
SimilarityLab: Molecular Similarity for SAR Exploration and Target Prediction on the Web


School of Biological Sciences, University of Edinburgh, The King’s Buildings, Edinburgh EH9 3BF, Scotland, UK
*
Authors to whom correspondence should be addressed.
Processes 2021, 9(9), 1520; https://0-doi-org.brum.beds.ac.uk/10.3390/pr9091520
Submission received: 31 July 2021
/
Revised: 19 August 2021
/
Accepted: 25 August 2021
/
Published: 27 August 2021
(This article belongs to the Special Issue Molecular Modeling: Computer-Aided Drug Design)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Exploration of chemical space around hit, experimental, and known active compounds is an important step in the early stages of drug discovery. In academia, where access to chemical synthesis efforts is restricted in comparison to the pharma-industry, hits from primary screens are typically followed up through purchase and testing of similar compounds, before further funding is sought to begin medicinal chemistry efforts. Rapid exploration of druglike similars and structure–activity relationship profiles can be achieved through our new webservice SimilarityLab. In addition to searching for commercially available molecules similar to a query compound, SimilarityLab also enables the search of compounds with recorded activities, generating consensus counts of activities, which enables target and off-target prediction. In contrast to other online offerings utilizing the USRCAT similarity measure, SimilarityLab’s set of commercially available small molecules is consistently updated, currently containing over 12.7 million unique small molecules, and not relying on published databases which may be many years out of date. This ensures researchers have access to up-to-date chemistries and synthetic processes enabling greater diversity and access to a wider area of commercial chemical space. All source code is available in the SimilarityLab source repository.
1. Introduction
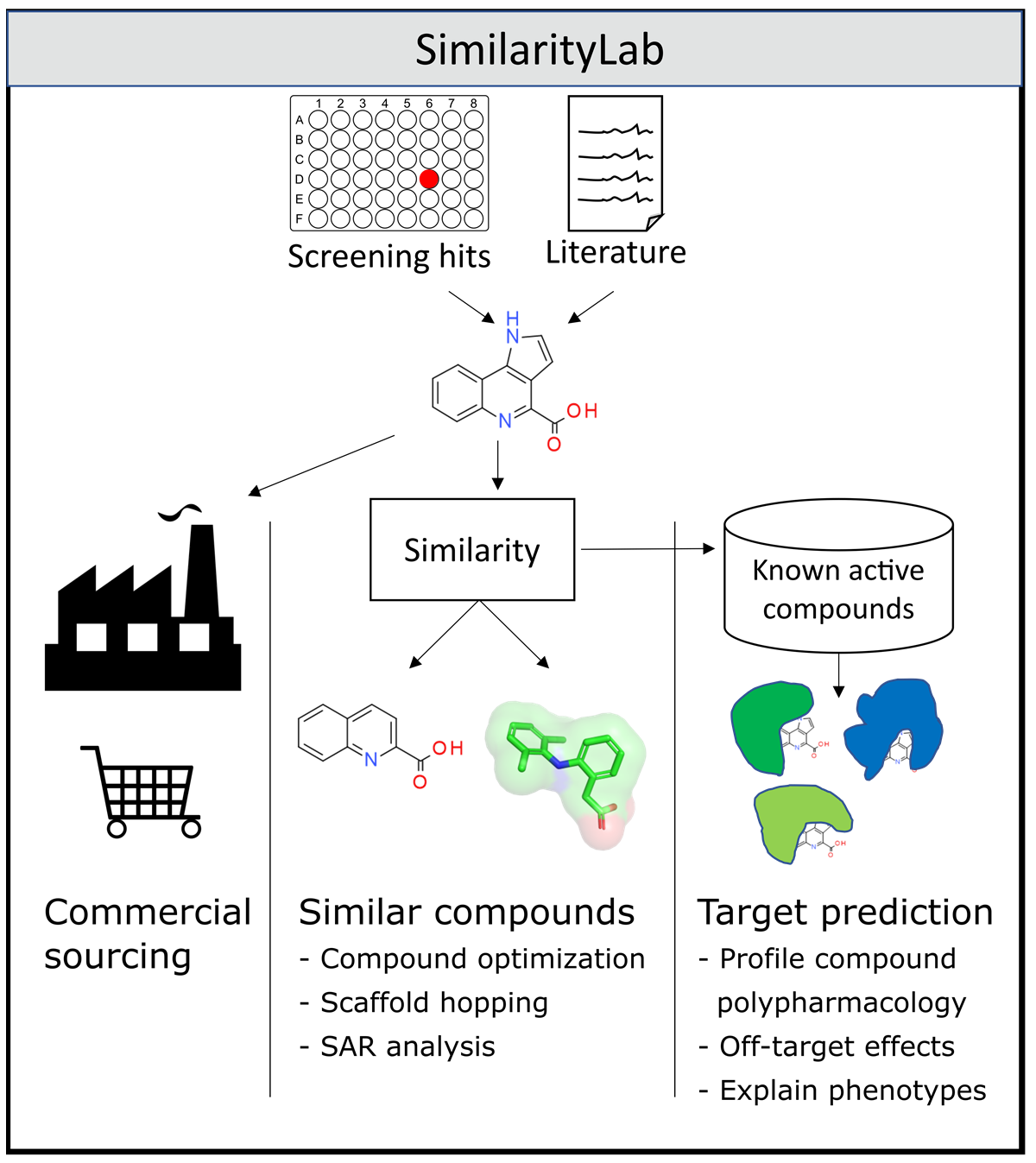
Academic groups running primary screens rely heavily on strong preliminary results to build a case for further funding to progress their drug and medicines discovery efforts. Whilst initial hits provide good starting points, some knowledge of the activity landscape can greatly help with medicinal chemistry feasibility and requirements. Activity cliffs [1,2,3,4] or areas of flat SAR (structure–activity relationships) [5,6,7] with little synthetic potential