In-Cell Determination of Lactate Dehydrogenase Activity in a Luminal Breast Cancer Model – ex vivo Investigation of Excised Xenograft Tumor Slices Using dDNP Hyperpolarized [1-13C]pyruvate

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals
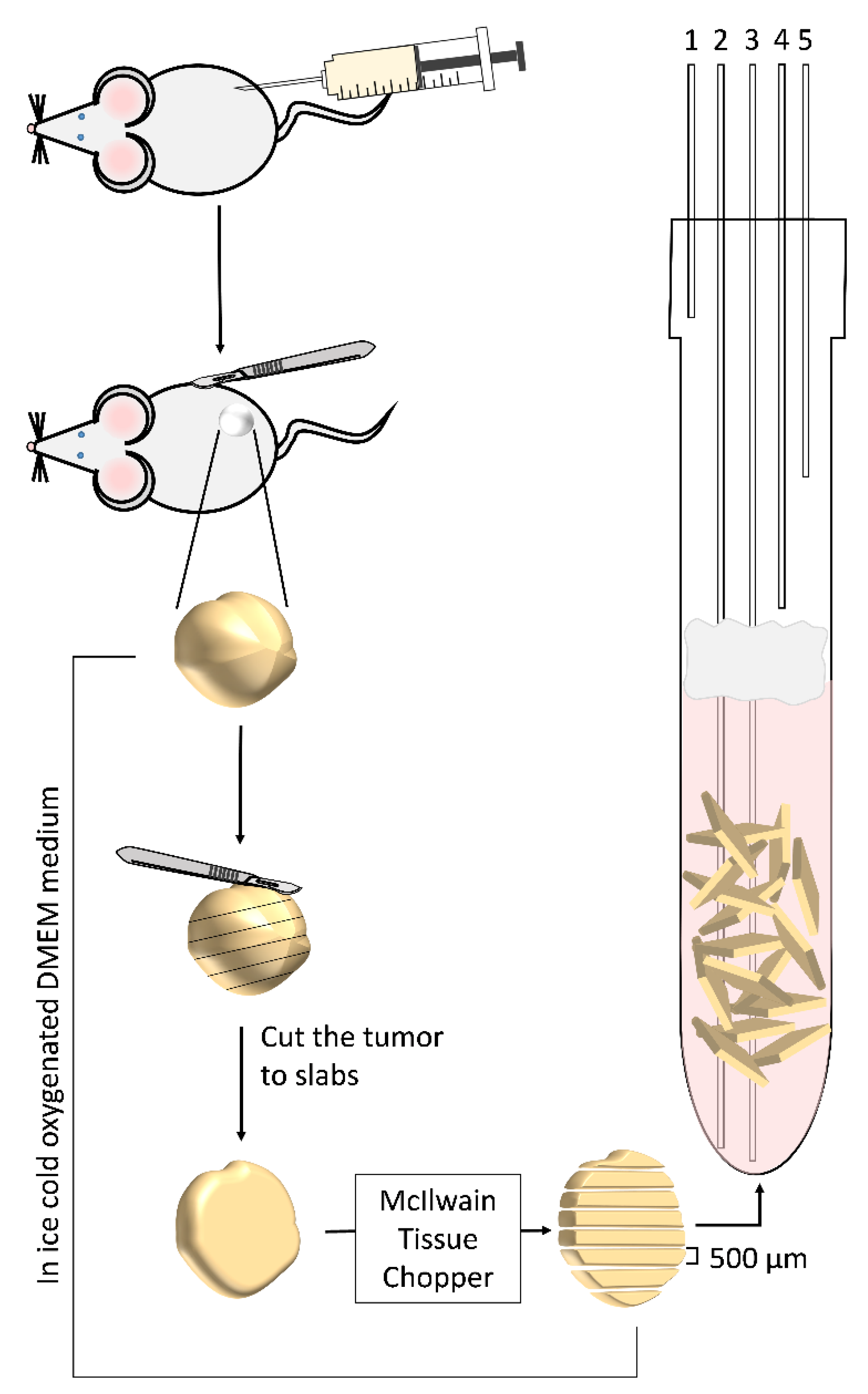
2.3. Surgical Procedure and Slice Preparation
2.4. Perfusion Media and the Perfusion System
2.5. Spin Polarization and Dissolution
2.6. Experimental Design: Hyperpolarized Media Injection and Acquisition Approach
- (1)
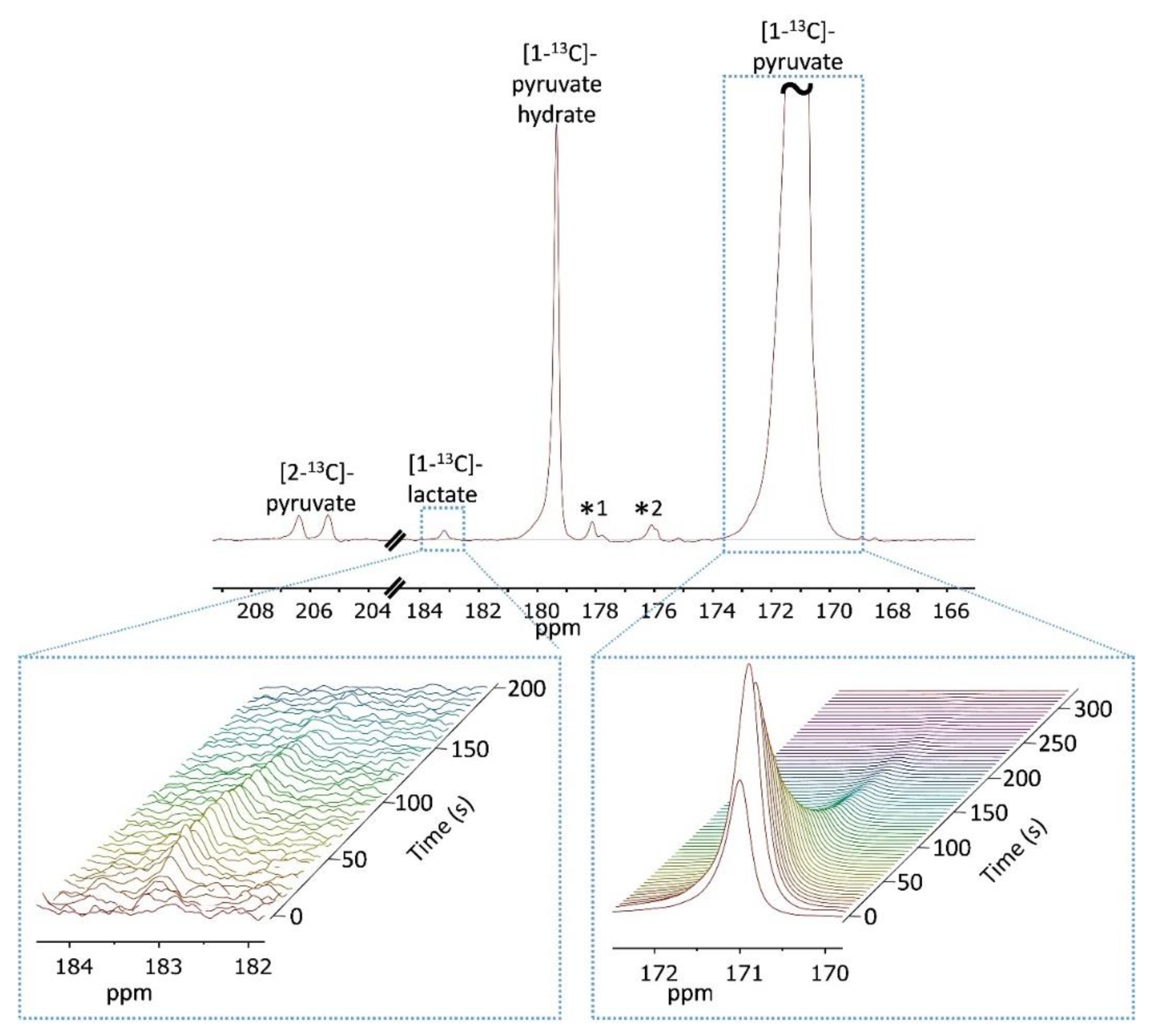
- Manual bolus injection during perfusion arrest and non-selective RF pulses: This is the most common hyperpolarized medium administration method and data acquisition approach in dDNP studies carried out in NMR tubes [27,29,30,51,52,54]. In this approach, the hyperpolarized solution was manually transferred via a manifold (connected to the in-flow line and made of a combination of medical grade 3-way valves and syringes) from the conical tube directly to the bottom of the NMR tube containing the tumor slices as previously described [52]. Altogether, the duration of hyperpolarized media transfer in this system was completed within 15 s from the start of the dissolution process. The hyperpolarized medium was injected gently to minimize tissue displacement and care was taken to avoid the introduction of air bubbles that could interfere with magnetic field homogeneity (due to the large difference in magnetic susceptibility between air and water). In this setup, the perfusion was stopped ~30 s before the injection of the hyperpolarized solution and was resumed only after the acquisition of the hyperpolarized spectra was completed (maximum of 4 min). This was done in order to characterize the metabolism of a constant concentration of [1-13C]pyruvate without the effects of wash-in and wash-out of the hyperpolarized medium.
- (2)
