
Competing Endogenous RNAs (ceRNAs) and Application of Their Regulatory Networks in Complex Traits and Diseases of Ruminants

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Literature Search Strategy to Identify Studies Associated with ceRNA Networks in Ruminants
3. Differential Gene Expression Analysis and Its Role in Economically Complex Traits and Diseases
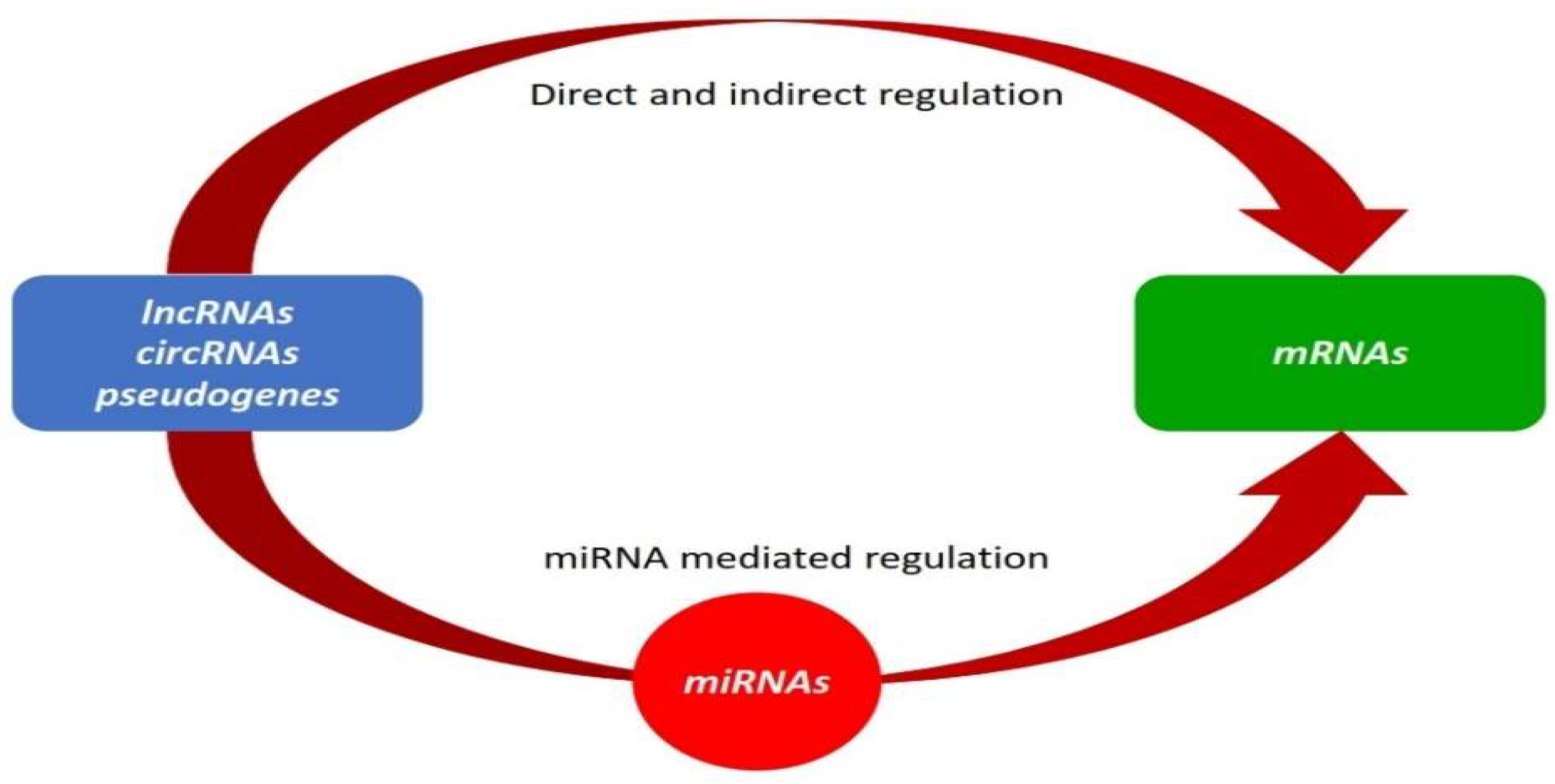
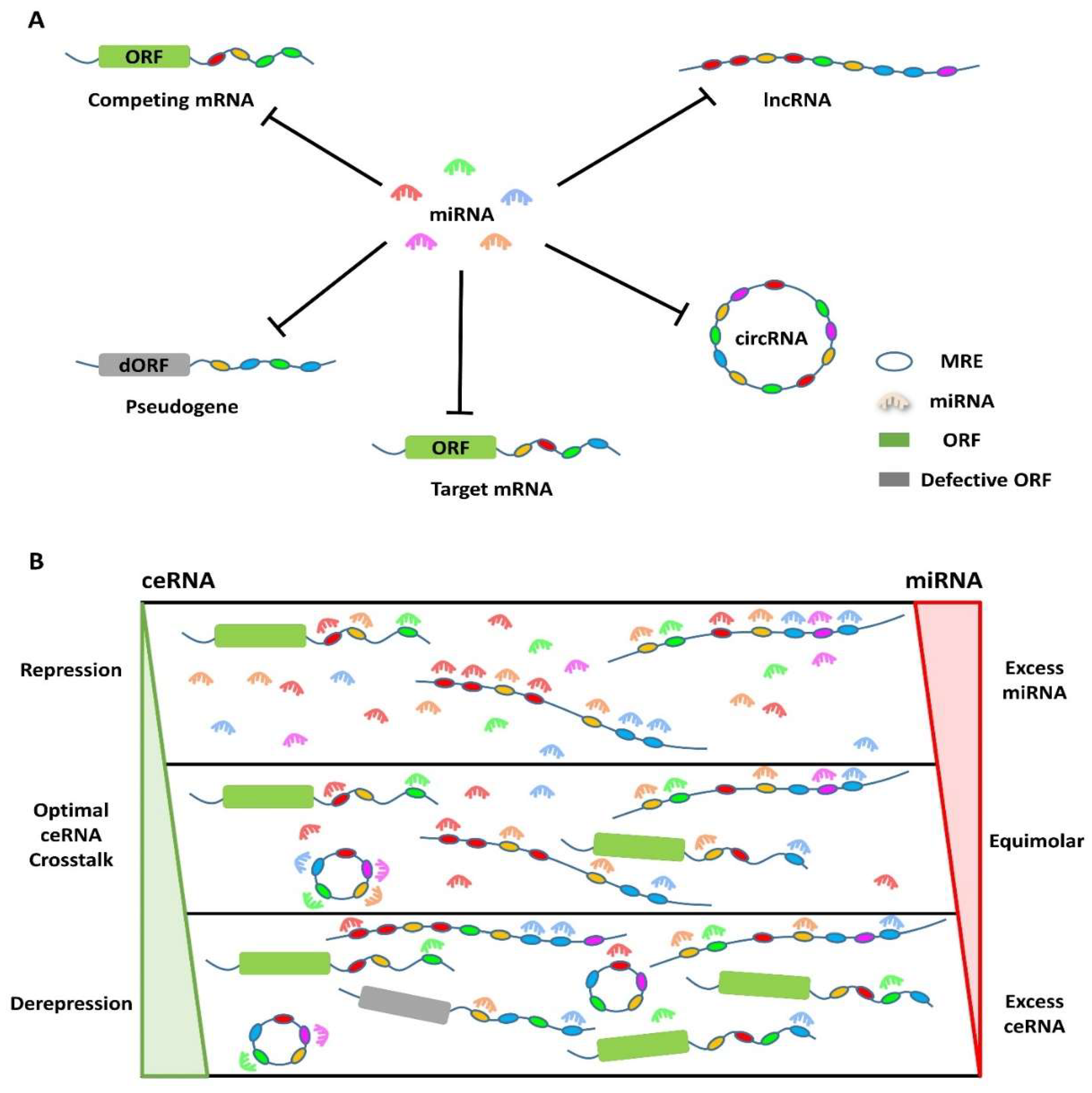
4. Competing Endogenous RNA (ceRNA) Theory
- Rates of production and turnover of miRNAs, their target RNAs, and how ceRNAs can determine how much and for how long genes are regulated. Therefore, there must be significant variations in the expression of ceRNAs to ease miRNA repression of target mRNAs.
- How the expression level of sequestered miRNAs (in very low or abundant conditions) can override competition.
- How competition among ceRNAs is affected by various factors such as the number of miRNAs they can sponge, their subcellular distribution, and their interactions with RNA-binding proteins and ribosomes. For competition to occur, ceRNAs and miRNAs must be concurrently present in the same tissue, cell type, or cell compartment.
- How the nucleotide composition of MREs on ceRNAs alters the efficiency of binding a specific miRNA.

