Poly(Glycerol-Co-Diacid)s Synthesized in Toluene. Polymers 2012, 4, 396–407. [Google Scholar] [CrossRef]
- Nilsson, G.S.; Bergquist, K.-E.; Nilsson, U.; Gorton, L. Determination of the Degree of Branching in Normal and Amylopectin Tye Potato Starch with 1H-NMR: Improved Resolution and Two-Dimensional Spectroscopy. Starch/Stärke 1996, 10, 352–357. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, L.; Chen, Y.; Yan, D. Using 2D NMR to Determine the Degree of Branching of Complicated Hyperbranched Polymers. Sci. China Ser. B Chem. 2008, 51, 1057–1065. [Google Scholar] [CrossRef]
- Markoski, L.J.; Thompson, J.L.; Moore, J.S. Indirect Method for Determining Degree of Branching in Hyperbranched Polymers. Macromolecules 2002, 35, 1599–1603. [Google Scholar] [CrossRef]
- Liu, J.M.; Chang, C.S.; Tsiang, R.C.C. Method of Determining the Degree of Branching from the Gel Permeation Chromatogram for Star-Shaped SBS Thermoplastic Block Copolymers. J. Polym. Sci. Part A Polym. Chem. 1997, 35, 3393–3401. [Google Scholar] [CrossRef]
- Mange, S.; Dever, C.; De Bruyn, H.; Gaborieau, M.; Castignolles, P.; Gilbert, R.G. Grafting of Oligosaccharides Onto Synthetic Polymer Colloids. Biomacromolecules 2007, 8, 1816–1823. [Google Scholar] [CrossRef] [PubMed]
- Baijun, W.; Hui, X.; Yonghong, Z. Study on Graft Emulsion Copolymerization of Styrene/Butyl Acrylate Onto Starch. Spec. Petrochem. 2005, 2. [Google Scholar]
- Hwang, J.H.; Ryu, H.; Cho, U.R. A Study on Starch-Acrylic Graft Copolymerization by Emulsion Polymerization. Elastomer 2008, 43, 221–229. [Google Scholar]
- Chu, H.J.; Wei, H.L.; Zhu, J. Ultrasound Enhanced Radical Graft Polymerization of Starch and Butyl Acrylate. Chem. Eng. Process. Process Intensif. 2015, 90, 1–5. [Google Scholar] [CrossRef]
- Peng, B.L.; Dhar, N.; Liu, H.L.; Tam, K.C. Chemistry and Applications of Nanocrystalline Cellulose and its Derivatives: A Nanotechnology Perspective. Can. J. Chem. Eng. 2011, 89, 1191–1206. [Google Scholar] [CrossRef]
- Kweon, M.; Slade, L.; Levine, H. Role of Glassy and Crystalline Transitions in the Responses of Corn Starches to Heat and High Pressure Treatments: Prediction of Solute-Induced Barostability from Solute-Induced Thermostability. Carbohydr. Polym. 2008, 72, 293–299. [Google Scholar] [CrossRef]
- Cranston, E.D.; Gray, D.G. Birefringence in Spin-Coated Films Containing Cellulose Nanocrystals. Colloids Surfaces A Physicochem. Eng. Asp. 2008, 325, 44–51. [Google Scholar] [CrossRef]
- Cheng, S.; Zhang, Y.; Wu, Y. Preparation and Characterization of Enzymatically Degraded Starch-g-Poly(Styrene-Co-Butyl Acrylate) Latex for Paper Coating. Polym. Plast. Technol. Eng. 2014, 53, 1811–1816. [Google Scholar] [CrossRef]
- Nikfarjam, N.; Taheri Qazvini, N.; Deng, Y. Cross-Linked Starch Nanoparticles Stabilized Pickering Emulsion Polymerization of Styrene In W/O/W System. Colloid Polym. Sci. 2014, 292, 599–612. [Google Scholar] [CrossRef]
- Jiang, B.; Tsavalas, J.G.; Sundberg, D.C. Water Whitening of Polymer Films: Mechanistic Studies and Comparisons Between Water and Solvent Borne Films. Prog. Org. Coat. 2017, 105, 56–66. [Google Scholar] [CrossRef]
- Rodehed, C.; Rånby, B. Structure and Molecular Properties of Saponified Starch–Graft-polyacrylonitrile. J. Appl. Polym. Sci. 1986, 32, 3323–3333. [Google Scholar] [CrossRef]
- Glenn, G.M.; Orts, W.; Imam, S.; Chiou, B.; Wood, D.F. Starch Polymers, 1st ed.; Halley, P., Averous, L., Eds.; Elsevier: New York, NY, USA, 2014; pp. 421–452. [Google Scholar]
- Mostafa, K.M. Graft Polymerization of Acrylic Acid Onto Starch Using Potassium Permanganate Acid (Redox System). J. Appl. Polym. Sci. 1995, 56, 263–269. [Google Scholar] [CrossRef]
- Cheng, S.; Xu, J.; Wu, Y. Preparation and Characterization of Oxidized Starch-graft-Poly(styrene-butyl acrylate) Latex Via Emulsion Polymerization. J. Polym. Eng. 2014, 34, 611–616. [Google Scholar] [CrossRef]
- Jiang, S.; Dai, L.; Xiong, L.; Sun, Q. Preparation and Characterization of Octenyl Succinic Anhydride Modified Taro Starch Nanoparticles. PLoS ONE 2016, 11, e0150043. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Toro, R.; Bonilla, J.; Talens, P.; Chiralt, A. Starch-Based Materials in Food Packaging: Processing, Characterization and Applications, 1st ed.; Villar, M.A., Barbosa, S.E., Garcia, A.M., Castillo, L.A., López, O.V., Eds.; Academic Press: London, UK, 2017; pp. 257–312. [Google Scholar]
- Ulu, A.; Koytepe, S.; Ates, B. Design of Starch Functionalized Biodegradable P(MMA-co-MMA) as Carrier Matrix for l-asparaginase Immobilization. Carbohydr. Polym. 2016, 153, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Ulu, A.; Koytepe, S.; Ates, B. Synthesis and Characterization of Biodegradable pHEMA-starc Composites for Immobilization of l-asparaginase. Polym. Bull. 2016, 73, 1891. [Google Scholar] [CrossRef]
- Garcia-Valdez, O.; Champagne, P.; Cunningham, M.F. Graft Modification of Natural Polysaccharides via Reversible Deactivation Radical Polymerization. Prog. Polym. Sci. 2018, 76, 151–173. [Google Scholar] [CrossRef]
- Glasing, J.; Champagne, P.; Cunningham, M.F. Graft Modification of Chitosan, Cellulose and Alginate Using Reversible Deactivation Radical Polymerization (RDRP). Curr. Opin. Green Sustain. Chem. 2016, 2, 15–21. [Google Scholar] [CrossRef]
- Chen, Q.; Yu, H.; Wang, L.; Ul Abdin, Z.; Chen, Y.; Wang, J.; Zhou, W.; Yang, X.; Khan, R.U.; Zhang, H.; et al. Recent Progress in Chemical Modification of Starch and Its Applications. RSC Adv. 2015, 5, 67459–67474. [Google Scholar] [CrossRef]
- Lu, D.; Duan, P.; Guo, R.; Yang, L.; Zhang, H. Synthesis and Characterization of Starch Graft Biodegradable Polyester and Polyesteramide by Direct Polycondensation. Polym. Plast. Technol. Eng. 2013, 52, 200–205. [Google Scholar] [CrossRef]
- Maiti, S.; Ranjit, S.; Sa, B. Polysaccharide-Based Graft Copolymers in Controlled Drug Delivery. Int. J. 2010, 2, 1350–1358. [Google Scholar]
- Djordjevic, S.; Nikolic, L.; Kovacevic, S.; Miljkovic, M.; Djordjevic, D. Graft Copolymerization of Acrylic Acid Onto Hydrolyzed Potato Starch Using Various Initiators. Period. Polytech. Chem. Eng. 2013, 57, 55–61. [Google Scholar] [CrossRef]
- Lai, S.M.; Don, T.M.; Liu, Y.H.; Chiu, W.Y. Graft Polymerization of Vinyl Acetate Onto Granular Starch: Comparison on The Potassium Persulfate and Ceric Ammonium Nitrate Initiated System. J. Appl. Polym. Sci. 2006, 102, 3017–3027. [Google Scholar] [CrossRef]
- Lanthong, P.; Nuisin, R.; Kiatkamjornwong, S. Graft Copolymerization, Characterization, and Degradation of Cassava Starch-g-Acrylamide/Itaconic Acid Superabsorbents. Carbohydr. Polym. 2006, 66, 229–245. [Google Scholar] [CrossRef]
- Shi, Z.; Reddy, N.; Shen, L.; Hou, X.; Yang, Y. Effects of Monomers and Homopolymer Contents on the Dry and Wet Tensile Properties of Starch Films Grafted with Various Methacrylates. J. Agric. Food Chem. 2014, 62, 4668–4676. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.B.; Lv, C.F.; Han, M.N. Synthesis and Performance Study of Polybasic Starch Graft Copolymerization Function Materials. Adv. Mater. Res. 2009, 79–82, 43–46. [Google Scholar] [CrossRef]
- Xue, Y.; Cheng, S.; We, Y. Study on Preparation of Graft Copolymer Emulsion of Zymolytic Starch-Styrene/Butyl Acrylate. Appl. Chem. Ind. 2012, 2. [Google Scholar]
- Lee, M.; Ryu, H.; Cho, U.R. A Study on The Synthesis of Starch-Acrylic Polymer by Emulsion Polymerization. Polymer 2010, 34, 58–62. [Google Scholar] [CrossRef]