- Manual bolus injection during perfusion arrest and selective RF pulses: here, the introduction of the hyperpolarized solution was the same as in approach 1. However, the acquisition was performed using hyperpolarized product-selective saturating RF excitations, termed hereafter selective excitations, which fully excited the metabolite of interest while the precursor (pyruvate) is excited to a much lower degree. Therefore, only newly synthetized metabolites were detected in the consecutive excitation (see the Supplementary Materials). A series of at least 4 such selective excitations was performed immediately after the end of the bolus injection with a repetition time of 1 to 5 s. This was done to quench the signals from [1-13C]pyruvate impurities that resonate close to the metabolite signals [56]. Then, the same pulses were applied with a repetition time of 8 to 16 s to record the metabolism.
- (3)
- Administration of the hyperpolarized medium in a continuous flow and selective RF pulses: in this approach, the hyperpolarized agent was introduced to the tissue via a bypass system. This involved combining the 4 mL of dissolution medium with 8 mL of heated and oxygenated phosphate buffer awaiting in the conical tube used for hyperpolarized media collection (described above). Then, these 12 mL of hyperpolarized medium were loaded into a heated extension tube (bypass line) connected to the perfusion system with a manifold made out of medical grade 3-way valves. Then, the content of the bypass line (12 mL) was infused into the NMR tube containing the slices at a rate of 4 mL/min using the perfusion system. The purpose of the bypass system was to ensure continuous delivery of well-oxygenated hyperpolarized medium throughout the metabolic investigation. An additional benefit of this perfusion system was avoiding turbulence and tissue movement that could be caused by the bolus injection. In this approach, acquisition was started following the loading of the hyperpolarized medium into the bypass line (prior to arrival of the hyperpolarized medium to the slices).