5. Biological Networks: From PPI Networks to WGCNA and ceRNA Regulatory Networks
6. Applications of ceRNA Regulatory Network in Animal Biosciences
6.1. Dairy Cattle
6.2. Beef Cattle
6.3. Sheep
6.4. Goat
6.5. Buffalo
6.6. Camel
7. Conclusions
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brosius, J.; Raabe, C.A. What is an RNA? A top layer for RNA classification. RNA Biol. 2016, 13, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Dahariya, S.; Paddibhatla, I.; Kumar, S.; Raghuwanshi, S.; Pallepati, A.; Gutti, R.K. Long non-coding RNA: Classification, biogenesis and functions in blood cells. Mol. Immunol. 2019, 112, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.S.; Wahlestedt, C.; Kapranov, P. The landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018, 46, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Rao, M.R.S. Long noncoding RNAs in pluripotency of stem cells and cell fate specification. Adv. Exp. Med. Biol. 2017, 1008, 223–252. [Google Scholar] [PubMed]
- Flynn, R.A.; Chang, H.Y. Long noncoding RNAs in cell-fate programming and reprogramming. Cell Stem Cell 2014, 14, 752–761. [Google Scholar] [CrossRef]
- Degirmenci, U.; Lei, S. Role of lncRNAs in cellular aging. Front. Endocrinol. 2016, 7, 151. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Thakkar, N.; Chhatai, J.; Pal Bhadra, M.; Bhadra, U. Long non-coding RNA: Functional agent for disease traits. RNA Biol. 2017, 14, 522–535. [Google Scholar] [CrossRef]
- Ghafouri, F.; Sedeghi, M.; Bahrami, A. Long non-coding RNAs (LncRNAs): Roles, functions, and mechanisms. Genet. Eng. Biosaf. J. 2018, 7, 245–266. [Google Scholar]
- Tutar, Y. Pseudogenes. Int. J. Genom. 2012, 2012, 424526. [Google Scholar] [CrossRef]
- Pu, M.; Chen, J.; Tao, Z.; Miao, L.; Qi, X.; Wang, Y.; Ren, J. Regulatory network of miRNA on its target: Coordination between transcriptional and post-transcriptional regulation of gene expression. Cell Mol. Life Sci. 2019, 76, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri, F.; Bahrami, A.; Sadeghi, M.; Miraei-Ashtiani, S.R.; Bakherad, M.; Barkema, H.W.; Larose, S. Omics multi-layers networks provide novel mechanistic and functional insights into fat storage and lipid metabolism in poultry. Front. Genet. 2021, 12, 646297. [Google Scholar] [CrossRef] [PubMed]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [PubMed]
- Ala, U.; Karreth, F.A.; Bosia, C.; Pagnani, A.; Taulli, R.; Léopold, V.; Tay, Y.; Provero, P.; Zecchina, R.; Pandolfi, P.P. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc. Natl. Acad. Sci. USA 2013, 110, 7154–7159. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Yu, X.; Hu, S.; Yu, J. A brief review on the mechanisms of miRNA regulation. Genom. Proteom. Bioinform. 2009, 7, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, X.; Cao, Y.H.; Pokharel, K.; Hu, X.J.; Chen, Z.H.; Xu, S.S.; Peippo, J.; Honkatukia, M.; Kantanen, J.; et al. Comparative mRNA and miRNA expression in European mouflon (Ovis musimon) and sheep (Ovis aries) provides novel insights into the genetic mechanisms for female reproductive success. Heredity 2019, 122, 172–186. [Google Scholar] [CrossRef]
- Gao, L.; Zhao, Y.; Ma, X.; Zhang, L. Integrated analysis of lncRNA–miRNA–mRNA ceRNA network and the potential prognosis indicators in sarcomas. BMC Med. Genom. 2021, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Ala, U. Competing endogenous RNAs, non-coding RNAs and diseases: An intertwined story. Cells 2020, 9, 1574. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Liu, X.; Bai, X.; Xiao, C.; Dong, Y. Different expression of lipid metabolism-related genes in Shandong black cattle and Luxi cattle based on transcriptome analysis. Sci. Rep. 2020, 10, 21915. [Google Scholar] [CrossRef]
- Dehghanian Reyhan, V.; Ghafouri, F.; Sadeghi, M.; Miraei-Ashtiani, S.R.; Kastelic, J.P.; Barkema, H.W.; Shirali, M. Integrated Comparative Transcriptome and circRNA-lncRNA-miRNA-mRNA ceRNA Regulatory Network Analyses Identify Molecular Mechanisms Associated with Intramuscular Fat Content in Beef Cattle. Animals 2023, 13, 2598. [Google Scholar] [CrossRef]