- Haaj, S.B.; Bou, S. Starch Nanoparticles Produced Via Ultrasonication As A Sustainable Stabilizer In Pickering Emulsion Polymerization. RSC Adv. 2014, 4, 42638–42646. [Google Scholar] [CrossRef]
- Pei, X.; Tan, Y.; Xu, K.; Liu, C.; Lu, C.; Wang, P. Pickering Polymerization of Styrene Stabilized by Starch-Based Nanospheres. Polym. Chem. 2016, 7, 3325–3333. [Google Scholar] [CrossRef]
- Haaj, S.B.; Thielemans, W.; Magnin, A.; Boufi, S. Starch Nanocrystal Stabilized Pickering Emulsion Polymerization for Nanocomposites with Improved Performance. ACS Appl. Mater. Interfaces 2014, 6, 8263–8273. [Google Scholar] [CrossRef] [PubMed]
- Angellier, H.; Choisnard, L.; Molina-Boisseau, S.; Ozil, P.; Dufresne, A. Optimization of the Preparation of Aqueous Suspensions of Waxy Maize Starch Nanocrystals Using a Response Surface Methodology. Biomacromolecules 2004, 5, 1545–1551. [Google Scholar] [CrossRef] [PubMed]
- Namazi, H.; Dadkhah, A.; Mosadegh, M. New Biopolymer Nanocomposite of Starch-Graft Polystyrene/Montmorillonite Clay Prepared Through Emulsion Polymerization Method. J. Polym. Environ. 2012, 20, 794–800. [Google Scholar] [CrossRef]
- Ulu, A.; Koytepe, S.; Ates, B. Synthesis and Characterization of PMMA Composites Activated With Starch for Immobilization of L-Asparaginase. J. Appl. Polym. Sci. 2016, 133, 1–11. [Google Scholar] [CrossRef]
- Xu, J.; Long, L.; Hu, H. Preparation of Starch-Based Styrene Acrylate Emulsion Used As Surface-Treatment Agent for Decorative Base Paper. J. Polym. Eng. 2013, 33, 323–330. [Google Scholar] [CrossRef]
- Wang, R.-M.; Wang, X.-W.; Guo, J.-F.; He, Y.-F.; Jiang, M.-L. Crosslinkable Potato Starch-Based Graft Copolymer Emulsion for Humidity Controlling Coatings. Mater. Res. 2013, 16, 1246–1253. [Google Scholar] [CrossRef]
- Terpstra, R. Potato Starch Stabilized Synthetic Latexes. Ph.D. Thesis, University of Groningen, Groningen, The Netherlands, 2015. [Google Scholar]
- Brockway, C.E.; Seaberg, P.A. Grafting of Polyacrylonitrile to Granular Corn Starch. J. Polym. Sci. Part A-1 1967, 5, 1313–1326. [Google Scholar] [CrossRef]
- Misra, B.N.; Dogra, R.; Kaur, I.; Sood, D. Grafting onto Starch. II. Graft Copolymerization of Vinyl Acetate on to Starch by Radical Initiator. J. Polym. Sci. Polym. Chem. Ed. 1980, 18, 341–344. [Google Scholar] [CrossRef]
- Misra, B.N.; Dogra, R. Grafting onto Starch. IV. Graft Copolymerization of Methyl Methacrylate by Use of AIBN as Radical Initiator. J. Macromol. Sci. 2006, 14, 763–770. [Google Scholar] [CrossRef]
- Pei, X.; Zhai, K.; Tan, Y.; Xu, K.; Lu, C.; Wang, P.; Wang, T.; Chen, C.; Tao, Y.; Dai, L.; et al. Synthesis of Monodisperse Starch-Polystyrene Core-Shell Nanoparticles Via Seeded Emulsion Polymerization without Stabilizer. Polymer 2016, 108, 78–86. [Google Scholar] [CrossRef]
- Abo-Shosha, M.H.; Ibrahim, N.A. Synthesis and Characterization of Starch-Glycidyl Methacrylate-Acrylic Acid Cation Exchange Composites. Starch/Stärke 1992, 44, 466–471. [Google Scholar] [CrossRef]
- Wu, G.; Limei, Z. Study on KPS-Na2S2O3 Initiated Graft Copolymerization of Butyl Acrylate Onto Corn Starch. Petrochemical Technol. 1993, 5. [Google Scholar]
- Hebeish, A.; Beliakova, M.K.; Bayazeed, A. Improved Synthesis of Poly (MAA)—Starch. Jourbal Appl. Polym. Sci. 1997, 68, 1709–1715. [Google Scholar] [CrossRef]
- Beliakova, M.K.; Aly, A.A.; Abdel-Mohdy, F.A. Grafting of Poly(Methacrylic Acid) on Starch and Poly(Vinyl Alcohol). Starch/Stärke 2004, 56, 407–412. [Google Scholar] [CrossRef]
- Ikhuoria, E.U.; Folayan, A.S.; Okieimen, F.E. Studies in the Graft Copolymerization of Acrylonitrile onto Cassava Starch by Ceric Ion Induced Initiation. Int. J. Biotechnol. Mol. Biol. Res. 2010, 1, 10–14. [Google Scholar]
- Burr, R.C.; Fanta, G.F.; Russell, C.R.; Rist, C.E. Influence of Swelling and Disruption of The Starch Granule on The Composition of The Starch-Polyacrylonitrile Copolymer. J. Macromol. Sci. Part A Chem. 1967, 7, 1381–1385. [Google Scholar] [CrossRef]
- Fanta, G.F.; Burr, R.C.; Russell, C.R.; Rist, C.E. Copolymers of Starch and Polyacrylonitrile. Influence of Granule Swelling on Copolymer Composition under Various Reaction Conditions. J. Macromol. Sci. Part A Chem. 1970, 4, 331–339. [Google Scholar] [CrossRef]
- Han, T.L.; Kumar, R.N.; Rozman, H.D.; Noor, M.A.M. GMA Grafted Sago Starch as a Reactive Component in Ultra Violet Radiation Curable Coatings. Carbohydr. Polym. 2003, 54, 509–516. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhuo, R. Controlled Release of Carboxylic-Containing Herbicides by Starch-g-Poly(Butyl Acrylate). J. Appl. Polym. Sci. 2001, 81, 1535–1543. [Google Scholar] [CrossRef]
- Li, M.-C.; Lee, J.K.; Cho, U.R. Synthesis, Characterization, and Enzymatic Degradation of Starch-Grafted Poly(Methyl Methacrylate) Copolymer Films. Polym. Polym. Compos. 2013, 21, 449–456. [Google Scholar] [CrossRef]
- Sangramsingh, N.M.; Patra, B.N.; Singh, B.C.; Patra, C.M. Graft Copolymerization of Methyl Methacrylate onto Starch Using a Ce (IV)—Glucose Initiator System. J. Appl. Ploymer Sci. 2003, 91, 981–990. [Google Scholar] [CrossRef]
- Okieimen, F.E.; Said, O.B. Studies on The Graft Copolymerization of Methyl Mathacrylate onto Starch. Acta Polym. 1989, 40, 708–710. [Google Scholar] [CrossRef]
- Ray-Chaudhuri, D.K. Grafted Starch Acrylates and Their Properties. Starch/Stärke 1969, 21, 47–52. [Google Scholar] [CrossRef]
- Fares, M.M.; El-Faqeeh, A.S.; Osman, M.E. Graft Copolymerization onto Starch-I. Synthesis and Optimization of Starch Grafted with N-Tert-Butylacrylamide Copolymer and Its Hydrogels. J. Polym. Res. 2003, 10, 119–125. [Google Scholar] [CrossRef]
- Zou, W.; Yu, L.; Liu, X.; Chen, L.; Zhang, X.; Qiao, D.; Zhang, R. Effects of Amylose/Amylopectin Ratio on Starch-Based Superabsorbent Polymers. Carbohydr. Polym. 2012, 87, 1583–1588. [Google Scholar] [CrossRef]
- Singh, O.P.; Sandle, N.K.; Varma, I.K. Graft-Copolymerization of Starch With Acrylamide, III. Physical Properties and Enzymatic Degradation. Die Angew. Makromol. Chemie 1984, 122, 193–201. [Google Scholar] [CrossRef]
- Gugliemelli, L.A.; Doane, W.M.; Russell, C.R. Preparation of Soapless Latexes by Sonification of Starch-based Poly(Isoprene–Co–Acrylonitrile) Graft Reaction Mixtures. J. Appl. Polym. Sci. 1979, 23, 635–644. [Google Scholar] [CrossRef]
- Weaver, M.O. Starch-g-Poly(Methyl Acrylate) Latexes for Stabilizing Soil to Water Erosion: Extending the Range of Polymer Add-On. Starch/Stärke 1989, 41, 106–110. [Google Scholar] [CrossRef]
- Gaborieau, M.; de Bruyn, H.; Mange, S.; Castignolles, P.; Brockmeyer, A.; Gilbert, R.G. Synthesis and Characterization of Synthetic Polymer Colloids Colloidally Stabilized by Cationized Starch Oligomers. J. Polym. Sci. Part A 2009, 47, 1836–1852. [Google Scholar] [CrossRef]
- Gugliemelli, L.A.; Swanson, C.L.; Baker, F.L.; Doane, W.M.; Russell, C.R. Cationic Starch-Polyacrylonitrile Graft. J. Polym. Sci. Polym. Chem. Ed. 1974, 12, 2683–2692. [Google Scholar] [CrossRef]
- Patel, N.; Patel, K.C.; Patel, R.D. The Effect of Acrylonitrile on Starch Gelatinization. Morphological Study. Starch/Stärke 1985, 37, 201–205. [Google Scholar] [CrossRef]
- Ghosh, P.; Paul, S.K. Graft Copolymerization of Methyl Methacrylate on Potato Starch Using Potassium Trioxalatomanganate, K3[Mn(C2O4)3], as Initiator. J. Macromol. Sci. 2006, 20, 179–188. [Google Scholar] [CrossRef]