2.7. NMR Spectroscopy
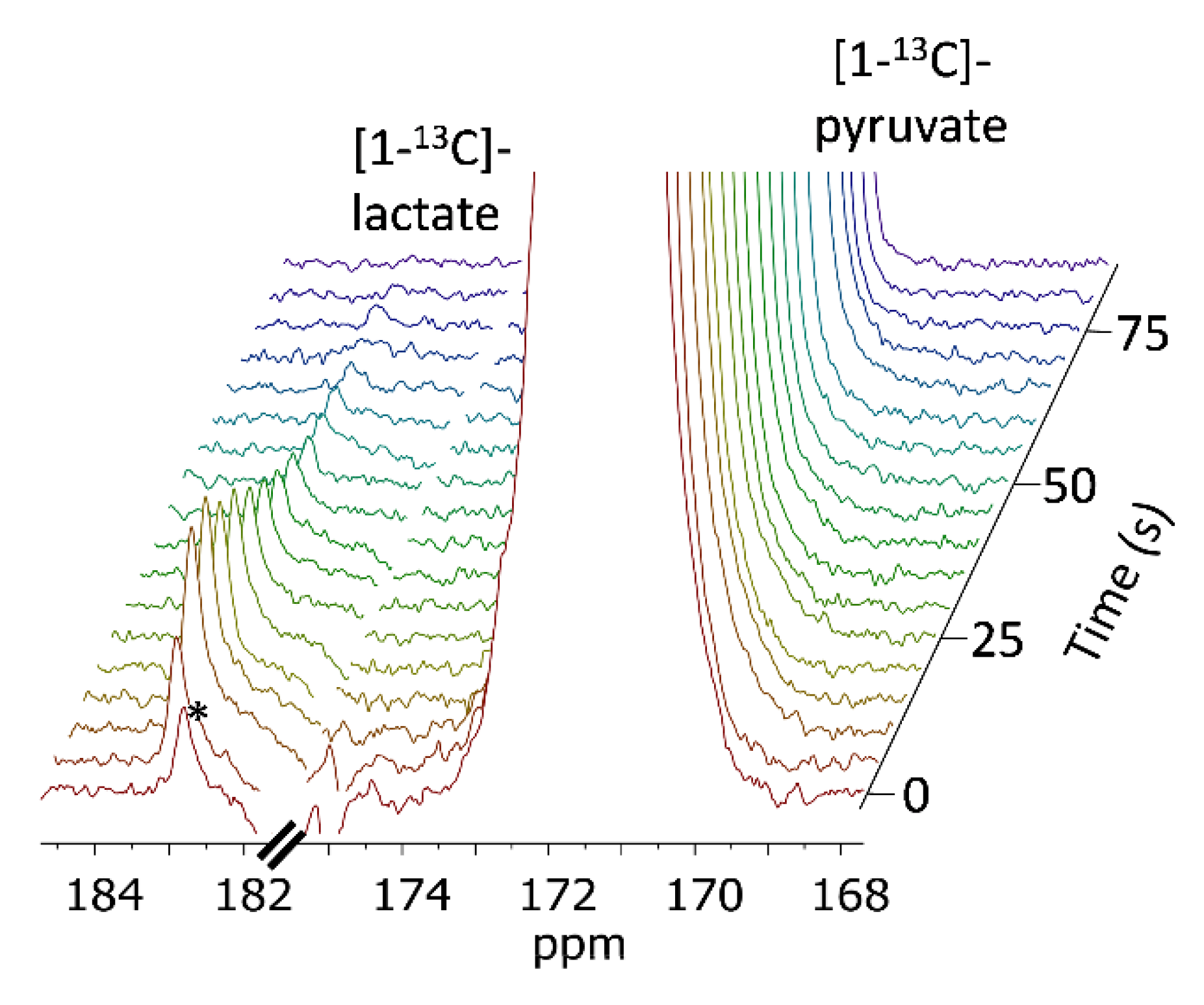
2.7.1. 13C-NMR Acquisitions with Non-selective RF Pulses
2.7.2. 13C-NMR Acquisitions with Selective RF Pulses
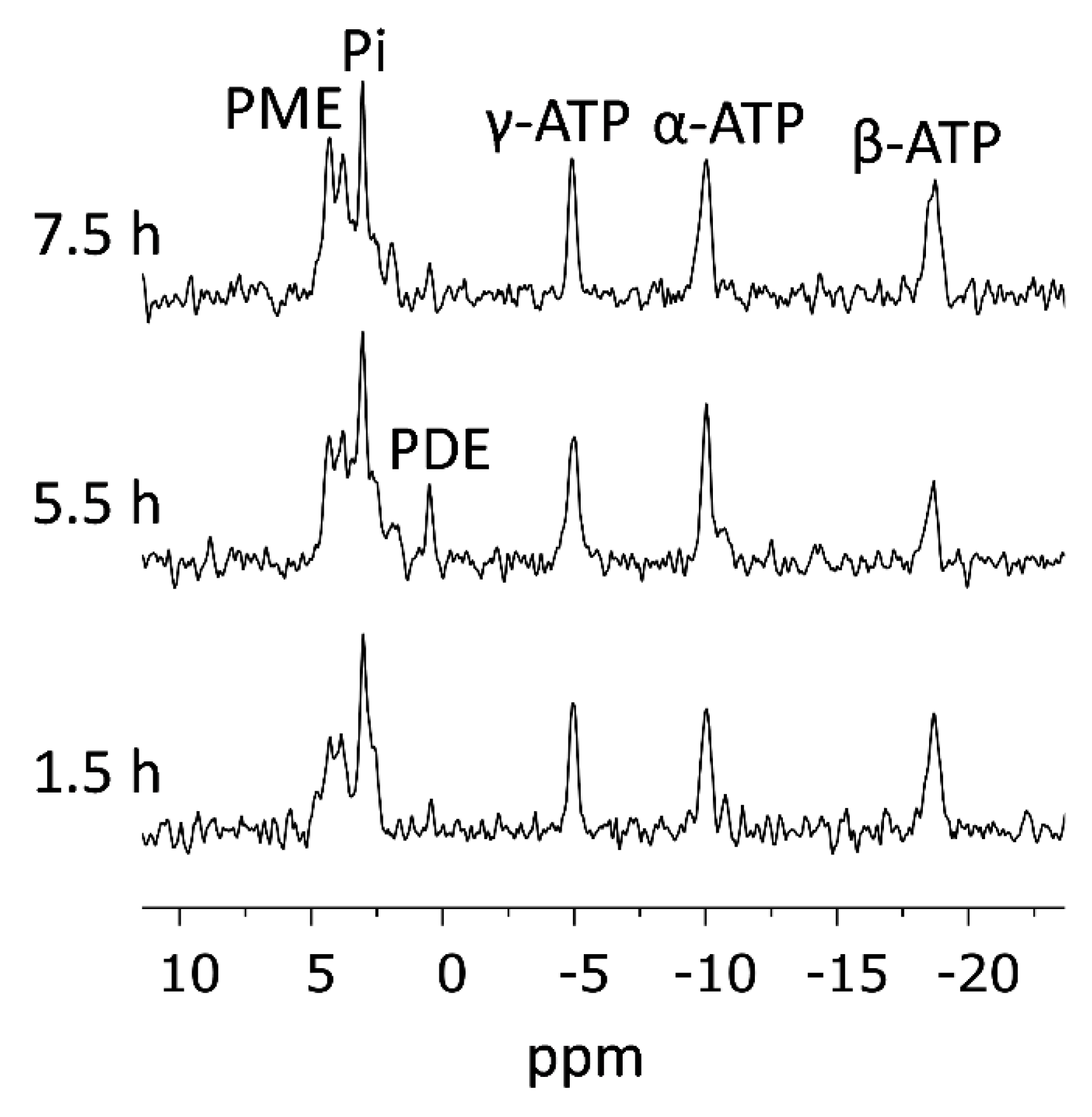
2.7.3. 31P-NMR Spectroscopy
2.7.4. Alternating 31P- and 13C-NMR Acquisitions
2.8. Validation of Viability
2.9. Processing and Data Analysis
2.9.1. Software
2.9.2. Determination of [1-13C]pyruvate T1 and Utilization of This Value
2.9.3. T1 Correction of the Hyperpolarized [1-13C]pyruvate Signal and Characterization of [1-13C]pyruvate Concentration in the Medium
2.9.4. ATP Concentration
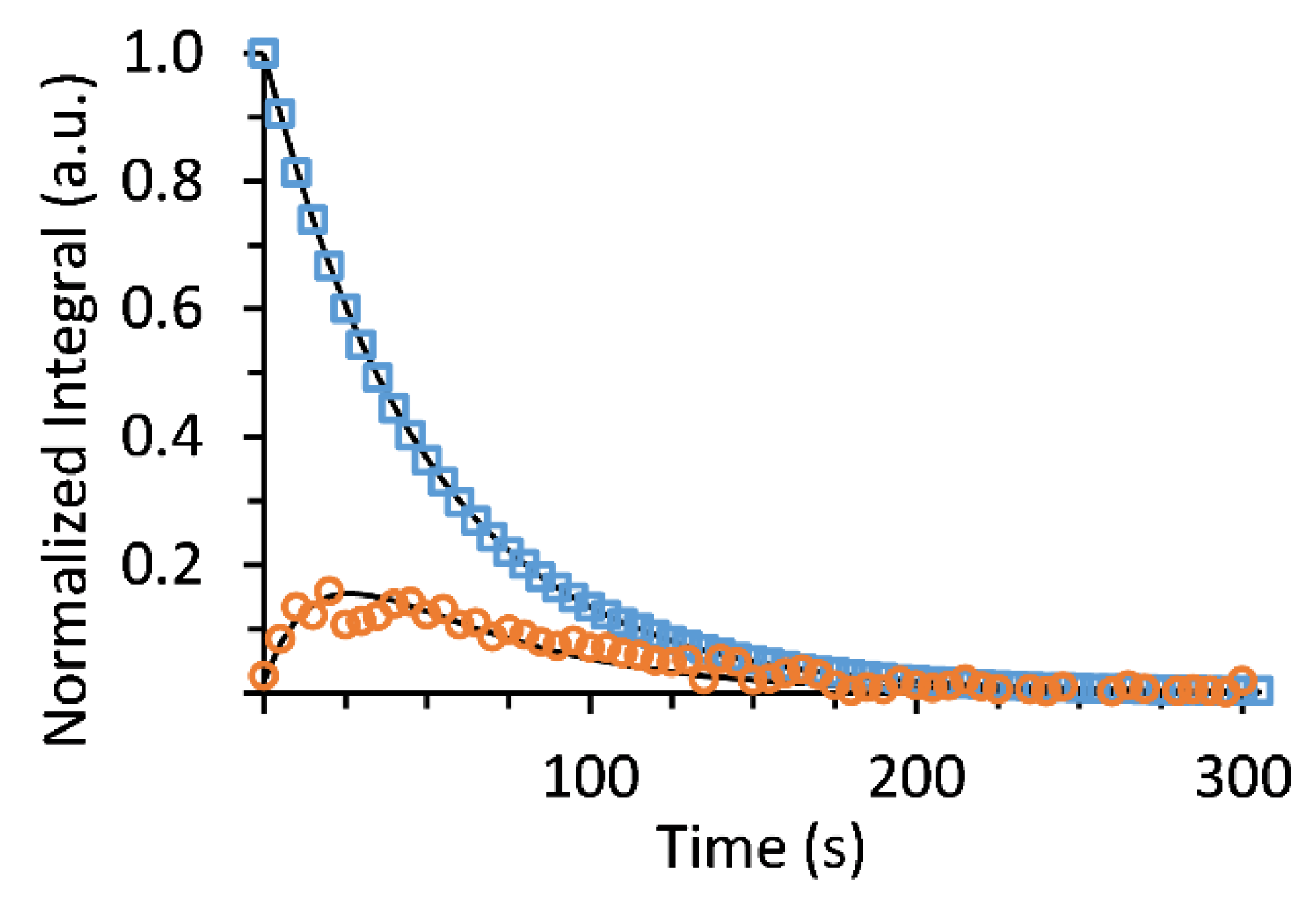
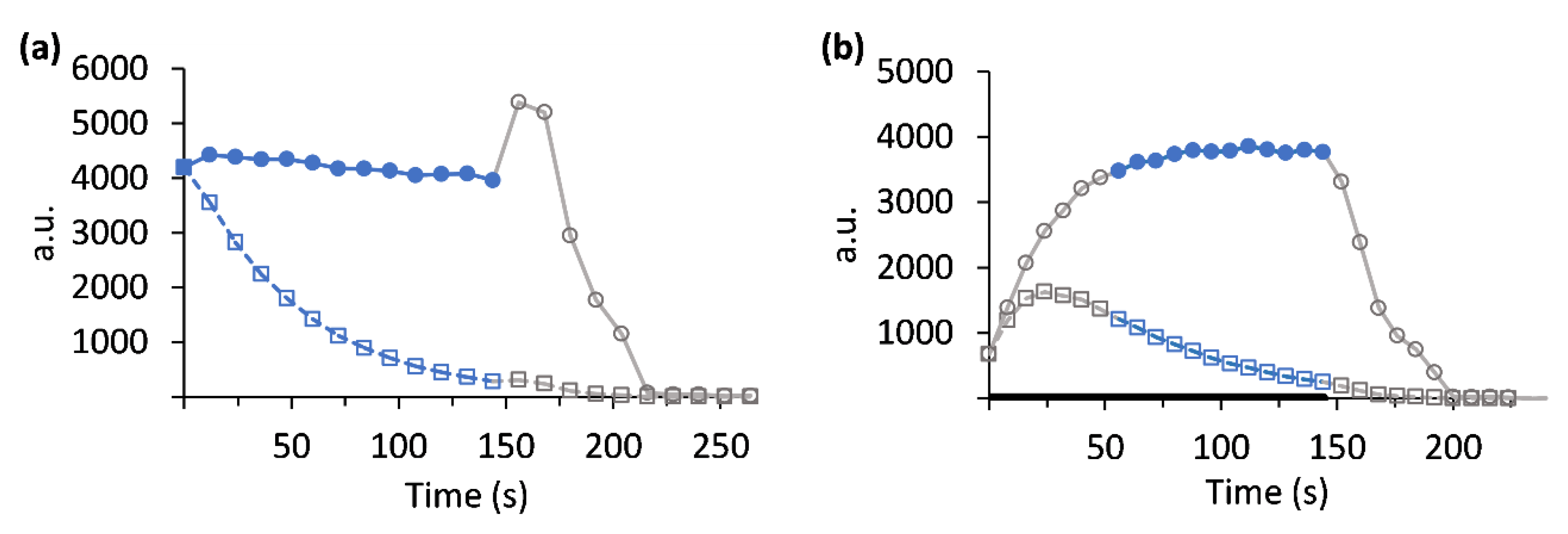
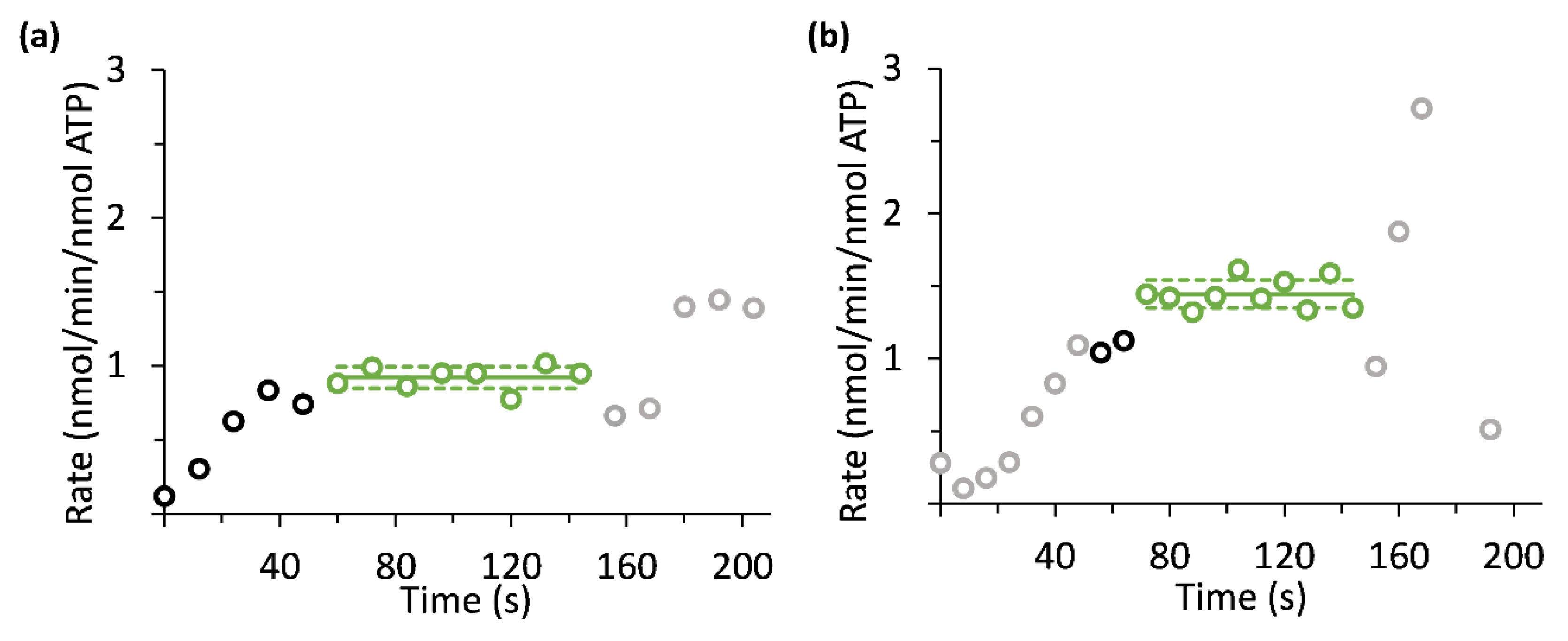
2.9.5. Metabolic Rate Calculation
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Gnant, M.; Harbeck, N.; Thomssen, C. St. Gallen/Vienna 2017: A brief summary of the consensus discussion about escalation and de-escalation of primary breast cancer treatment. Breast Care 2017, 12, 102–107. [Google Scholar] [CrossRef]
- Coates, A.S.; Winer, E.P.; Goldhirsch, A.; Gelber, R.D.; Gnant, M.; Piccart-Gebhart, M.; Thurlimann, B.; Senn, H.J. Tailoring therapies--improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann. Oncol. 2015, 26, 1533–1546. [Google Scholar] [CrossRef] [PubMed]
- Mann, R.M.; Balleyguier, C.; Baltzer, P.A.; Bick, U.; Colin, C.; Cornford, E.; Evans, A.; Fallenberg, E.; Forrai, G.; Fuchsjäger, M.H.; et al. Breast MRI: EUSOBI recommendations for women’s information. Eur. Radiol. 2015, 25, 3669–3678. [Google Scholar] [CrossRef]
- Liberman, L.; Abramson, A.F.; Squires, F.B.; Glassman, J.R.; Morris, E.A.; Dershaw, D.D. The breast imaging reporting and data system: positive predictive value of mammographic features and final assessment categories. Am. J. Roentgenol. 1998, 171, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Eun, N.L.; Son, E.J.; Gweon, H.M.; Youk, J.H.; Kim, J.A. The value of breast MRI for BI-RADS category 4B mammographic microcalcification: based on the 5th edition of BI-RADS. Clin. Radiol. 2018, 73, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Pinker-Domenig, K.; Bogner, W.; Gruber, S.; Bickel, H.; Duffy, S.; Schernthaner, M.; Dubsky, P.; Pluschnig, U.; Rudas, M.; Trattnig, S.; et al. High resolution MRI of the breast at 3 T: which BI-RADS® descriptors are most strongly associated with the diagnosis of breast cancer? Eur. Radiol. 2012, 22, 322–330. [Google Scholar] [CrossRef]
- Morrow, M.; Waters, J.; Morris, E. MRI for breast cancer screening, diagnosis, and treatment. Lancet 2011, 378, 1804–1811. [Google Scholar] [CrossRef]
- Huang, W.; Tudorica, L.A.; Li, X.; Thakur, S.B.; Chen, Y.; Morris, E.A.; Tagge, I.J.; Korenblit, M.E.; Rooney, W.D.; Koutcher, J.A.; et al. Discrimination of benign and malignant breast lesions by using shutter-speed dynamic contrast-enhanced MR imaging. Radiology 2011, 261, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, K.; Sun, X.; Wang, K.; Sun, Y.; Zhang, G.; Shen, B. Parameters of dynamic contrast-enhanced MRI as imaging markers for angiogenesis and proliferation in human breast cancer. Med. Sci. Monit. 2015, 21, 376–382. [Google Scholar] [CrossRef]