and no options for scaffold hopping [8,9] can quickly discount hits and induce failing fast and early, thereby saving time, money and effort. Such good practice contributes to avoiding the currently disastrously high attrition rates in drug discovery [10,11]. In this short communication, we wish to highlight our most recently developed web-service, SimilarityLab (https://similaritylab.bio.ed.ac.uk accessed on 5 June 2021) [12], giving all researchers in the field a quick way to source and purchase similar compounds which are mostly used to explore SAR around their own hit compounds, as well as those from the literature (see Figure 1). SimilarityLab makes extensive use of the USRCAT [13] 3D molecular similarity measure to query a local, processed version of the eMolecules database [14], currently containing over 12.7 million commercially available, unique druglike small molecules. Of crucial importance is the up-to-date nature of this commercial chemical space explorable with SimilarityLab, achieved through consistent updates of new compounds and removal of those no longer available. This is in contrast with existing online offerings such as USR-VS [15], which allows querying of a database last updated with molecules from the 2013 ZINC database [16] and an estimated commercial availability of around 50%. A similar story regards comparable tools and websites, with many utilizing out-of-date compound archives [17,18]. Integration of new molecules into SimilarityLab requires low-energy 3D conformations to be generated. This step, along with the efficient rebuilding of new updates into the commercial chemical space, is handled in a compute and data-efficient manner, greatly reducing the burden of updating the commercially available chemical space.
Alongside the main use of SimilarityLab for finding 3D similar molecules to users’ input queries, a secondary database can also be queried in a mode which enables prediction of protein targets for small molecules. We believe that the implemented approach which retrieves known active 3D similars from the ChEMBL [19] database will have an impact when integrated with phenotypic screening campaigns and used to guide target deconvolution.
2. Materials and Methods
All code generated for the SimilarityLab website and supporting codes for dataset preparation, including 3D conformer and descriptor generation, are available within the SimilarityLab source repository under an open-source license on GitHub [20].