- Zheng, Y.; Chen, J.; Yang, Y.; Wang, Q.; Yu, Q. Metagenomic and transcriptomic analyses reveal the differences and associations between the gut microbiome and muscular genes in Angus and Chinese Simmental cattle. Front. Microbiol. 2022, 13, 815915. [Google Scholar] [CrossRef]
- Wei, X.; Zhu, Y.; Zhao, X.; Zhao, Y.; Jing, Y.; Liu, G.; Wang, S.; Li, H.; Ma, Y. Transcriptome profiling of mRNAs in muscle tissue of Pinan cattle and Nanyang cattle. Gene 2022, 825, 146435. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Bai, Y.; Liu, J.; Cai, W.; Liu, L.; He, Y.; Song, J. Metabolic regulations by lncRNA, miRNA, and ceRNA under grass-fed and grain-fed regimens in Angus beef cattle. Front. Genet. 2021, 12, 579393. [Google Scholar] [CrossRef]
- Huang, C.; Ge, F.; Ma, X.; Dai, R.; Dingkao, R.; Zhaxi, Z.; Burenchao, G.; Bao, P.; Wu, X.; Guo, X.; et al. Comprehensive analysis of mRNA, lncRNA, circRNA, and miRNA expression profiles and Their ceRNA networks in the longissimus dorsi muscle of Cattle-Yak and Yak. Front. Genet. 2021, 12, 772557. [Google Scholar] [CrossRef] [PubMed]
- Yue, B.; Liu, M.; Li, M.; Chen, H. Characterization of lncRNA–miRNA–mRNA network to reveal potential functional ceRNAs in bovine skeletal muscle. Front. Genet. 2019, 10, 430773. [Google Scholar] [CrossRef]
- Wang, K.; Cheng, Y.; Guo, T.; Guo, X.; Zhang, H.; Ma, X.; Pan, Y.; Kebreab, E.; Wang, D.; Lyu, L. Analyzing the interactions of mRNAs, miRNAs and lncRNAs to predict ceRNA networks in bovine cystic follicular granulosa cells. Front. Vet. Sci. 2022, 9, 1028867. [Google Scholar] [CrossRef]
- Feng, X.; Cai, Z.; Mu, T.; Yu, B.; Wang, Y.; Ma, R.; Liu, J.; Wang, C.; Zhang, J.; Gu, Y. CircRNA screening and ceRNA network construction for milk fat metabolism in dairy cows. Front. Vet. Sci. 2022, 9, 995629. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Xia, H.; Wang, X.; Wang, Y.; Fang, J.; Li, S.; Zhai, Y.; Han, Z. Comprehensive profiling of ceRNA (circRNA-miRNA-mRNA) networks in hypothalamic-pituitary-mammary gland axis of dairy cows under heat stress. Int. J. Mol. Sci. 2023, 24, 888. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A.R.; Salazar, N.A.; Ayoola, A.O.; Memili, E.; Thomas, B.N.; Morenikeji, O.B. Regulatory network of miRNA, lncRNA, transcription factor and target immune response genes in bovine mastitis. Sci. Rep. 2021, 11, 21899. [Google Scholar] [CrossRef]
- Liang, R.; Han, B.; Li, Q.; Yuan, Y.; Li, J.; Sun, D. Using RNA sequencing to identify putative competing endogenous RNAs (ceRNAs) potentially regulating fat metabolism in bovine liver. Sci. Rep. 2017, 7, 6396. [Google Scholar] [CrossRef]
- Mu, T.; Hu, H.; Feng, X.; Ma, Y.; Wang, Y.; Zhang, J.; Gu, Y. Screening and conjoint analysis of key lncRNAs for milk fat metabolism in dairy cows. Front. Genet. 2022, 13, 772115. [Google Scholar] [CrossRef] [PubMed]
- Yousuf, S.; Malik, W.A.; Feng, H.; Liu, T.; Xie, L.; Miao, X. Genome wide identification and characterization of fertility associated novel CircRNAs as ceRNA reveal their regulatory roles in sheep fecundity. J. Ovarian Res. 2023, 16, 115. [Google Scholar] [CrossRef] [PubMed]
- Cui, R.; Kang, X.; Liu, Y.; Liu, X.; Chan, S.; Wang, Y.; Li, Z.; Ling, Y.; Feng, D.; Li, M.; et al. Integrated analysis of the whole transcriptome of skeletal muscle reveals the ceRNA regulatory network related to the formation of muscle fibers in Tan sheep. Front. Genet. 2022, 13, 991606. [Google Scholar] [CrossRef]
- Sadeghi, M.; Bahrami, A.; Hasankhani, A.; Kioumarsi, H.; Nouralizadeh, R.; Abdulkareem, S.A.; Ghafouri, F.; Barkema, H.W. lncRNA–miRNA–mRNA ceRNA Network Involved in Sheep Prolificacy: An Integrated Approach. Genes 2022, 13, 1295. [Google Scholar] [CrossRef]