- Baré, M.; Torà, N.; Salas, D.; Sentís, M.; Ferrer, J.; Ibáñez, J.; Zubizarreta, R.; Sarriugarte, G.; Barata, T.; Domingo, L.; et al. Mammographic and clinical characteristics of different phenotypes of screen-detected and interval breast cancers in a nationwide screening program. Breast Cancer Res. Treat. 2015, 154, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Boisserie-Lacroix, M.; Bullier, B.; Hurtevent-Labrot, G.; Ferron, S.; Lippa, N.; Mac Grogan, G. Correlation between imaging and prognostic factors: Molecular classification of breast cancers. Diagn. Interv. Imaging 2014, 95, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Irshad, A.; Leddy, R.; Pisano, E.; Baker, N.; Lewis, M.; Ackerman, S.; Campbell, A. Assessing the role of ultrasound in predicting the biological behavior of breast cancer. Am. J. Roentgenol. 2013, 200, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Rashmi, S.; Kamala, S.; Murthy, S.S.; Kotha, S.; Rao, Y.S.; Chaudhary, K.V. Predicting the molecular subtype of breast cancer based on mammography and ultrasound findings. Indian. J. Radiol. Imaging 2018, 28, 354–361. [Google Scholar] [CrossRef]
- Lazarus, E.; Mainiero, M.B.; Schepps, B.; Koelliker, S.L.; Livingston, L.S. BI-RADS lexicon for US and mammography: Interobserver variability and positive predictive value. Radiology 2006, 239, 385–391. [Google Scholar] [CrossRef]
- Timmers, J.M.H.; van Doorne-Nagtegaal, H.J.; Zonderland, H.M.; van Tinteren, H.; Visser, O.; Verbeek, A.L.M.; den Heeten, G.J.; Broeders, M.J.M. The breast imaging reporting and data system (BI-RADS) in the dutch breast cancer screening programme: its role as an assessment and stratification tool. Eur. Radiol. 2012, 22, 1717–1723. [Google Scholar] [CrossRef]
- Zonderland, H.M.; Pope, T.L.; Nieborg, A.J. The positive predictive value of the breast imaging reporting and data system (BI-RADS) as a method of quality assessment in breast imaging in a hospital population. Eur. Radiol. 2004, 14, 1743–1750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, J.; Xiao, Y.; Cui, H.; Du, G.; Wang, Y.; Li, Z.; Wu, T.; Li, X.; Tian, J. Identifying ultrasound and clinical features of breast cancer molecular subtypes by ensemble decision. Sci. Rep. 2015, 5, 11085. [Google Scholar] [CrossRef]
- Ardenkjaer-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Hansson, L.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.Z.; Harzstark, A.L.; Ferrone, M.; van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef]
- Cunningham, C.H.; Lau, J.Y.; Chen, A.P.; Geraghty, B.J.; Perks, W.J.; Roifman, I.; Wright, G.A.; Connelly, K.A. Hyperpolarized 13C metabolic MRI of the human heart: initial experience. Circ. Res. 2016, 119, 1177–1182. [Google Scholar] [CrossRef]