Backend technologies used to serve SimilarityLab which currently runs on the University of Edinburgh’s Eleanor cloud service include the Python Flask web framework (version 1.1.2, Pocoo, distributed opensource project), gunicorn (version 20.1.0, distributed opensource project) and Nginx (version 1.18.0, 5F, Seattle, Washington, United States), and a backend job queue controlled by Cellery (version 5.0.5, distributed opensource project) utilizing a Redis (version 5.0.3, Redis Labs, Mountain View, California, USA) database. The RDKit [21] package (version 2020.09.1.0, distributed opensource project) is used extensively by the backend to process molecules, generate conformers and perform molecular similarity calculations. Web pages served by the backend make use of Bootstrap (version 5.0.0, distributed opensource project) for styling, Kekule.js [22] (version 0.9.3, distributed opensource project) for user entry of 2D chemical structures, SmilesDrawer [23] (version 1.2.0, distributed opensource project) for drawing molecules to HTML canvas elements. When commercially available, compound databases are updated, and the QED [24] measure of druglikeness is applied with a cut-off of less than 0.67 to remove non-druglike small molecules. These molecules then have a single low-energy conformer generated, using the protocol outlined by Ebjner [25], which is then used to generate USRCAT descriptors which are stored by the backend (see repository for code listing). The same protocol is followed when a user draws a query molecule (without the druglike filter), with a single conformer being generated as an intermediary step before descriptor generation and comparison against commercially available small molecules, whereby the top similars are returned. The number of returned similars is user-definable, allowing concise SAR exploration with 100–200 molecules or larger datasets of up to 2000 molecules to be generated for further use in docking, virtual screening and cheminformatics studies.
Target prediction is achieved using a similar approach to commercial chemical space exploration, whereby the USRCAT molecular similarity technique is applied to "active" molecules within ChEMBL [19] (version 29). Active in this sense is defined as having a recorded IC50 or KD of minimally 10 µM against protein targets. The top 100 similar active molecules then have their activities against all protein targets counted. The protein targets are then sorted by the number of times they are hit by this 100-compound similar list, and this list of targets is returned to the user as a ranked list of likely targets, along with the IDs of known active compounds for each target, which may be further explored and evaluated as to their similarity to the user’s supplied query compound.