- Bao, G.; Zhao, F.; Wang, J.; Liu, X.; Hu, J.; Shi, B.; Wen, Y.; Zhao, L.; Luo, Y.; Li, S. Characterization of the circRNA–miRNA–mRNA network to reveal the potential functional ceRNAs associated with dynamic changes in the meat quality of the longissimus thoracis muscle in Tibetan sheep at different growth stages. Front. Vet. Sci. 2022, 9, 803758. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Wang, Y.; He, J. Transcriptomic analysis reveals the involvement of lncRNA–miRNA–mRNA networks in hair follicle induction in Aohan fine wool sheep skin. Front. Genet. 2020, 11, 533225. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Liu, Y.; He, X.; Fang, M.; Chu, M. Uterus proliferative period ceRNA network of Yunshang black goat reveals candidate genes on different kidding number trait. Front. Endocrinol. 2023, 14, 1165409. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Wang, J.; Mao, M.; Zhao, X.; Li, Q.; Xuan, R.; Li, F.; Chao, T. Analyses of lncRNAs, circRNAs, and the Interactions between ncRNAs and mRNAs in Goat Submandibular Glands Reveal Their Potential Function in Immune Regulation. Genes 2023, 14, 187. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri, F.; Sadeghi, M.; Bahrami, A.; Naserkheil, M.; Reyhan, V.D.; Javanmard, A.; Miraei-Ashtiani, S.R.; Ghahremani, S.; Barkema, H.W.; Abdollahi-Arpanahi, R.; et al. Construction of a circRNA–lincRNA–lncRNA–miRNA–mRNA ceRNA regulatory network identifies genes and pathways linked to goat fertility. Front. Genet. 2023, 14, 1195480. [Google Scholar] [CrossRef]
- Shang, F.; Ma, R.; Rong, Y.; Pan, J.; Wang, M.; Niu, S.; Qi, Y.; Li, Y.; Wang, Z.; Lv, Q.; et al. Construction and functional analysis of ceRNA regulatory network related to the development of secondary hair follicles in Inner Mongolia cashmere goats. Front. Vet. Sci. 2022, 9, 959952. [Google Scholar] [CrossRef]
- Jin, M.; Fan, W.; Lyu, S.; Cong, L.; Xue, T. Screening and bioinformatics analysis of a potential ceRNA network in melatonin-induced cashmere growth in Liaoning cashmere goats. Arch. Anim. Breed. 2024, 67, 97–109. [Google Scholar] [CrossRef]
- Pan, Y.; Yang, S.; Cheng, J.; Lv, Q.; Xing, Q.; Zhang, R.; Liang, J.; Shi, D.; Deng, Y. Whole-Transcriptome Analysis of LncRNAs Mediated ceRNA Regulation in Granulosa Cells Isolated from Healthy and Atresia Follicles of Chinese Buffalo. Front. Vet. Sci. 2021, 8, 680182. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Feng, T.; Qian, Q.; Liu, Q. Comparison of long non-coding RNA expression profiles of cattle and buffalo differing in muscle characteristics. Front. Genet. 2020, 11, 492141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Pan, J.; Zhou, H.; Cao, Y. Evidence from ileum and liver transcriptomes of resistance to high-salt and water-deprivation conditions in camel. Zool. Lett. 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Cassar-Malek, I.; Picard, B.; Bernard, C.; Hocquette, J.F. Application of gene expression studies in livestock production systems: A European perspective. Aust. J. Exp. Agric. 2008, 48, 701–710. [Google Scholar] [CrossRef]
- Cesar, A.S.; Regitano, L.C.; Reecy, J.M.; Poleti, M.D.; Oliveira, P.S.; de Oliveira, G.B.; Moreira, G.C.; Mudadu, M.A.; Tizioto, P.C.; Koltes, J.E.; et al. Identification of putative regulatory regions and transcription factors associated with intramuscular fat content traits. BMC Genom. 2018, 19, 1–20. [Google Scholar] [CrossRef]
- Loging, W.; Harland, L.; Williams-Jones, B. High-throughput electronic biology: Mining information for drug discovery. Nat. Rev. Drug Discov. 2007, 6, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.J.; Gaulton, A.; Marshall, J.; Bichko, D.; Martin, S.; Brouwer, C.; Harland, L. Visualizing the drug target landscape. Drug Disco. Today 2010, 15, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Reyhan, V.D.; Sadeghi, M.; Miraei-Ashtiani, S.R.; Ghafouri, F.; Kastelic, J.P.; Barkema, H.W. Integrated transcriptome and regulatory network analyses identify candidate genes and pathways modulating ewe fertility. Gene Rep. 2022, 28, 101659. [Google Scholar] [CrossRef]
- Jansen, R.C.; Nap, J.P. Genetical genomics: The added value from segregation. Trends Genet. 2001, 17, 388–391. [Google Scholar] [CrossRef]
- Fairfax, B.P.; Knight, J.C. Genetics of gene expression in immunity to infection. Curr. Opin. Immunol. 2014, 30, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Cloney, R. Integrating gene variation and expression to understand complex traits. Nat. Rev. Genet. 2016, 17, 194. [Google Scholar] [CrossRef] [PubMed]
- Essa, B.; Al-Sharif, M.; Abdo, M.; Fericean, L.; Ateya, A. New insights on nucleotide sequence variants and mRNA levels of candidate genes assessing resistance/susceptibility to mastitis in Holstein and montbéliarde dairy cows. Vet. Sci. 2023, 10, 35. [Google Scholar] [CrossRef]