- Miloushev, V.Z.; Granlund, K.L.; Boltyanskiy, R.; Lyashchenko, S.K.; DeAngelis, L.M.; Mellinghoff, I.K.; Brennan, C.W.; Tabar, V.; Yang, T.J.; Holodny, A.I.; et al. Metabolic imaging of the human brain with hyperpolarized 13C pyruvate demonstrates 13C lactate production in brain tumor patients. Cancer Res. 2018, 78, 3755–3760. [Google Scholar] [CrossRef]
- Aggarwal, R.; Vigneron, D.B.; Kurhanewicz, J. Hyperpolarized 1-[13C]-pyruvate magnetic resonance imaging detects an early metabolic response to androgen ablation therapy in prostate cancer. Eur. Urol. 2017, 72, 1028–1029. [Google Scholar] [CrossRef]
- Grist, J.T.; McLean, M.A.; Riemer, F.; Schulte, R.F.; Deen, S.S.; Zaccagna, F.; Woitek, R.; Daniels, C.J.; Kaggie, J.D.; Matys, T.; et al. Quantifying normal human brain metabolism using hyperpolarized [1–13C]pyruvate and magnetic resonance imaging. NeuroImage 2019, 189, 171–179. [Google Scholar] [CrossRef]
- Park, I.; Larson, P.E.Z.; Gordon, J.W.; Carvajal, L.; Chen, H.-Y.; Bok, R.; Van Criekinge, M.; Ferrone, M.; Slater, J.B.; Xu, D.; et al. Development of methods and feasibility of using hyperpolarized carbon-13 imaging data for evaluating brain metabolism in patient studies. Magn. Reson. Med. 2018, 80, 864–873. [Google Scholar] [CrossRef]
- Lodi, A.; Woods, S.M.; Ronen, S.M. Treatment with the MEK inhibitor U0126 induces decreased hyperpolarized pyruvate to lactate conversion in breast, but not prostate, cancer cells. NMR Biomed. 2013, 26, 299–306. [Google Scholar] [CrossRef]
- Harris, T.; Degani, H.; Frydman, L. Hyperpolarized 13C NMR studies of glucose metabolism in living breast cancer cell cultures. NMR Biomed. 2013, 26, 1831–1843. [Google Scholar] [CrossRef]
- Witney, T.H.; Kettunen, M.I.; Brindle, K.M. Kinetic modeling of hyperpolarized 13C label exchange between pyruvate and lactate in tumor cells. J. Biol. Chem. 2011, 286, 24572–24580. [Google Scholar] [CrossRef]
- Ward, C.S.; Venkatesh, H.S.; Chaumeil, M.M.; Brandes, A.H.; VanCriekinge, M.; Dafni, H.; Sukumar, S.; Nelson, S.J.; Vigneron, D.B.; Kurhanewicz, J.; et al. Noninvasive detection of target modulation following phosphatidylinositol 3-kinase inhibition using hyperpolarized 13C magnetic resonance spectroscopy. Cancer Res. 2010. [Google Scholar] [CrossRef]
- Harris, T.; Eliyahu, G.; Frydman, L.; Degani, H. Kinetics of hyperpolarized 13C1-pyruvate transport and metabolism in living human breast cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 18131–18136. [Google Scholar] [CrossRef]
- Bohndiek, S.E.; Kettunen, M.I.; Hu, D.E.; Brindle, K.M. Hyperpolarized 13C spectroscopy detects early changes in tumor vasculature and metabolism after VEGF neutralization. Cancer Res. 2012, 72, 854–864. [Google Scholar] [CrossRef]
- Kettunen, M.I.; Kennedy, B.W.C.; Hu, D.-e.; Brindle, K.M. Spin echo measurements of the extravasation and tumor cell uptake of hyperpolarized [1-13C]lactate and [1-13C]pyruvate. Magn. Reson. Med. 2013, 70, 1200–1209. [Google Scholar] [CrossRef]
- Golman, K.; in’t Zandt, R.; Lerche, M.; Pehrson, R.; Ardenkjaer-Larsen, J.H. Metabolic imaging by hyperpolarized 13C magnetic resonance imaging for in vivo tumor diagnosis. Cancer Res. 2006, 66, 10855–10860. [Google Scholar] [CrossRef]