3. Results
SimilarityLab presents a fast, user-friendly interface for fast molecular similarity calculations (See Figure 2). With an emphasis on speed and near instant results, it is envisioned that SimilarityLab will play a major role on not only research but also teaching, allowing large groups the ability to progress cheminformatics experiments, retrieving compounds which are then used as input to a variety of different tools, models and simulations.
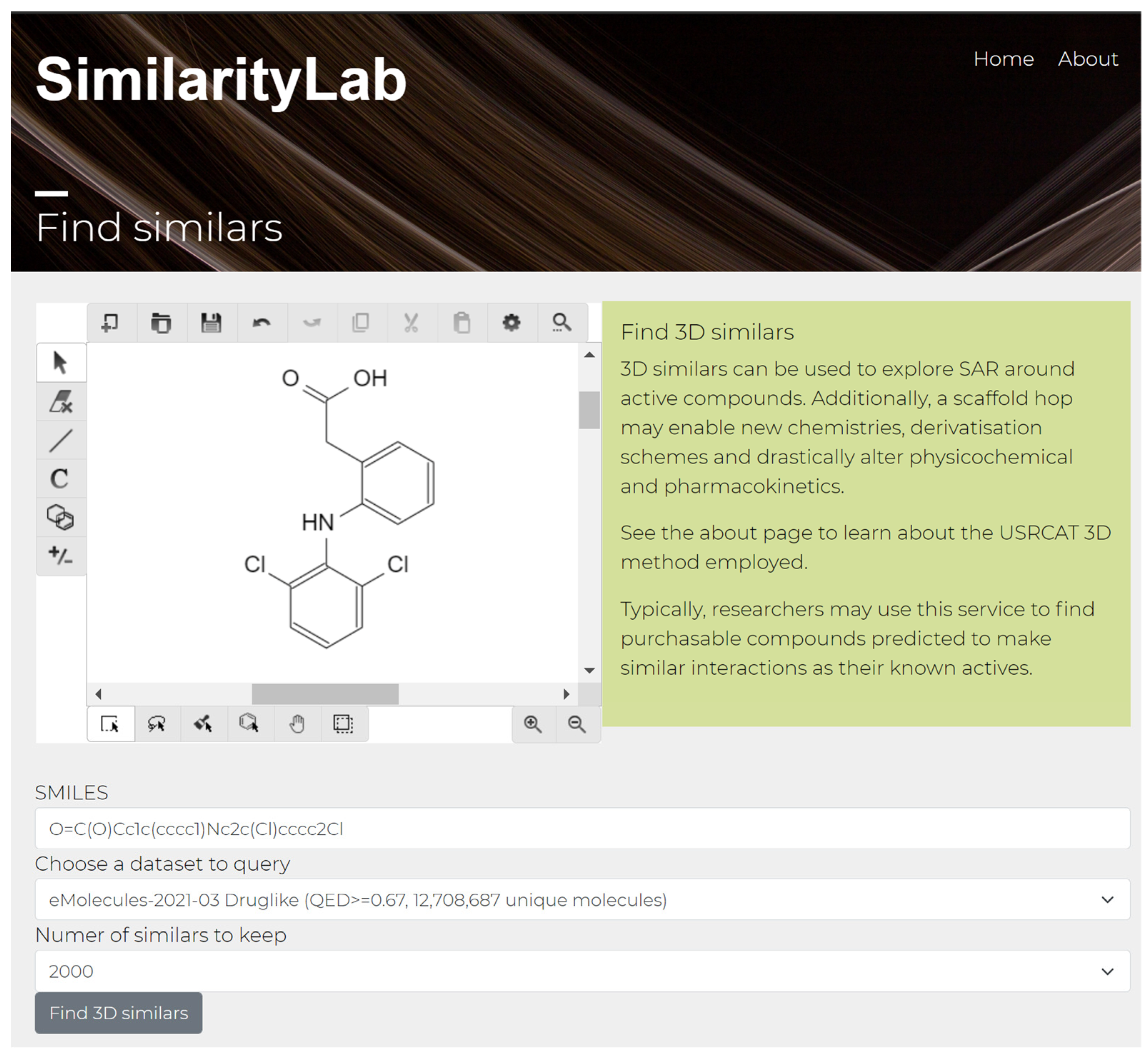
The educational applications of SimilarityLab are strengthened through an intuitive interface, allowing input of molecules using the 2D drawing capabilities of the Kekule.js editor interface, with live automatic updating of the query in the SMILES molecular format shown below (See Figure 3).
Querying for similar molecules is achieved through the “Find similars” link displayed on the landing page in Figure 2. Following this link leads to the “Find similars” page displayed in Figure 3, which allows drawing of query molecules such as diclofenac shown above using the Kekule.js drawing applet. Standard chemical file formats such as SDF are supported by the applet which translates uploaded files into 2D, before submission to the SimilarityLab backend as SMILES for 3D conformer generation using the method outlined by Ebejer [25] and molecular similarity calculations. The database of small molecules assessed against the supplied query is user-selectable, along with the number of requested top similars which are to be returned up to a limit of 2000. A similar process is used to assess the targets of diclofenac and suggest possible modes of action. From the landing page in Figure 2, the “Predict targets” link can be followed to arrive at an interface similar to that shown in Figure 3, without the ability to choose a small-molecule database. Drawing in diclofenac again to this interface and clicking predict targets takes the user to a page containing top-noted targets for close similars for diclofenac, with the two top targets being Cyclooxygenase-2 and Alpha-1a adrenergic receptor, hit by nine and eight close similars to diclofenac, respectively. This is in agreement with the literature, which documents the role of cyclooxygenase-2 in acute pain and pain relief achieved through its inhibition [26] and the role of adrenergic receptors in pain [27].