- Carlsohn, H.; Hartmann, M. Mangan(1V)-Initiierte Pfropfcopolymerisation von Methylmethacrylat Und Anderen Acrylmonomeren Auf Starke Und Dextran. Acta Polym. 1982, 33, 640–643. [Google Scholar] [CrossRef]
- Mehrotra, R.; Ranby, B. Graft Copolymerization onto Starch. I. Complexes of Mn3 as Initiators. J. Appl. Polym. Sci. 1977, 21, 1647–1654. [Google Scholar] [CrossRef]
- Qu, B.; Li, H.; Niu, Y. Graft Copolymerization of Poly(Vinyl Acetate) onto Starch Using KMnO4-H2SO4 Redox System. J. Polym. Eng. 2013, 33, 521–526. [Google Scholar] [CrossRef]
- Xiao, L.; Huigen, Y.; Shunying, T.; Qiyun, Z.; Renyun, P. Study on the Graft Copolymerization of Styrene and Binary Monomers Onto Corn Starch by Manganese Pyrophosphate Initiation. China Synth. Resin Plast. 1995, 3. [Google Scholar]
- Chen, S.; Tang, G.D. Emulsion Graft Copolymerization of St/MMA Onto Starch. J. Nanjing Univ. 2001, 6. [Google Scholar]
- Tian, X.; Huang, K.; Ma, T. Study on Graft Copolymerization of Styrene on Corn Starch with Manganese Pyrophosphate as Initiator. Chem. Adhes. 2004, 3. [Google Scholar]
- Trimnell, D.; Fanta, G.F.; Salch, J.H. Graft Polymerization of Methyl Acrylate onto Granular Starch: Comparison of the Fe+2/H2O2 and Ceric Initiating Systems. J. Appl. Polym. Sci. 1996, 60, 285–292. [Google Scholar] [CrossRef]
- Aravindakshan, P.; Bhatt, A.; Kumar, V.G. Matrix-Induced Variation in Kinetics and Control of Molecular Weight of Methacrylic Acid Polymers During Graft Copolymerization with Starch. J. Appl. Polym. Sci. 1997, 66, 397–403. [Google Scholar] [CrossRef]
- Fanta, G.F.; Burr, R.C.; Doane, W.M.; Russell, C.R. Graft Copolymers of Starch with Mixtures of Acrylamide and the Nitric Acid Salt of Dimethylaminoethyl Methacrylate. J. Appl. Polym. Sci. 1972, 16, 2835–2845. [Google Scholar] [CrossRef]
- Witono, J.R.; Noordergraaf, I.W.; Heeres, H.J.; Janssen, L.P.B.M. Graft Copolymerization of Acrylic Acid to Cassava Starch—Evaluation of The Influences of Process Parameters by an Experimental Design Method. Carbohydr. Polym. 2012, 90, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Herold, B.R.; Fouassier, J. Photochemical Grafting of Vinyl Monomers Onto Starch. Starch/Stärke 1981, 33, 90–97. [Google Scholar] [CrossRef]
- Kaur, I.; Sharma, M. Synthesis, Characterization and Applications of Graft Copolymers of Sago and Acrylamide. Trends Carbohydr. Res. 2014, 6, 51–66. [Google Scholar]
- El-Sayed, M.M.; Sorour, M.H.; Abd El Moneem, N.; Shalaan, H.; El Marsafy, S. Grafting of Acrylamide onto Polysaccharides Blend Using Photo-Initiators. J. Polym. Environ. 2017, 25, 402–407. [Google Scholar] [CrossRef]
- Comer, C.; Jessop, J. Photoinitiated Emulsion Graft Polymerization of Synthetic Monomers to Starch. In Proceedings of the RadTech Conference & Exhibition, The Hague, The Netherlands, 14–15 September 2006. [Google Scholar]
- Comer, C.M.; Jessop, J.L.P. Evaluation of Novel Back-Flush Filtration for Removal of Homopolymer From Starch-g-PMMA. Starch/Stärke 2008, 60, 335–339. [Google Scholar] [CrossRef]
- Suwanmala, P.; Hemvichian, K.; Hoshina, H.; Srinuttrakul, W.; Seko, N. Preparation of Metal Adsorbent from Poly(Methyl Acrylate)-Grafted-Cassava Starch via Gamma Irradiation. Radiat. Phys. Chem. 2012, 81, 982–985. [Google Scholar] [CrossRef]
- Kaur, I.; Sharma, M. Synthesis and Characterization of Graft Copolymers of Sago Starch and Acrylic Acid. Starch/Stärke 2012, 64, 441–451. [Google Scholar] [CrossRef]
- Zhang, Y.N.; Xu, S.A. Effects of Amylose/Amylopectin Starch on Starch-Based Superabsorbent Polymers Prepared by γ-Radiation. Starch/Stärke 2017, 69, 1–9. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, W.; Wang, H.; Qi, W.; Yue, L.; Ye, Q. Synthesis and Characterisation of Starch Grafted Superabsorbent via 10 MeV Electron-Beam Irradiation. Carbohydr. Polym. 2014, 101, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cunningham, M.F.; Smeets, N.M.B.; Dubé, M.A. Starch Nanoparticle Incorporation in Latex-Based Adhesives. Eur. Polym. J. 2018, 106, 128–138. [Google Scholar] [CrossRef]
- Nikolic, V.; Velickovic, S.; Antonovic, D.; Popovic, A. Biodegradation of Starch-Graft-Polystyrene and Starch-Graft- Poly(Methacrylic Acid) Copolymers in Model River Water. J. Serbian Chem. Soc. 2013, 78, 1425–1441. [Google Scholar] [CrossRef]
- Samaha, S.H.; Nasr, H.E.; Hebeish, A. Synthesis and Characterization of Starch-Poly(Vinyl Acetate) Graft Copolymers and Their Saponified for m. J. Polym. Res. 2005, 12, 343–353. [Google Scholar] [CrossRef]
- De Graaf, R.A.; Janssen, L.P.B.M. Production of a New Partially Biodegradable Starch Plastic by Reactive Extrusion. Polym. Eng. Sci. 2000, 40, 2086–2094. [Google Scholar] [CrossRef]
- McDowall, D.J.; Gupta, B.S.; Stannett, V.T. Grafting of Vinyl Monomers to Cellulose by Ceric Ion Initiation. Prog. Polym. Sci. 1984, 10, 1–50. [Google Scholar] [CrossRef]
- Kalia, S.; Sabaa, M.W. Polysaccharide Based Graft Copolymers, 1st ed.; Kalia, S., Sabaa, M.W., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- De Bruyn, H.; Sprong, E.; Gaborieau, M.; David, G.; Roper, J.A., III; Gilbert, R.G. Starch-Graft-Copolymer Latexes Initiated and Stabilized by Ozonolyzed Amylopectin. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 5832–5845. [Google Scholar] [CrossRef]
- de Bruyn, H.; Sprong, E.; Gaborieau, M.; Roper III, J.A.; Gilbert, R.G. Starch-graft-(synthetic copolymer) Latexes Initiated with Ce4+ and Stabilized by Amylopectin. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 4185–4192. [Google Scholar] [CrossRef]
- Nikolic, V.; Velickovic, S.; Popovic, A. Influence of Amine Activators and Reaction Parameters on Grafting Reaction Between Polystyrene and Starch. J. Polym. Res. 2014, 21, 363. [Google Scholar] [CrossRef]
- BelHaaj, S.; Ben Mabrouk, A.; Thielemans, W.; Boufi, S. A One-Step Miniemulsion Polymerization Route Towards The Synthesis of Nanocrystal Reinforced Acrylic Nanocomposites. Soft Matter 2013, 9, 1975–1984. [Google Scholar] [CrossRef]
- Lu, D.; Xiao, C.; Sun, F. Controlled Grafting of Poly(Vinyl Acetate) onto Starch via RAFT Polymerization. J. Appl. Polym. Sci. 2012, 124, 3450–3455. [Google Scholar] [CrossRef]
- Rotureau, E.; Raynaud, J.; Choquenet, B.; Marie, E.; Nouvel, C.; Six, J.L.; Dellacherie, E.; Durand, A. Application of Amphiphilic Polysaccharides as Stabilizers in Direct and Inverse Free-Radical Miniemulsion Polymerization. Colloids Surf. A Physicochem. Eng. Asp. 2008, 331, 84–90. [Google Scholar] [CrossRef]
- Wang, S.; Xu, J.; Wang, Q.; Fan, X.; Yu, Y.; Wang, P.; Zhang, Y.; Yuan, J.; Cavaco-Paulo, A. Preparation and Rheological Properties of Starch-g-Poly(Butyl Acrylate) Catalyzed by Horseradish Peroxidase. Process Biochem. 2017, 59, 104–110. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Q.; Fan, X.; Xu, J.; Zhang, Y.; Yuan, J.; Jin, H.; Cavaco-Paulo, A. Synthesis and Characterization of Starch-Poly(Methyl Acrylate) Graft Copolymers Using Horseradish Peroxidase. Carbohydr. Polym. 2016, 136, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Shogren, R.L.; Willett, J.L.; Biswas, A. HRP-Mediated Synthesis of Starch-Polyacrylamide Graft Copolymers. Carbohydr. Polym. 2009, 75, 189–191. [Google Scholar] [CrossRef]
- Lv, S.; Sun, T.; Zhou, Q.; Liu, J.; Ding, H. Synthesis of Starch-g-p(DMDAAC) Using HRP Initiation and the Correlation of Its Structure and Sludge Dewaterability. Carbohydr. Polym. 2014, 103, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Kalra, B.; Gross, R.A. HRP-Mediated Polymerizations of Acrylamide and Sodium Acrylate. Green Chem. 2002, 4, 174–178. [Google Scholar] [CrossRef]