- Day, S.E.; Kettunen, M.I.; Gallagher, F.A.; Hu, D.E.; Lerche, M.; Wolber, J.; Golman, K.; Ardenkjaer-Larsen, J.H.; Brindle, K.M. Detecting tumor response to treatment using hyperpolarized 13C magnetic resonance imaging and spectroscopy. Nat. Med. 2007, 13, 1382–1387. [Google Scholar] [CrossRef]
- Asghar Butt, S.; Sogaard, L.V.; Ardenkjaer-Larsen, J.H.; Lauritzen, M.H.; Engelholm, L.H.; Paulson, O.B.; Mirza, O.; Holck, S.; Magnusson, P.; Akeson, P. Monitoring mammary tumor progression and effect of tamoxifen treatment in MMTV-PymT using MRI and magnetic resonance spectroscopy with hyperpolarized [1-13C]pyruvate. Magn. Reson. Med. 2015, 73, 51–58. [Google Scholar] [CrossRef]
- Albers, M.J.; Bok, R.; Chen, A.P.; Cunningham, C.H.; Zierhut, M.L.; Zhang, V.Y.; Kohler, S.J.; Tropp, J.; Hurd, R.E.; Yen, Y.F.; et al. Hyperpolarized 13C lactate, pyruvate, and alanine: noninvasive biomarkers for prostate cancer detection and grading. Cancer Res. 2008, 68, 8607–8615. [Google Scholar] [CrossRef]
- Von Morze, C.; Larson, P.E.; Hu, S.; Yoshihara, H.A.; Bok, R.A.; Goga, A.; Ardenkjaer-Larsen, J.H.; Vigneron, D.B. Investigating tumor perfusion and metabolism using multiple hyperpolarized 13C compounds: HP001, pyruvate and urea. Magn. Reson. Imaging 2012, 30, 305–311. [Google Scholar] [CrossRef]
- Su, J.S.; Woods, S.M.; Ronen, S.M. Metabolic consequences of treatment with AKT inhibitor perifosine in breast cancer cells. NMR Biomed. 2012, 25, 379–388. [Google Scholar] [CrossRef]
- Lumata, L.; Yang, C.; Ragavan, M.; Carpenter, N.; DeBerardinis, R.J.; Merritt, M.E. Chapter two - hyperpolarized 13C magnetic resonance and Its use in metabolic assessment of cultured cells and perfused organs. In Meth. Enzymol.; Metallo, C.M., Ed.; Academic Press: New York, NY, USA, 2015; pp. 73–106. [Google Scholar]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Serganova, I.; Cohen, I.J.; Vemuri, K.; Shindo, M.; Maeda, M.; Mane, M.; Moroz, E.; Khanin, R.; Satagopan, J.; Koutcher, J.A.; et al. LDH-A regulates the tumor microenvironment via HIF-signaling and modulates the immune response. PLoS ONE 2018, 13, e0203965. [Google Scholar] [CrossRef]
- Yang, D.Q.; Freund, D.M.; Harris, B.R.; Wang, D.; Cleary, M.P.; Hegeman, A.D. Measuring relative utilization of aerobic glycolysis in breast cancer cells by positional isotopic discrimination. FEBS Lett. 2016, 590, 3179–3187. [Google Scholar] [CrossRef]
- Rosado, A.; Morris, H.P.; Weinhouse, S. Lactate dehydrogenase subunits in normal and neoplastic tissues of the rat. Cancer Res. 1969, 29, 1673. [Google Scholar]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef]
- Wespi, P.; Steinhauser, J.; Kwiatkowski, G.; Kozerke, S. Overestimation of cardiac lactate production caused by liver metabolism of hyperpolarized [1-13C]pyruvate. Magn. Reson. Med. 2018, 80, 1882–1890. [Google Scholar] [CrossRef]
- Ball, D.R.; Cruickshank, R.; Carr, C.A.; Stuckey, D.J.; Lee, P.; Clarke, K.; Tyler, D.J. Metabolic imaging of acute and chronic infarction in the perfused rat heart using hyperpolarised [1-13C]pyruvate. NMR Biomed. 2013, 26, 1441–1450. [Google Scholar] [CrossRef]