4. Discussion
SimilarityLab being publicly available represents a major resource and fills a need present for mainly academic groups in the early stages of drug discovery. Now more than ever, funding for drug discovery efforts is scarce and difficult to consistently achieve without commercial funding, carrying IP restrictions and other constraints. It is hoped that SimilarityLab will be used to capitalize on results from primary screens in academia, allowing SAR exploration by non-specialists without access to computational chemists or cheminformaticians. With SAR landscapes understood or looking promising, this strengthens further funding cases. The high rates of attrition in drug discovery point to the need for more novel and agile techniques, moving away from industry standard approaches; the ultimate solution may lay in hits identified by smaller, more specialist groups which are then independently progressed to lead status. It should also be stated here that SimilarityLab holds the potential to become a standard resource of information in basic research, particularly in the field of chemical biology and for the generation of tool compounds. Chemical molecules used as tools to study biological function are employed as standard repertoire these days to progress the fundamental understanding of biology. Researchers might ask the questions what other molecules are available to investigate their biological systems. With a quick query on the SimilarityLab website, they will obtain these required answers.
Author Contributions
Conceptualization, S.S. and M.A.; methodology, S.S.; programming, S.S.; investigation, S.S.; resources, M.A.; data curation, S.S.; writing—original draft preparation, S.S.; writing—review and editing, S.S. and M.A.; supervision, M.A.; project administration, M.A.; funding acquisition, M.A. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge financial support from the Wellcome Trust (ISSF3-SimilarityLab), the Scottish Universities Life Sciences Alliance (SULSA- http://www.sulsa.ac.uk accessed on 5 June 2021) and the Medical Research Council (MRC- www.mrc.ac.uk accessed on 5 June 2021, J54359) Strategic Grant.
Data Availability Statement
All code generated for the SimilarityLab website and supporting codes for dataset preparation, including 3D conformer and descriptor generation, are available within the SimilarityLab source repository under an open-source license, available at https://github.com/stevenshave/SimilarityLab (accessed on 5 June 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Guha, R.; van Drie, J.H. Structure−Activity landscape index: Identifying and quantifying activity cliffs. J. Chem. Inf. Modeling 2008, 48, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Stumpfe, D.; Bajorath, J.R. Exploring activity cliffs in medicinal chemistry: Miniperspective. J. Med. Chem. 2012, 55, 2932–2942. [Google Scholar] [CrossRef] [PubMed]
- Stumpfe, D.; Hu, H.; Bajorath, J.R. Evolving concept of activity cliffs. ACS Omega 2019, 4, 14360–14368. [Google Scholar] [CrossRef]
- Bajorath, J. Representation and identification of activity cliffs. Expert Opin. Drug Discov. 2017, 12, 879–883. [Google Scholar] [CrossRef]
- Esposito, E.X.; Hopfinger, A.J.; Madura, J.D. Methods for applying the quantitative structure-activity relationship paradigm. In Chemoinformatics; Springer: Berlin/Heidelberg, Germany, 2004; pp. 131–213. [Google Scholar]
- Perkins, R.; Fang, H.; Tong, W.; Wesh, W.J. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ. Toxicol. Chem. Int. J. 2003, 22, 1666–1679. [Google Scholar] [CrossRef]
- Wassermann, A.M.; Wawer, M.; Bajorath, J.R. Activity landscape representations for structure−Activity relationship analysis. J. Med. Chem. 2010, 53, 8209–8223. [Google Scholar] [CrossRef]
- Böhm, H.-J.; Flohr, A.; Stahl, M. Scaffold hopping. Drug Discov. Today Technol. 2004, 1, 217–224. [Google Scholar] [CrossRef]
- Schneider, G.; Schneider, P.; Renner, S. Scaffold-hopping: How far can you jump? Qsar Comb. Sci. 2006, 25, 1162–1171. [Google Scholar] [CrossRef]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Shih, H.-P.; Zhang, X.; Aronov, A.M. Drug discovery effectiveness from the standpoint of therapeutic mechanisms and indications. Nat. Rev. Drug Discov. 2018, 17, 19–33. [Google Scholar] [CrossRef]
- Shave, S.; Auer, M. SimilarityLab. Available online: https://similaritylab.bio.ed.ac.uk/ (accessed on 5 June 2021).
- Schreyer, A.M.; Blundell, T. USRCAT: Real-time ultrafast shape recognition with pharmacophoric constraints. J. Cheminformatics 2012, 4, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- eMolecules. 2021. Available online: https://www.emolecules.com/ (accessed on 5 June 2021).