- Pino-Ramos, V.; Meléndez-Ortiz, I.; Ramos-Ballesteros, A.; Bucio, E. Biopolymer Grafting: Applications, 1st ed.; Thakur, V.K., Ed.; Elsevier Inc.: London, UK, 2018; pp. 205–250. [Google Scholar]
- Dao, V.H.; Cameron, N.R.; Saito, K. Synthesis, Properties and Performance of Organic Polymers Employed in Flocculation Applications. Polym. Chem. 2016, 7, 11–25. [Google Scholar] [CrossRef]
- Mishra, S.; Mukul, A.; Sen, G.; Jha, U. Microwave Assisted Synthesis of Polyacrylamide Grafted Starch (St-g-PAM) and Its Applicability as Flocculant for Water Treatment. Int. J. Biol. Macromol. 2011, 48, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Nath, L.K.; Guha, M. Microwave Assisted Synthesis and Characterization of Sago Starch-g-Acrylamide. Starch/Stärke 2011, 63, 740–745. [Google Scholar] [CrossRef]
- Singh, V.; Tiwari, A.; Pandey, S.; Singh, S.K. Peroxydisulfate Initiated Synthesis of Potato Starch-Graft-Poly(Acrylonitrile) Under Microwave Irradiation. Express Polym. Lett. 2007, 1, 51–58. [Google Scholar] [CrossRef]
- Singh, V.; Tiwari, A.; Pandey, S.; Singh, S.K. Microwave Accelerated Synthesis and Characterization of Potato Starch-g-Poly(Acrylamide). Starch/Stärke 2006, 58, 536–543. [Google Scholar] [CrossRef]
- Cummings, S.; Cunningham, M.; Dubé, M.A. The Use of Amylose-Rich Starch Nanoparticles In Emulsion Polymerization. J. Appl. Polym. Sci. 2018, 135, 46485. [Google Scholar] [CrossRef]
- Cummings, S.; Trevino, E.; Zhang, Y.; Cunningham, M.; Dubé, M.A. Incorporation of Modified Regenerated Starch Nanoparticles in Emulsion Polymer Latexes. Starch/Stärke 2018. [Google Scholar] [CrossRef]
- Pei, X.; Zhai, K.; Liang, X.; Deng, Y.; Xu, K.; Tan, Y.; Yao, X.; Wang, P. Fabrication of Shape-Tunable Macroparticles by Seeded Polymerization of Styrene Using Non-Cross-Linked Starch-Based Seed. J. Colloid Interface Sci. 2018, 512, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Rotureau, E.; Marie, E.; Dellacherie, E.; Durand, A. From Polymeric Surfactants to Colloidal Systems (3): Neutral and Anionic Polymeric Surfactants Derived from Dextran. Colloids Surf. A Physicochem. Eng. Asp. 2007, 301, 229–238. [Google Scholar] [CrossRef]
- Agama-Acevedo, E.; Bello-Perez, L.A. Starch as an Emulsions Stability: The Case of Octenyl Succinic Anhydride (OSA) Starch. Curr. Opin. Food Sci. 2017, 13, 78–83. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, G.; Wang, P.; Guo, L.; Qu, Q.; Li, J.; Wu, G. Preparation and Characterization of Starch Graft Soap-Free Styrene-Acrylate Copolymer Emulsion. Trans. China Pulp Pap. 2013, 28, 32–37. [Google Scholar]
- Ortega-Ojeda, F.E.; Larsson, H.; Eliasson, A.C. Gel for mation in Mixtures of Hydrophobically Modified Potato and High Amylopectin Potato Starch. Carbohydr. Polym. 2005, 59, 313–327. [Google Scholar] [CrossRef]
- Jonhed, A.; Järnström, L. Influence of Polymer Charge, Temperature, and Surfactants on Surface Sizing of Liner and Greaseproof Paper with Hydrophobically Modified Starch. Tappi J. 2009, 33–38. [Google Scholar]
- Cheng, L.; Guo, H.; Gu, Z.; Li, Z.; Hong, Y. Effects of Compound Emulsifiers on Properties of Wood Adhesive With High Starch Content. Int. J. Adhes. Adhes. 2017, 72, 92–97. [Google Scholar] [CrossRef]
- Jovanović, R.; Dubé, M.A. Emulsion-Based Pressure-Sensitive Adhesives: A Review. J. Macromol. Sci. Part C Polym. Rev. 2004, 44, 1–51. [Google Scholar] [CrossRef]
- Ye, F.; Miao, M.; Jiang, B.; Hamaker, B.R.; Jin, Z.; Zhang, T. Characterizations of Oil-in-Water Emulsion Stabilized by Different Hydrophobic Maize Starches. Carbohydr. Polym. 2017, 166, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiao, J.; Huang, Q. Pickering Emulsions Stabilized by Media-Milled Starch Particles. Food Res. Int. 2018, 105, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Ettelaie, R.; Holmes, M.; Chen, J.; Farshchi, A. Steric Stabilising Properties of Hydrophobically Modified Starch: Amylose vs. Amylopectin. Food Hydrocoll. 2016, 58, 364–377. [Google Scholar] [CrossRef]
- Kraak, A. Industrial Applications of Potato Starch Products. Ind. Crops Prod. 1993, 1, 107–112. [Google Scholar] [CrossRef]
- Athawale, V.D.; Lele, V. Recent Trends in Hydrogels Based on Starch-Graft-Acrylic Acid: A Review. Starch/Stärke 2001, 53, 7–13. [Google Scholar] [CrossRef]
- Guo, C.; Zhou, L.; Lv, J. Effects of Expandable Graphite and Modified Ammonium Polyphosphate on the Flame-Retardant and Mechanical Properties of Wood Flour-Polypropylene Composites. Polym. Polym. Compos. 2013, 21, 449–456. [Google Scholar] [CrossRef]
- Labet, M.; Thielemans, W.; Dufresne, A. Polymer Grafting onto Starch Nanocrystals. Biomacromolecules 2007, 8, 2916–2927. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ai, F.; Dufresne, A.; Gao, S.; Huang, J.; Chang, P.R. Structure and Mechanical Properties of Poly(Lactic Acid) Filled with (Starch Nanocrystal)-Graft-Poly(ε-Caprolactone). Macromol. Mater. Eng. 2008, 293, 763–770. [Google Scholar] [CrossRef]
- Brune, D.; Eben-Worllée, R. Von. Starch-Based Graft Polymer, Process for Its Preparation, and Use Thereof In Printing Inks and Operprint Varnishes. U.S. Patent 6,423,775, 23 July 2002. [Google Scholar]
- Baruch-Teblum, E.; Mastai, Y.; Landfester, K. Miniemulsion Polymerization of Cyclodextrin Nanospheres for Water Purification from Organic Pollutants. Eur. Polym. J. 2010, 46, 1671–1678. [Google Scholar] [CrossRef]
- Satheesh Kumar, M.N.; Siddaramaiah. Studies on Corn Starch Filled Poly (Styrene-Co-Butyl Acrylate) Latex Reinforced Polyester Nonwoven Fabric Composites. Autex Res. J. 2005, 5, 227–234. [Google Scholar]
- Liu, G.; Gu, Z.; Hong, Y.; Cheng, L.; Li, C. Trends in Food Science & Technology Structure, Functionality and Applications of Debranched Starch: A Review. Trends Food Sci. Technol. 2017, 63, 70–79. [Google Scholar]
- Alcázar-alay, S.C.; Angela, M.; Meireles, A. Physicochemical Properties, Modifications and Applications of Starches from Different Botanical Sources. Food Sci. Technol (Campinas) 2015, 35, 215–236. [Google Scholar] [CrossRef]
- Versino, F.; Lopez, O.V.; Garcia, M.A.; Zaritzky, N.E. Starch-Based Films and Food Coatings: An Overview. Starch/Stärke 2016, 1026–1037. [Google Scholar] [CrossRef]
- Li, G.; Zhu, F. Quinoa Starch: Structure, Properties, and Applications. Carbohydr. Polym. 2018, 181, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, A. Processing of Polymer Nanocomposites Reinforced With Polysaccharide Nanocrystals. Molecules 2010, 15, 4111–4128. [Google Scholar] [CrossRef] [PubMed]
- Samsudin, H.; Hani, N.M. Use of Starch in Food Packaging. Starch-Based Mater. Food Packag. Process. Charact. Appl. 2017, 2019, 229–256. [Google Scholar]
- Shevkani, K.; Singh, N.; Bajaj, R.; Kaur, A. Wheat Starch Production, Structure, Functionality and Applications-A Review. Int. J. Food Sci. Technol. 2017, 52, 38–58. [Google Scholar] [CrossRef]
- Masina, N.; Choonara, Y.E.; Kumar, P.; du Toit, L.C.; Govender, M.; Indermun, S.; Pillay, V. A Review of the Chemical Modification Techniques of Starch. Carbohydr. Polym. 2017, 157, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Bloembergen, S.; McLennan, I.J.; Lee, D.I. Use of Biobased Sugar Monomers in Vinyl Copolymers as Latex Binders and Compositions Based Thereon. U.S. Patent 9,080,290, 14 July 2015. [Google Scholar]
Figure 1.
Mechanisms of the three intervals of an emulsion polymerization.