- Merritt, M.E.; Harrison, C.; Storey, C.; Jeffrey, F.M.; Sherry, A.D.; Malloy, C.R. Hyperpolarized 13C allows a direct measure of flux through a single enzyme-catalyzed step by NMR. Proc. Natl. Acad. Sci. USA 2007, 104, 19773–19777. [Google Scholar] [CrossRef]
- Moreno, K.X.; Sabelhaus, S.M.; Merritt, M.E.; Sherry, A.D.; Malloy, C.R. Competition of pyruvate with physiological substrates for oxidation by the heart: implications for studies with hyperpolarized [1-13C]pyruvate. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1556–H1564. [Google Scholar] [CrossRef]
- Schroeder, M.A.; Atherton, H.J.; Ball, D.R.; Cole, M.A.; Heather, L.C.; Griffin, J.L.; Clarke, K.; Radda, G.K.; Tyler, D.J. Real-time assessment of Krebs cycle metabolism using hyperpolarized 13C magnetic resonance spectroscopy. FASEB J. 2009, 23, 2529–2538. [Google Scholar] [CrossRef]
- Moreno, K.X.; Satapati, S.; DeBerardinis, R.J.; Burgess, S.C.; Malloy, C.R.; Merritt, M.E. Real-time detection of hepatic gluconeogenic and glycogenolytic states using hyperpolarized [2-13C]dihydroxyacetone. J. Biol. Chem. 2014, 289, 35859–35867. [Google Scholar] [CrossRef]
- Lev-Cohain, N.; Sapir, G.; Harris, T.; Azar, A.; Gamliel, A.; Nardi-Schreiber, A.; Uppala, S.; Sosna, J.; Gomori, J.M.; Katz-Brull, R. Real-time ALT and LDH activities determined in viable precision-cut mouse liver slices using hyperpolarized [1-13C]pyruvate – implications for studies on biopsied liver tissues. NMR Biomed. 2019, 32, e4043. [Google Scholar] [CrossRef]
- Harris, T.; Azar, A.; Sapir, G.; Gamliel, A.; Nardi-Schreiber, A.; Sosna, J.; Gomori, J.M.; Katz-Brull, R. Real-time ex-vivo measurement of brain metabolism using hyperpolarized [1-13C]pyruvate. Sci. Rep. 2018, 8, 9564. [Google Scholar] [CrossRef]
- Keshari, K.R.; Sriram, R.; Van Criekinge, M.; Wilson, D.M.; Wang, Z.J.; Vigneron, D.B.; Peehl, D.M.; Kurhanewicz, J. Metabolic reprogramming and validation of hyperpolarized 13C lactate as a prostate cancer biomarker using a human prostate tissue slice culture bioreactor. Prostate 2013, 73, 1171–1181. [Google Scholar] [CrossRef]
- Sriram, R.; Van Criekinge, M.; Delos Santos, J.; Keshari, K.R.; Wilson, D.M.; Peehl, D.; Kurhanewicz, J.; Wang, Z.J. Non-Invasive Differentiation of Benign Renal Tumors from Clear Cell Renal Cell Carcinomas Using Clinically Translatable Hyperpolarized 13C Pyruvate Magnetic Resonance. Tomography 2016, 2, 35–42. [Google Scholar] [CrossRef]
- Whittle, J.R.; Lewis, M.T.; Lindeman, G.J.; Visvader, J.E. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015, 17, 17. [Google Scholar] [CrossRef]
- Harris, T.; Gamliel, A.; Sosna, J.; Gomori, J.M.; Katz-Brull, R. Impurities of [1-13C] pyruvic acid and a method to minimize their signals for hyperpolarized pyruvate metabolism studies. Appl. Magn. Reson. 2018, 1–14. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Allouche-Arnon, H.; Hovav, Y.; Friesen-Waldner, L.; Sosna, J.; Moshe Gomori, J.; Vega, S.; Katz-Brull, R. Quantification of rate constants for successive enzymatic reactions with DNP hyperpolarized MR. NMR Biomed. 2014, 27, 656–662. [Google Scholar] [CrossRef]