- Rau, A.; Marot, G.; Jaffrézic, F. Differential meta-analysis of RNA-seq data from multiple studies. BMC Bioinform. 2014, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Keel, B.N.; Lindholm-Perry, A.K. Recent developments and future directions in meta-analysis of differential gene expression in livestock RNA-Seq. Front. Genet. 2022, 13, 983043. [Google Scholar] [CrossRef] [PubMed]
- Lindholm-Perry, A.K.; Meyer, A.M.; Kern-Lunbery, R.J.; Cunningham-Hollinger, H.C.; Funk, T.H.; Keel, B.N. Genes involved in feed efficiency identified in a meta-analysis of rumen tissue from two populations of beef steers. Animals 2022, 12, 1514. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri, F.; Mehrabani Yeganeh, H.; Mohamadian Jeshvaghani, S. Big data and the role of high-throughput technologies in livestock and poultry breeding. Prof. J. Domest. 2020, 20, 34–40. [Google Scholar]
- Ghafouri, F.; Alipour, S.; Mohamadian Jeshvaghani, S. Application of machine learning approach and its subset algorithms in estimating genomic breeding values. Prof. J. Domest. 2020, 20, 19–29. [Google Scholar]
- Denzler, R.; Agarwal, V.; Stefano, J.; Bartel, D.P.; Stoffel, M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol. Cell 2014, 54, 766–776. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Sumazin, P.; Yang, X.; Chiu, H.S.; Chung, W.J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 2011, 147, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.S.; Martínez, M.R.; Bansal, M.; Subramanian, A.; Golub, T.R.; Yang, X.; Sumazin, P.; Califano, A. High-throughput validation of ceRNA regulatory networks. BMC Genom. 2017, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Cao, F.; Gu, X.; Chen, J.; Xu, R.; Huang, Y.; Ying, L. LncRNA XIST, as a ceRNA of miR-204, aggravates lipopolysaccharide-induced acute respiratory distress syndrome in mice by upregulating IRF2. Int. J. Clin. Exp. Pathol. 2019, 12, 2425. [Google Scholar] [PubMed]
- Arvey, A.; Larsson, E.; Sander, C.; Leslie, C.S.; Marks, D.S. Target mRNA abundance dilutes microRNA and siRNA activity. Mol. Syst. Biol. 2010, 6, 363. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Zhang, D.H.; Wu, N.; Xiao, J.H.; Wang, X.; Ma, W. ceRNA in cancer: Possible functions and clinical implications. J. Med. Genet. 2015, 52, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Wahlstedt, H.; Daniel, C.; Ensterö, M.; Öhman, M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009, 19, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Zipeto, M.A.; Jiang, Q.; Melese, E.; Jamieson, C.H. RNA rewriting, recoding, and rewiring in human disease. Trends Mol. Med. 2015, 21, 549–559. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E.Y. A-to-I RNA editing—Immune protector and transcriptome diversifier. Nat. Rev. Genet. 2018, 19, 473–490. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, X.; Jin, Y. ADAR-mediated RNA editing in non-coding RNA sequences. Sci. China Life Sci. 2013, 56, 944–952. [Google Scholar] [CrossRef]
- Hwang, T.; Park, C.K.; Leung, A.K.; Gao, Y.; Hyde, T.M.; Kleinman, J.E.; Rajpurohit, A.; Tao, R.; Shin, J.H.; Weinberger, D.R. Dynamic regulation of RNA editing in human brain development and disease. Nat. Neurosci. 2016, 19, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Lagergren, J.; Öhman, M. RNA editing of non-coding RNA and its role in gene regulation. Biochimie 2015, 117, 22–27. [Google Scholar] [CrossRef]
- Shevchenko, G.; Morris, K.V. All I’s on the RADAR: Role of ADAR in gene regulation. FEBS Lett. 2018, 592, 2860–2873. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Cui, C.; Liu, J.; Luo, Y.; Quan, F.; Jin, Y.; Zhang, Y. The growth and reproduction performance of TALEN-mediated β-lactoglobulin-knockout bucks. Transgenic Res. 2016, 25, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Wang, Y.; Zhang, Y.; Yang, M.; Lv, J.; Liu, J.; Zhang, Y. TALE nickase-mediated SP110 knockin endows cattle with increased resistance to tuberculosis. Proc. Natl. Acad. Sci. USA 2015, 112, 1530–1539. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, L.; Ren, G.; Li, Z.; Ren, C.; Zhang, T.; Xu, K.; Zhang, Z. Targeted disruption of the sheep MSTN gene by engineered zinc-finger nucleases. Mol. Biol. Rep. 2014, 41, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Song, Y.; Liu, J.; Ge, H.; Li, Q.; Huang, H.; Hu, L.; Zhu, H.; Jin, Y.; Zhang, Y. Gene targeting by TALEN-induced homologous recombination in goats directs production of β-lactoglobulin-free, high-human lactoferrin milk. Sci. Rep. 2015, 5, 10482. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Guo, W.; Chang, B.; Liu, J.; Guo, Z.; Quan, F.; Zhang, Y. Zinc-finger nickase-mediated insertion of the lysostaphin gene into the beta-casein locus in cloned cows. Nat. Commun. 2013, 4, 2565. [Google Scholar] [CrossRef] [PubMed]
- Jenko, J.; Gorjanc, G.; Cleveland, M.A.; Varshney, R.K.; Whitelaw, C.B.A.; Woolliams, J.A.; Hickey, J.M. Potential of promotion of alleles by genome editing to improve quantitative traits in livestock breeding programs. Genet. Sel. Evol. 2015, 47, 1–14. [Google Scholar]
- Wang, Y.; Hou, J.; He, D.; Sun, M.; Zhang, P.; Yu, Y.; Chen, Y. The emerging function and mechanism of ceRNAs in cancer. Trends Genet. 2016, 32, 211–224. [Google Scholar] [CrossRef]
- Chan, J.J.; Tay, Y. Noncoding RNA: RNA regulatory networks in cancer. Int. J. Mol. Sci. 2018, 19, 1310. [Google Scholar] [CrossRef]
- Dehghanian Reyhan, V.; Sadeghi, M.; Ghafouri, F. Method of weighted gene co-expression network analysis and its application in animal and poultry breeding and genetics. Prof. J. Domest. 2022, 22, 5–13. [Google Scholar]
- Walhout, M.; Vidal, M.; Dekker, J. (Eds) Handbook of Systems Biology: Concepts and Insights; Academic Press: New York, NY, USA, 2012. [Google Scholar]
- Zhao, X.; Wang, C.; Wang, Y.; Zhou, L.; Hu, H.; Bai, L.; Wang, J. Weighted gene co-expression network analysis reveals potential candidate genes affecting drip loss in pork. Anim. Genet. 2020, 51, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Li, Q.; Wu, Y.; Zhang, Y.; Zhang, B.; Zhang, H. Identification of candidate genes that specifically regulate subcutaneous and intramuscular fat deposition using transcriptomic and proteomic profiles in Dingyuan pigs. Sci. Rep. 2022, 12, 2844. [Google Scholar] [CrossRef]
- Ovens, K.; Eames, B.F.; McQuillan, I. Comparative analyses of gene co-expression networks: Implementations and applications in the study of evolution. Front. Genet. 2021, 12, 695399. [Google Scholar] [CrossRef] [PubMed]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, Y.; Xin, S.; Yang, L.; Jiang, M.; Xin, Y.; Wang, Y.; Yang, J.; Lu, J. Insight into LncRNA-and CircRNA-Mediated CeRNAs: Regulatory Network and Implications in Nasopharyngeal Carcinoma—A Narrative Literature Review. Cancers 2022, 14, 4564. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Qiu, X.; He, Q.; Fu, X.; Ji, F.; Tian, X. CCND1-associated ceRNA network reveal the critical pathway of TPRG1-AS1-hsa-miR-363-3p-MYO1B as a prognostic marker for head and neck squamous cell carcinoma. Sci. Rep. 2023, 13, 11831. [Google Scholar] [CrossRef]
- Ma, S.C.; Li, Q.; Peng, J.Y.; Zhouwen, J.L.; Zhang, D.N.; Zhang, C.B.; Jiang, W.G.; Jia, W. CLDN5 affects lncRNAs acting as ceRNA dynamics contributing to regulating blood-brain barrier permeability in tumor brain metastasis. Oncol. Rep. 2018, 39, 1441–1453. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, B.; Huang, J.; Li, W.; Yi, P.; Yi, M.; Peng, W. Identification of the protective effect of Polygonatum sibiricum polysaccharide on d-galactose-induced brain ageing in mice by the systematic characterization of a circular RNA-associated ceRNA network. Pharm. Biol. 2021, 59, 345–364. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, L.; Yang, H.; Chen, T.; Wu, X.; Lv, K. Analyzing the lncRNA, miRNA, and mRNA-associated ceRNA networks to reveal potential prognostic biomarkers for glioblastoma multiforme. Cancer Cell Int. 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.R.; Zhang, Y.B.; Zheng, S.F.; Xu, Y.W.; Lin, P.; Shang-Guan, H.C.; Lin, Y.X.; Kang, D.Z.; Yao, P.S. Decreased SPTBN2 expression regulated by the ceRNA network is associated with poor prognosis and immune infiltration in low-grade glioma. Exp. Ther. Med. 2023, 25, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Wang, X.; Li, W.; Liu, J.; Fan, Y.; Zhang, H.; Yang, J.; Gao, Y.; Liu, Y. Identification and characterization of lncRNAs expression profile related to goat skeletal muscle at different development stages. Animals 2022, 12, 2683. [Google Scholar] [CrossRef] [PubMed]