Figure 2.
Film formation mechanism of polymer latex.

Figure 3.
Structure of starch with carbon atoms labelled.

Figure 4.
Some examples of starch modification by small molecule chemistry.

Figure 5.
Example of anionic functionalized starch.

Figure 6.
Example of cationic functionalized starch.

Figure 7.
Visualization of the “grafting to” and “grafting from” mechanisms. “Grafting to” occurs when propagating or fully reacted polymer chains bond to the starch chains. “Grafting from” describes initiator complexing or radical deprotonation of the anhydroglucose repeat unit, which creates a radical from which monomer can polymerize.
Figure 7.
Visualization of the “grafting to” and “grafting from” mechanisms. “Grafting to” occurs when propagating or fully reacted polymer chains bond to the starch chains. “Grafting from” describes initiator complexing or radical deprotonation of the anhydroglucose repeat unit, which creates a radical from which monomer can polymerize.

Figure 8.
Initiation, propagation, chain transfer and termination mechanisms from the grafting of vinyl monomer to the starch backbone. Adapted from [119].
Figure 8.
Initiation, propagation, chain transfer and termination mechanisms from the grafting of vinyl monomer to the starch backbone. Adapted from [119].

Figure 9.
Mechanisms for initiation of starch graft polymerization.

Figure 10.
Mechanism of vinyl monomer “grafting from” starch by persulfate initiation. Adapted from [15].
Figure 10.
Mechanism of vinyl monomer “grafting from” starch by persulfate initiation. Adapted from [15].

Figure 11.
Possible mechanisms of Ceric ion initiation of starch. Adapted from [188].
Figure 11.
Possible mechanisms of Ceric ion initiation of starch. Adapted from [188].

Figure 12.
Ceric ion initiation of C1 hemiacetal at reducing end of starch chain. Adapted from [187].
Figure 12.
Ceric ion initiation of C1 hemiacetal at reducing end of starch chain. Adapted from [187].

Figure 13.
Radical generation in a potassium permanganate and sulphuric acid system. Adapted from [15].
Figure 13.
Radical generation in a potassium permanganate and sulphuric acid system. Adapted from [15].

Figure 14.
Direct radical formation of manganese initiators and starch. Adapted from [15].
Figure 14.
Direct radical formation of manganese initiators and starch. Adapted from [15].

Figure 15.
Fe2+/H2O2 initiation of vinyl monomer or starch. Adapted from [15].
Figure 15.
Fe2+/H2O2 initiation of vinyl monomer or starch. Adapted from [15].

Figure 16.
Asobisisobutryonitrile (AIBN) initiation of vinyl monomer or starch. Adapted from [11].
Figure 16.
Asobisisobutryonitrile (AIBN) initiation of vinyl monomer or starch. Adapted from [11].