- Perkins, R.J.; Shoemaker, R.K.; Carpenter, B.K.; Vaida, V. Chemical equilibria and kinetics in aqueous solutions of zymonic acid. J. Phys. Chem. A 2016, 120, 10096–10107. [Google Scholar] [CrossRef]
- Duwel, S.; Hundshammer, C.; Gersch, M.; Feuerecker, B.; Steiger, K.; Buck, A.; Walch, A.; Haase, A.; Glaser, S.J.; Schwaiger, M.; et al. Imaging of pH in vivo using hyperpolarized 13C-labelled zymonic acid. Nat. Commun. 2017, 8, 15126. [Google Scholar] [CrossRef]
- Katz-Brull, R.; Margalit, R.; Degani, H. Differential routing of choline in implanted breast cancer and normal organs. Magn. Reson. Med. 2001, 46, 31–38. [Google Scholar] [CrossRef]
- Contractor, K.B.; Kenny, L.M.; Stebbing, J.; Al-Nahha, A.; Palmieri, C.; Sinnett, D.; Lewis, J.S.; Hogben, K.; Osman, S.; Shousha, S.; et al. [11C] choline positron emission tomography in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2009, 15, 5503–5510. [Google Scholar] [CrossRef]
- Kenny, L.; Coombes, R.C.; Vigushin, D.M.; Al-Nahhas, A.; Shousha, S.; Aboagye, E.O. Imaging early changes in proliferation at 1 week post chemotherapy: a pilot study in breast cancer patients with 3’-deoxy-3’- [18F]fluorothymidine positron emission tomography. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1339–1347. [Google Scholar] [CrossRef]
- Smyczek-Gargya, B.; Fersis, N.; Dittmann, H.; Vogel, U.; Reischl, G.; Machulla, H.J.; Wallwiener, D.; Bares, R.; Dohmen, B.M. PET with [18F]fluorothymidine for imaging of primary breast cancer: a pilot study. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 720–724. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adler-Levy, Y.; Nardi-Schreiber, A.; Harris, T.; Shaul, D.; Uppala, S.; Sapir, G.; Lev-Cohain, N.; Sosna, J.; Goldberg, S.N.; Gomori, J.M.; et al. In-Cell Determination of Lactate Dehydrogenase Activity in a Luminal Breast Cancer Model – ex vivo Investigation of Excised Xenograft Tumor Slices Using dDNP Hyperpolarized [1-13C]pyruvate. Sensors 2019, 19, 2089. https://0-doi-org.brum.beds.ac.uk/10.3390/s19092089
Adler-Levy Y, Nardi-Schreiber A, Harris T, Shaul D, Uppala S, Sapir G, Lev-Cohain N, Sosna J, Goldberg SN, Gomori JM, et al. In-Cell Determination of Lactate Dehydrogenase Activity in a Luminal Breast Cancer Model – ex vivo Investigation of Excised Xenograft Tumor Slices Using dDNP Hyperpolarized [1-13C]pyruvate. Sensors. 2019; 19(9):2089. https://0-doi-org.brum.beds.ac.uk/10.3390/s19092089
Chicago/Turabian StyleAdler-Levy, Yael, Atara Nardi-Schreiber, Talia Harris, David Shaul, Sivaranjan Uppala, Gal Sapir, Naama Lev-Cohain, Jacob Sosna, Shraga Nahum Goldberg, J. Moshe Gomori, and et al. 2019. "In-Cell Determination of Lactate Dehydrogenase Activity in a Luminal Breast Cancer Model – ex vivo Investigation of Excised Xenograft Tumor Slices Using dDNP Hyperpolarized [1-13C]pyruvate" Sensors 19, no. 9: 2089. https://0-doi-org.brum.beds.ac.uk/10.3390/s19092089