- Boichard, D. Estimation of the economic value of conception rate in dairy cattle. Livest. Prod. Sci. 1990, 24, 187–204. [Google Scholar] [CrossRef]
- Dekkers, J.C.M. Estimation of economic values for dairy cattle breeding goals: Bias due to sub-optimal management policies. Livest. Prod. Sci. 1991, 29, 131–149. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| References | Phenotype | Animal | Breed | Country |
|---|---|---|---|---|
| [19,20] | Intramuscular fat (IMF) | Beef cattle | Shandong Black and Luxi | China |
| [20,21] | Intramuscular fat (IMF) | Beef cattle | Angus and Chinese Simmental | China |
| [20,22] | Intramuscular fat (IMF) | Beef cattle | Nanyang | China |
| [23] | Meat quality, growth rate, greenhouse emission, and animal welfare | Beef cattle | Angus | USA |
| [24] | Growth traits | Beef cattle | Cattle-yaks (Aberdeen Angus ♂ × Yak ♀) and Ashidan yaks | China |
| [25] | Skeletal muscle development | Beef cattle | Qinchuan | China |
| [26] | Fertility (ovarian cyst) | Dairy cattle | Hereford | China |
| [27] | Milk fat metabolism | Dairy cattle | Holstein | China |
| [28] | Hypothalamic–pituitarymammary gland (HPM) axis performance under heat stress (HS) | Dairy cattle | Holstein | China |
| [29] | Mastitis | Dairy cattle | - | - |
| [30] | Milk fat | Dairy cattle | Chinese Holstein | China |
| [31] | Milk fat | Dairy cattle | Holstein | China |
| [32] | Fertility (fecundity) | Sheep | Small Tail Han sheep and Dolang Sheep | China |
| [33] | Growth (development of muscle fibers) | Sheep | Tan and Dorper | China |
| [34] | Fertility | Sheep | Baluch and Romanov | Iran |
| [35] | Meat quality | Sheep | Tibetan | China |
| [36] | Wool diameter | Sheep | Aohan | China |
| [37] | Fertility (goat kidding numbers) | Goat | Yunshang black goat | China |
| [38] | Immunity | Goat | - | China |
| [39] | Fertility | Goat | Ji’ning Gray | China |
| [40] | Development of secondary hair follicles | Goat | Mongolia Cashmere goat | Mongolia |
| [41] | Cashmere growth | Goat | Liaoning Cashmere goat | China |
| [42] | Fertility (healthy and Atresia follicle) | Buffalo | Chinese Buffalo | China |
| [43] | Muscle characteristics | Buffalo | Chinese swamp buffalo and Guangxi native cattle | China |
| [44] | Resistance to high-salt and water-deprivation conditions | Camel | Alxa Bactrian | China |
| Networks | Molecule Types | Regulatory Mechanism | Level of Regulation | Capture Non-Coding RNAs | Reveal Novel Interactions | Functional Insights |
|---|---|---|---|---|---|---|
| ceRNA | mRNAs, lncRNAs, circRNAs | miRNA-mediated competition | RNA | Yes | Yes | RNA-level regulation |
| PPI | Proteins | Protein–protein interactions | Protein | No | Limited to proteins | Protein functions |
| WGC | mRNAs | Co-expression patterns | mRNA | Limited | Limited to co-expression | Gene co-expression modules |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghafouri, F.; Dehghanian Reyhan, V.; Sadeghi, M.; Miraei-Ashtiani, S.R.; Kastelic, J.P.; Barkema, H.W.; Shirali, M. Competing Endogenous RNAs (ceRNAs) and Application of Their Regulatory Networks in Complex Traits and Diseases of Ruminants. Ruminants 2024, 4, 165-181. https://0-doi-org.brum.beds.ac.uk/10.3390/ruminants4020011
Ghafouri F, Dehghanian Reyhan V, Sadeghi M, Miraei-Ashtiani SR, Kastelic JP, Barkema HW, Shirali M. Competing Endogenous RNAs (ceRNAs) and Application of Their Regulatory Networks in Complex Traits and Diseases of Ruminants. Ruminants. 2024; 4(2):165-181. https://0-doi-org.brum.beds.ac.uk/10.3390/ruminants4020011
Chicago/Turabian StyleGhafouri, Farzad, Vahid Dehghanian Reyhan, Mostafa Sadeghi, Seyed Reza Miraei-Ashtiani, John P. Kastelic, Herman W. Barkema, and Masoud Shirali. 2024. "Competing Endogenous RNAs (ceRNAs) and Application of Their Regulatory Networks in Complex Traits and Diseases of Ruminants" Ruminants 4, no. 2: 165-181. https://0-doi-org.brum.beds.ac.uk/10.3390/ruminants4020011