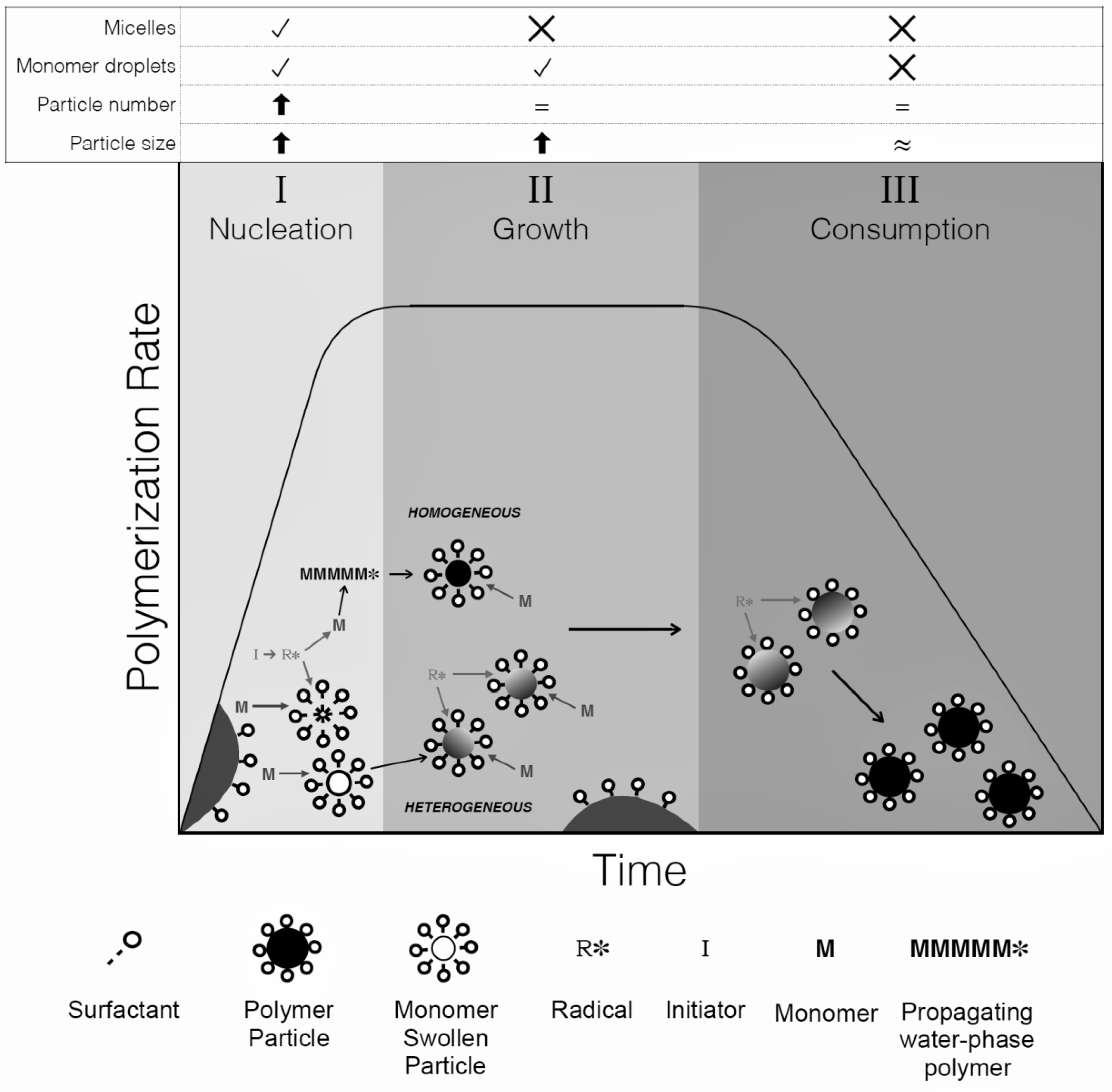{kind=link}
{kind=link}
{kind=link}
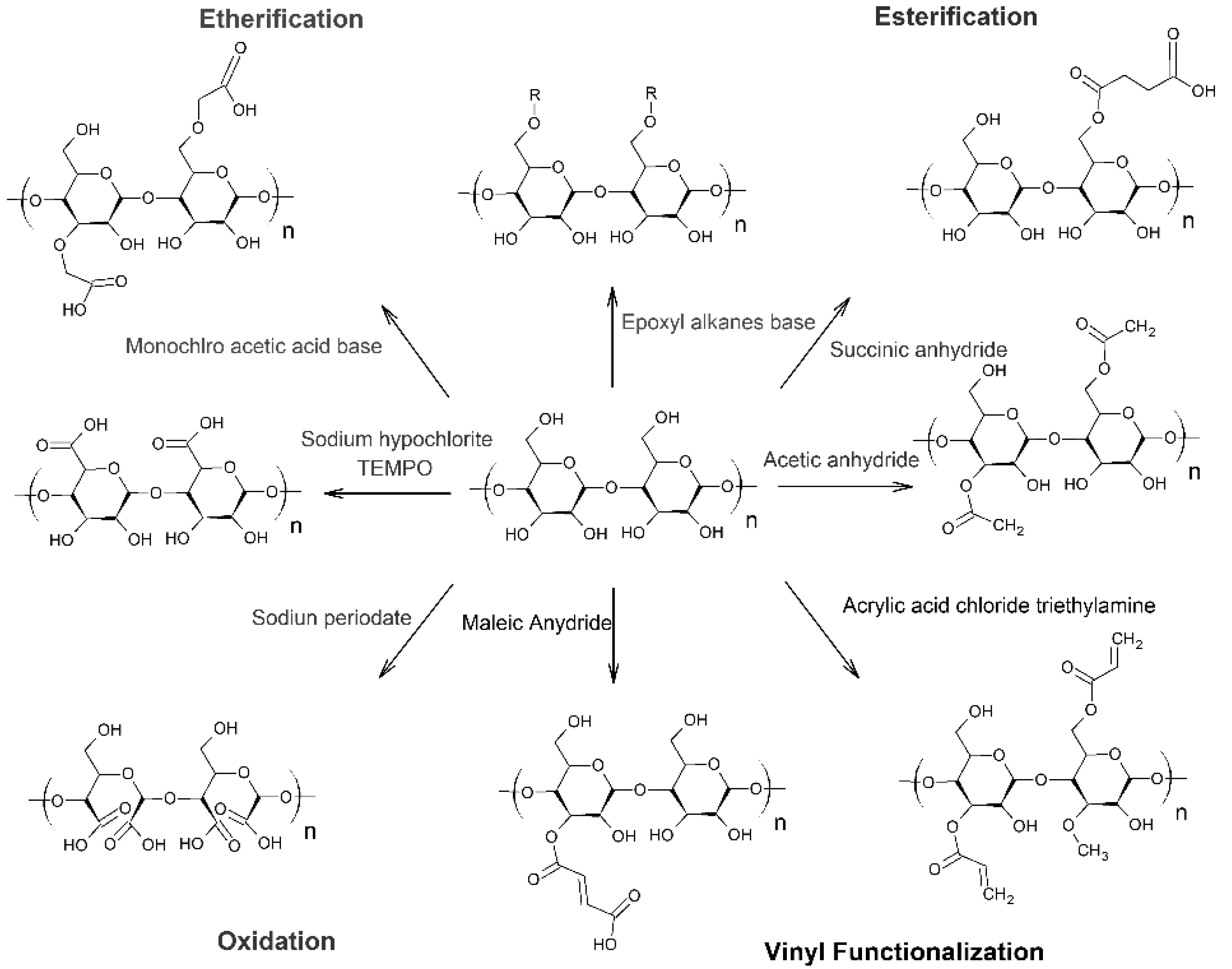{kind=link}
{kind=link}
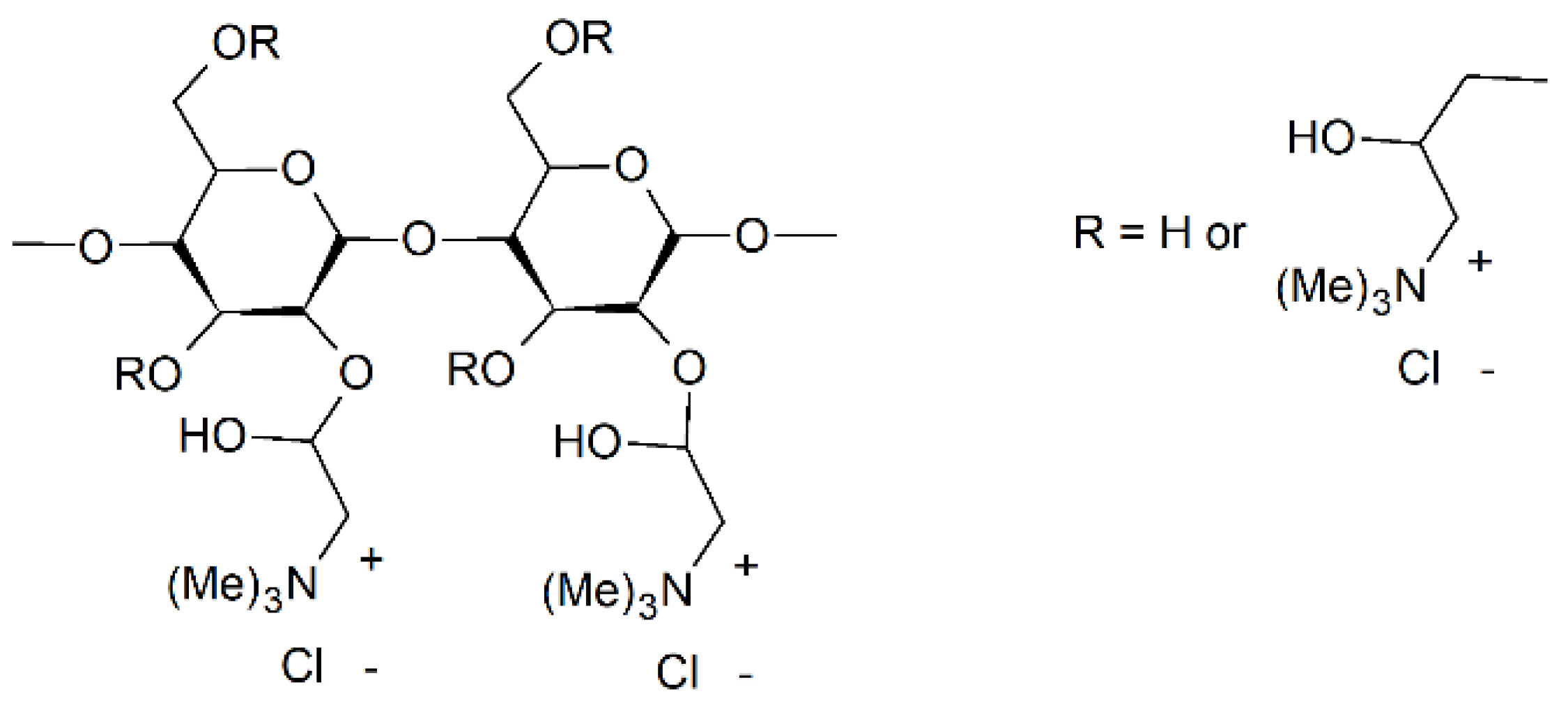{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
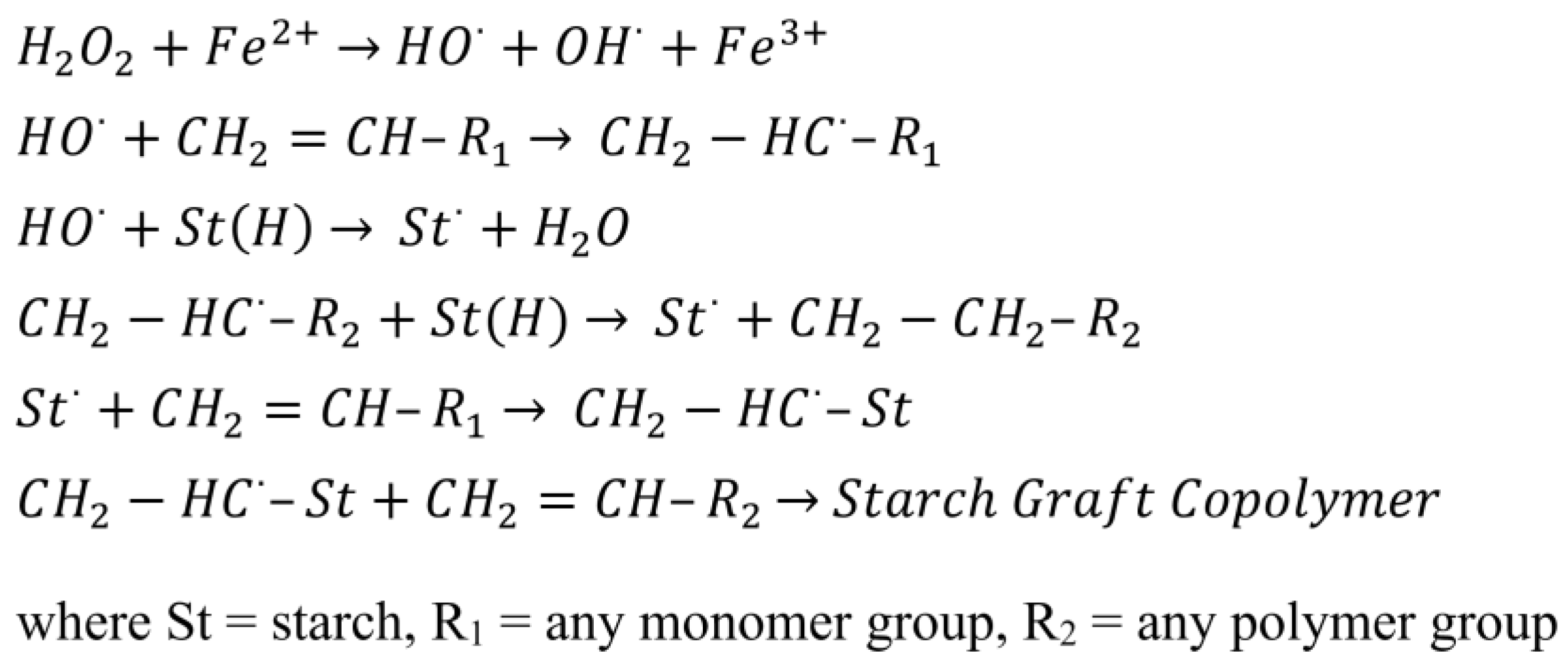{kind=link}
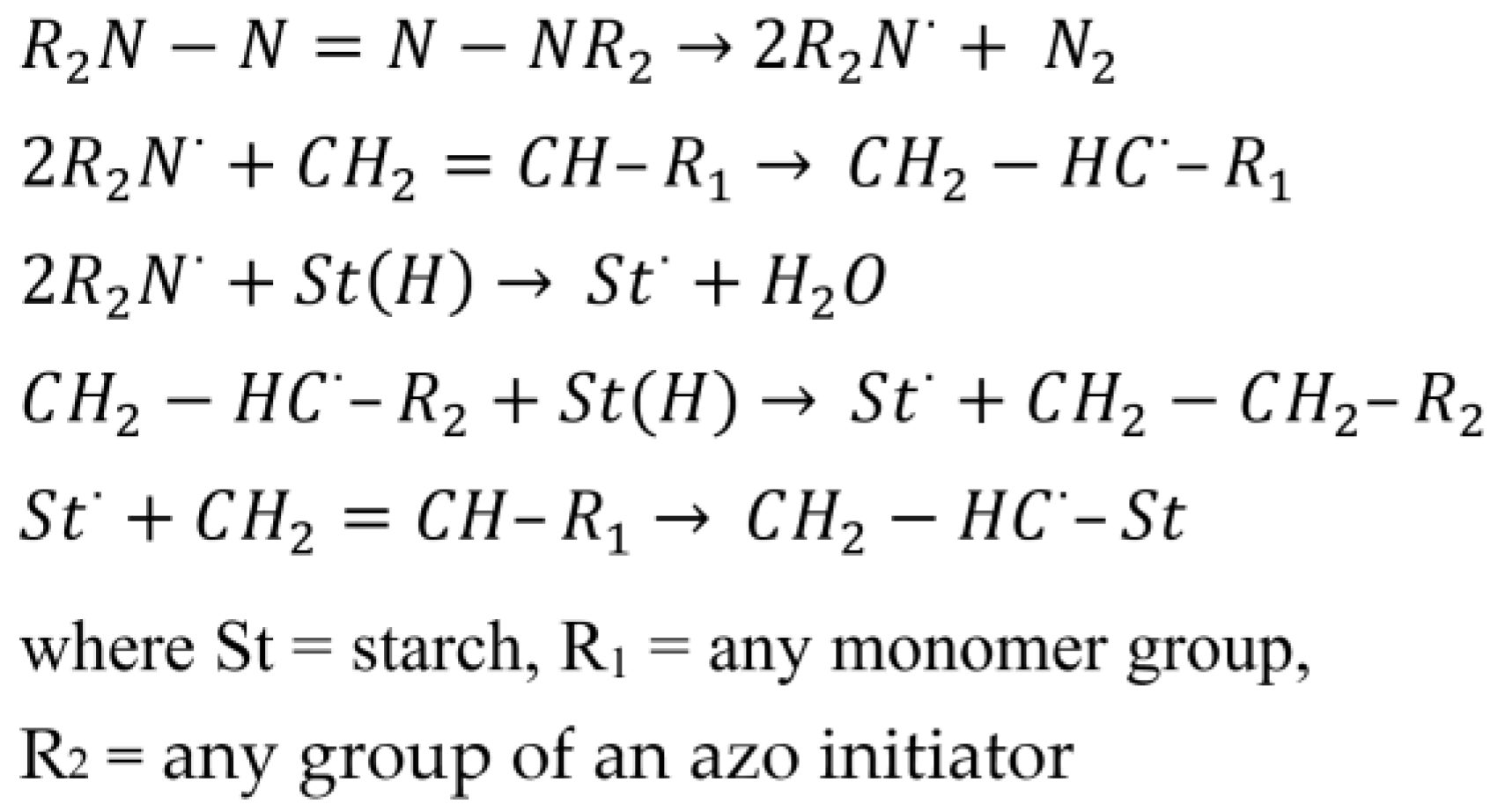{kind=link}
Table 1.
Summary of some starch physical and chemical modification pathways.
| Method | Pathways | Primary Modificationa | Product |
|---|---|---|---|
| Physical | |||
| Mechanical extrusion [67] | Heat treatment and mechanical work | Molecular weight | Amorphous pre-gelatinized starch |
| Microfluidization [68] | / | / | SNPb |
| Electric field [69] | / | Crystallinity | Amorphous pre-gelatinized starch |
| High pressure [70,71] | Hydrostatic processing | Crystallinity | Amorphous gelatinized starch |
| Supercritical medium | |||
| Homogenization | |||
| Microfluidization | |||
| Irradiation [14,69] | UV | Molecular weight | Lower molecular weight starch |
| λ | |||
| γ | |||
| Microwave | |||
| Ball milling [72] | / | Crystallinity Particle size | Amorphous partially gelatinized starch granules |
| Ultrasonication [73] | 24kHz treatment of starch aqueous solution | Crystallinity | Partially amorphous starch granules |
| Microwave [74] | Treatment of starch paste | Crystallinity | Partially amorphous starch granules |
| Thermal degradation [75] | Treatment of dried starch | Molecular weight | Lower molecular weight starch |
| Re-crystallization [76] | Hydrothermal | Crystallinity | Crystalline “resistant” starch |
| Heat-moisture treatment | SNCc | ||
| Chemical | |||
| Acid Hydrolysis [77] | Acid | Crystallinity Branching | SNC Debranched starch |
| Emulsion precipitation | Butanol complexing co-crystallization [78] | Crystallinity | SNC |
| Non-solvent precipitation crosslinking [79] | Degree of crosslinking | SNP | |
| Reactive extrusion [80] | Water-phase crosslinking | Crystallinity Molecular weight Crosslink density | SNP |
| Alkaline Treatment [72] | NaOH | Molecular weight | Low amylose content, lower molecular weight starch |
| Enzymatic | |||
| Hydrolysis [81] | Enzymatic | Crystallinity Branching | SNC Debranched starch |
| Re-crystallization [76] | Enzymatic/melt/crystallization | Crystallinity | SNC |
a Particle size is reduced in all methods. b Starch nano-particle (SNP): mostly amorphous, in some cases completely so. c Starch nano-crystal (SNC): mostly crystalline, with the amorphous regions degraded and removed to differing degrees.
Table 2.
Summary of measurements for grafting performance.
| Variable/Characteristic | Results | Extraction Methods | Analysis Methods |
|---|---|---|---|
| Grafting efficiency | wt.% of grafted polymer in total formulation | Soxhlet extraction Centrifugal solvent extraction Hydrolysis | FTIR 13C-NMR 1H-NMR Gravimetry |
| Percent Grafting (Ratio) | wt.% ratio of synthetic grafts to the starch to which they are bonded | ||
| Percent Grafting (Add-on) | wt.% of synthetic polymer in total grafted polymer | ||
| Homopolymerization ratio | wt.% of synthetic monomer that resulted only in homopolymer | ||