- Li, H.; Leung, K.S.; Wong, M.H.; Ballester, P.J. USR-VS: A web server for large-scale prospective virtual screening using ultrafast shape recognition techniques. Nucleic Acids Res. 2016, 44, W436–W441. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC-A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model 2005, 45, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Shave, S.; Blackburn, E.A.; Adie, J.; Houston, D.R.; Auer, M.; Webster, S.P.; Taylor, P.; Walkinshaw, M.D. UFSRAT: Ultra-fast shape recognition with atom types–The discovery of novel bioactive small molecular scaffolds for FKBP12 and 11βHSD1. PLoS ONE 2015, 10, e0116570. [Google Scholar] [CrossRef] [PubMed]
- Hsin, K.-Y.; Morgan, H.P.; Shave, S.R.; Hinton, A.C.; Taylor, P.; Walkinshaw, M.D. EDULISS: A small-molecule database with data-mining and pharmacophore searching capabilities. Nucleic Acids Res. 2011, 39 (Suppl. S1), D1042–D1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaulton, A.; Bellis, L.J.; Bento, P.A.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Shave, S. SimilarityLab; A Website for Running Molecular Similarity and Target Prediction. Available online: https://github.com/stevenshave/SimilarityLab (accessed on 5 June 2021).
- Landrum, G. RDKit: A Software Suite for Cheminformatics, Computational Chemistry, and Predictive Modeling; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Jiang, C.; Jin, X.; Dong, Y.; Chen, M. Kekule.js: An open source javascript chemoinformatics toolkit. J. Chem. Inf. Modeling 2016, 56, 1132–1138. [Google Scholar]
- Probst, D.; Reymond, J.-L. SmilesDrawer: Parsing and drawing SMILES-encoded molecular structures using client-side JavaScript. J. Chem. Inf. Modeling 2018, 58, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebejer, J.P.; Morris, G.M.; Deane, C.M. Freely Available Conformer Generation Methods: How Good Are They? J. Chem. Inf. Modeling 2012, 52, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rodriguez, C.; Dionne, R. The role of COX-2 in acute pain and the use of selective COX-2 inhibitors for acute pain relief. Curr. Pharm. Des. 2005, 11, 1737–1755. [Google Scholar]
- Perl, E.R. Causalgia, pathological pain, and adrenergic receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 7664–7667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic workflow of SimilarityLab functionality, enabling the sourcing of hit compounds or small molecules reported in the literature, their molecular similars for SAR exploration and target prediction.
Figure 1.
Schematic workflow of SimilarityLab functionality, enabling the sourcing of hit compounds or small molecules reported in the literature, their molecular similars for SAR exploration and target prediction.

Figure 2.
Landing page of the SimilarityLab website, allowing access to further interfaces for searching commercial chemical space for molecules highly similar to an input query compound, as well as target prediction for user’s supplied queries.
Figure 2.
Landing page of the SimilarityLab website, allowing access to further interfaces for searching commercial chemical space for molecules highly similar to an input query compound, as well as target prediction for user’s supplied queries.

Figure 3.
SimilarityLab 2D drawing interface for 3D similarity searching, shown with the approved drug diclofenac being queried.
Figure 3.
SimilarityLab 2D drawing interface for 3D similarity searching, shown with the approved drug diclofenac being queried.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Shave, S.; Auer, M. SimilarityLab: Molecular Similarity for SAR Exploration and Target Prediction on the Web. Processes 2021, 9, 1520. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9091520
AMA Style
Shave S, Auer M. SimilarityLab: Molecular Similarity for SAR Exploration and Target Prediction on the Web. Processes. 2021; 9(9):1520. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9091520
Chicago/Turabian StyleShave, Steven, and Manfred Auer. 2021. "SimilarityLab: Molecular Similarity for SAR Exploration and Target Prediction on the Web" Processes 9, no. 9: 1520. https://0-doi-org.brum.beds.ac.uk/10.3390/pr9091520
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.