| Molecular weight of grafts | Weight or number average g/mol of the grafted synthetic polymer chains | Enzyme or acid hydrolysis of starch within extracted graft polymer will release the grafted synthetic chains | GPC |
Table 3.
Common characterization methods for each component of a starch graft polymer.
| Sample Type | Characteristic | Method | Typical Goal of Study |
|---|---|---|---|
| Graft polymer | Composition | Gravimetry IR/NMR | Achieve target grafting performance or create a new hybrid starch-synthetic particle. |
| Morphology | (cryo)TEM/SEM | ||
| Crystallinity | XRD | ||
| Degree of substitution (max of 3.0) | Titration Elemental analysis 1H-NMR TGA-IR | ||
| MW of grafted chains | GPC | ||
| Homopolymer | Molecular weight Degree of branching | GPC HPLC | Effect of reaction variables, formulation or procedure on kinetics of homopolymerization in the presence of starch. |
| Unreacted starch | Morphology Crystallinity Particle size/density Molecular weight | TEM/SEM XRD Flow fractionation DLS GPC | Effect of reaction variables, formulation or procedure on structure of starch in the presence of initiator and vinyl monomer. |
Table 4.
Summary of characterization methods for determination of latex properties.
| Characteristic | Analysis Method |
|---|---|
| Viscosity | Viscometry |
| Particle size | Asymmetric flow field-flow fractionation coupled with DLS |
| Birefringence | Angle dependent optical reflectometry |
| pH | Electrical potential |
| Conductivity | |
| Zeta Potential | Electrophoretic light-scattering |
| Film formation | Minimum film formation temperature (MFFT) Visual |
| Shelf life | Visual |
| Grit | Gravimetry |
Table 5.
Summary of characterization methods for determination of film properties.
| Characteristic | Analysis Method |
|---|---|
| Hydrophobicity | Contact angle |
| Adhesive Properties | Tack, peel and shear |
| Opacity | Spectrophotometry |
| Water whitening | Spectrophotometry |
| Gel content | Solvent swelling, extraction, gravimetry |
| Solubility | Solvent swelling, extraction, gravimetry |
| Tensile strength | Instron tensile strength |
| Elasticity | |
| Water/oxygen barrier | Water and oxygen permeation |
| Glass transition temperature | DSC |
| Melting temperature | |
| Thermal stability | TGA |
| Biodegradability | Enzyme, culture, composting |
Table 6.
Summary of grafting results for different initiation methods in starch graft polymerization.
Table 6.
Summary of grafting results for different initiation methods in starch graft polymerization.
| Initiator | Maximum Grafting Efficiency (%) | Maximum Percentage Graftinga (%) | Monomers |
|---|---|---|---|
| Persulfate | Solution | ||
| 64 | 30 | Acrylic acid (AA) [122] | |
| 9 | GR = 17 | Vinyl acetate (VAc) [123] | |
| 84 | AO = 60 GR = 150 | Acrylamide (AM)/itaconic acid (IA) [124] | |
| 69 | - | Methyl methacrylate (MMA) [125] | |
| 65 | - | Ethyl methacrylate (EMA) [125] | |
| 60 | - | Butyl methacrylate (BMA) [125] | |
| 79 | 84 | Butyl acrylate (BA)/VAc [126] | |
| Emulsion | |||
| 37–74 | 104–301 | BA/St [88,100,127] | |
| - | - | 2-ethylhexyl acrylate (EHA) [101,128] | |
| - | - | Methacrylic acid (MAA) [101] | |
| 68 | 49 | BA [102,128] | |
| - | - | AA [101,128] | |
| - | - | BMA [129,130,131] | |
| 59–60 | PG = 60, GR = 57 | Styrene (St) [43,132,133] | |
| - | - | MMA [134,135] | |
| - | 350 | BA/MMA/diacetone acrylamide (DAAM) [136] | |
| - | - | BA/MMA/AA/ethyl acrylate (EA) [90] | |
| 52 | - | VAc [137] | |
| - | - | EA [135] | |
| AIBN (Azobisisobutyronitrile) | Solution | ||
| - | - | Acrylonitrile (AN) [138] | |
| 5 | 24 | VAc [139] | |
| 8 | 73 | MMA [140] | |
| 57 | 27 | AA [122] | |
| Emulsion | |||
| - | - | St [141] | |
| Thiosulfate + persulfate | Solution | ||
| 99 | - | N-Methylol acrylamide (NMA) [142] | |
| - | - | BA [143] | |
| 86 | - | Methyl acrylate (MA)/NMA [142] | |
| 68 | - | MA [142] | |
| - | GR = 30 | MAA [144,145] | |
| 41 | - | VAc [137] | |
| Emulsion | |||
| - | - | - | |
| Ceric | Solution | ||
| - | GR = 42 | AN [146,147,148] | |
| 99 | 50 | Glycidyl methacrylate (GMA) [149] | |
| 69 | 10 | BA [150] | |
| 65–95 | 75–110 | MMA [151,152,153] | |
| - | - | MA [154] | |
| - | - | EA [154] | |
| - | - | MAA [154] | |
| 33 | - | N-tert-butyl acrylamide (BAM) [155] | |
| 81 | GR = 162 | AM [156,157] | |
| 37 | 20 | VAc [123] | |
| Emulsion | |||
| - | - | AN/isoprene [158] | |
| - | AO = 45 | MA [159] | |
| 65 | - | MMA [160] | |
| Manganese | Solution | ||
| 96 | AO = 45−49 | AN [21,22,161,162] | |
| 8.5−100 | GR = 120 AO = 36−253 | MMA [23,93,99,163,164,165] | |
| 59 | AO = 19 | AM [165] | |
| <9 | AO = 38 | VAc [166] | |
| - | - | AA [111] | |
| Emulsion | |||
| 30−50 | - | St/AN [167] | |
| 30−50 | - | St/EA [167] | |
| 77 | 52 | St/MMA [168] | |
| 90 | 200 | St [167,169] | |
| Ferrous | Solution | ||
| 96 | - | AN [138] | |
| 98 | AO = 54 | MMA [138] | |
| 85 | - | MA [170] | |
| 85 | AO = 20 | MAA [171] | |
| 53 | AO = 18 | DMAEMA/AM [172] | |
| 44 | 17 | AA [173] | |
| 49 | 35 | St/BA [34] | |
| 44 | 20 | St/MMA [34] | |
| Emulsion | |||
| 36 | 59 | St/BA [91] | |
| Photoinitiator | Solution | ||
| - | 260 | MMA [174] | |
| - | 190 | AN [174] | |
| - | 30 | St [174] | |
| 72 | 91−907 | AM [174,175,176] | |
| - | ~350 | AA [174] | |
| - | 420 | MAA [174] | |
| Emulsion | |||
| 13 | - | Styrene [177] | |
| 81 | - | BA [177] | |
| 30 | - | MMA [178] | |
| Irradiation | Solution | ||
| - | 135 | MA [179] | |
| >99 | 158−243 | AA [180,181] | |
| ~85 | 200 | AM [182] | |
| Emulsion | |||
| - | - | - |
a If not specified as add-on (AO) or grafting ratio (GR), values were reported in the article as “percent grafting (PG),” which usually represents the grafting ratio.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cummings, S.; Zhang, Y.; Smeets, N.; Cunningham, M.; Dubé, M.A. On the Use of Starch in Emulsion Polymerizations. Processes 2019, 7, 140. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7030140
AMA Style
Cummings S, Zhang Y, Smeets N, Cunningham M, Dubé MA. On the Use of Starch in Emulsion Polymerizations. Processes. 2019; 7(3):140. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7030140
Chicago/Turabian StyleCummings, Shidan, Yujie Zhang, Niels Smeets, Michael Cunningham, and Marc A. Dubé. 2019. "On the Use of Starch in Emulsion Polymerizations" Processes 7, no. 3: 140. https://0-doi-org.brum.beds.ac.uk/10.3390/pr7030140
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.