AMPK Is the Crucial Target for the CDK4/6 Inhibitors Mediated Therapeutic Responses in PANC-1 and MIA PaCa-2 Pancreatic Cancer Cell Lines

 , ,
, ,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
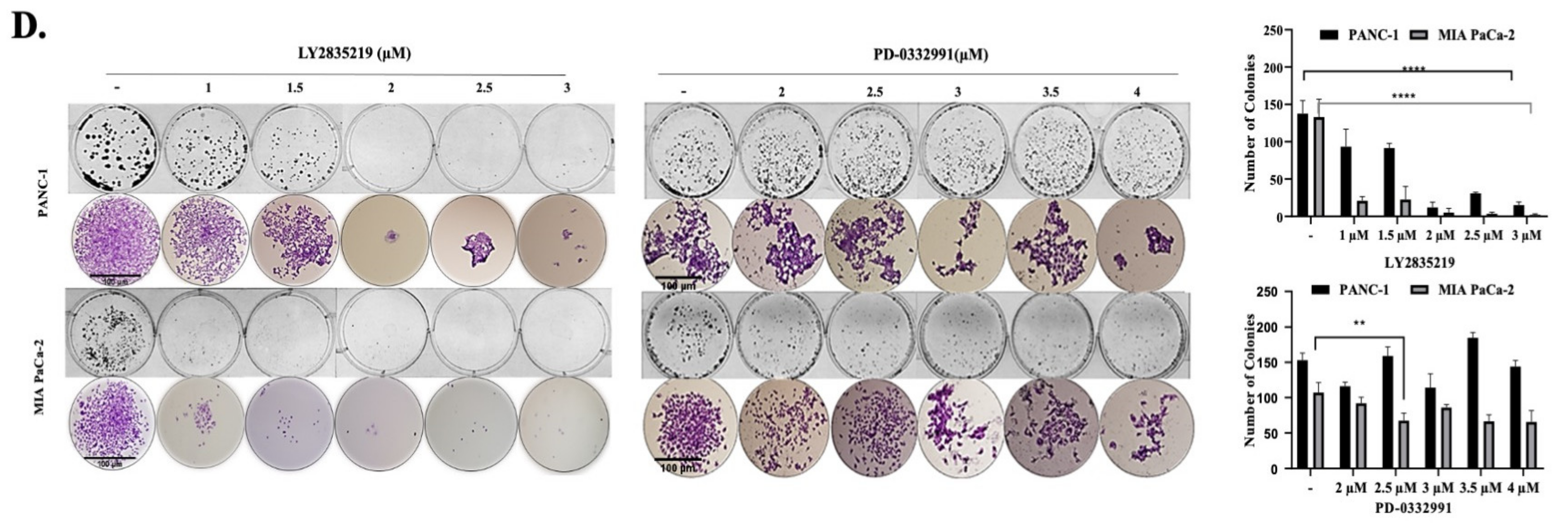
2.1. CDK4/6 Inhibitors Reduced Cell Viability and Proliferation in a Dose-Dependent Manner
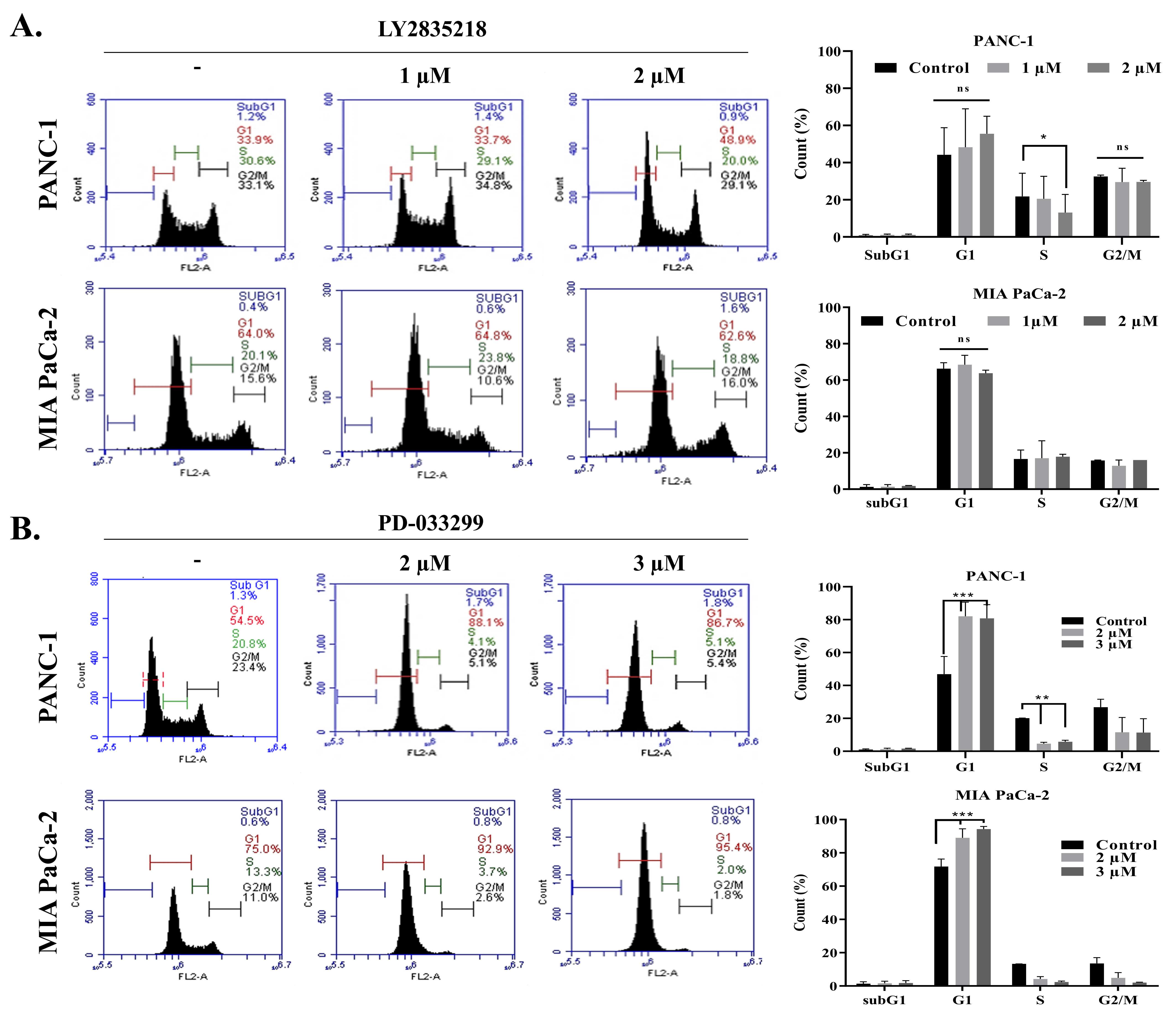
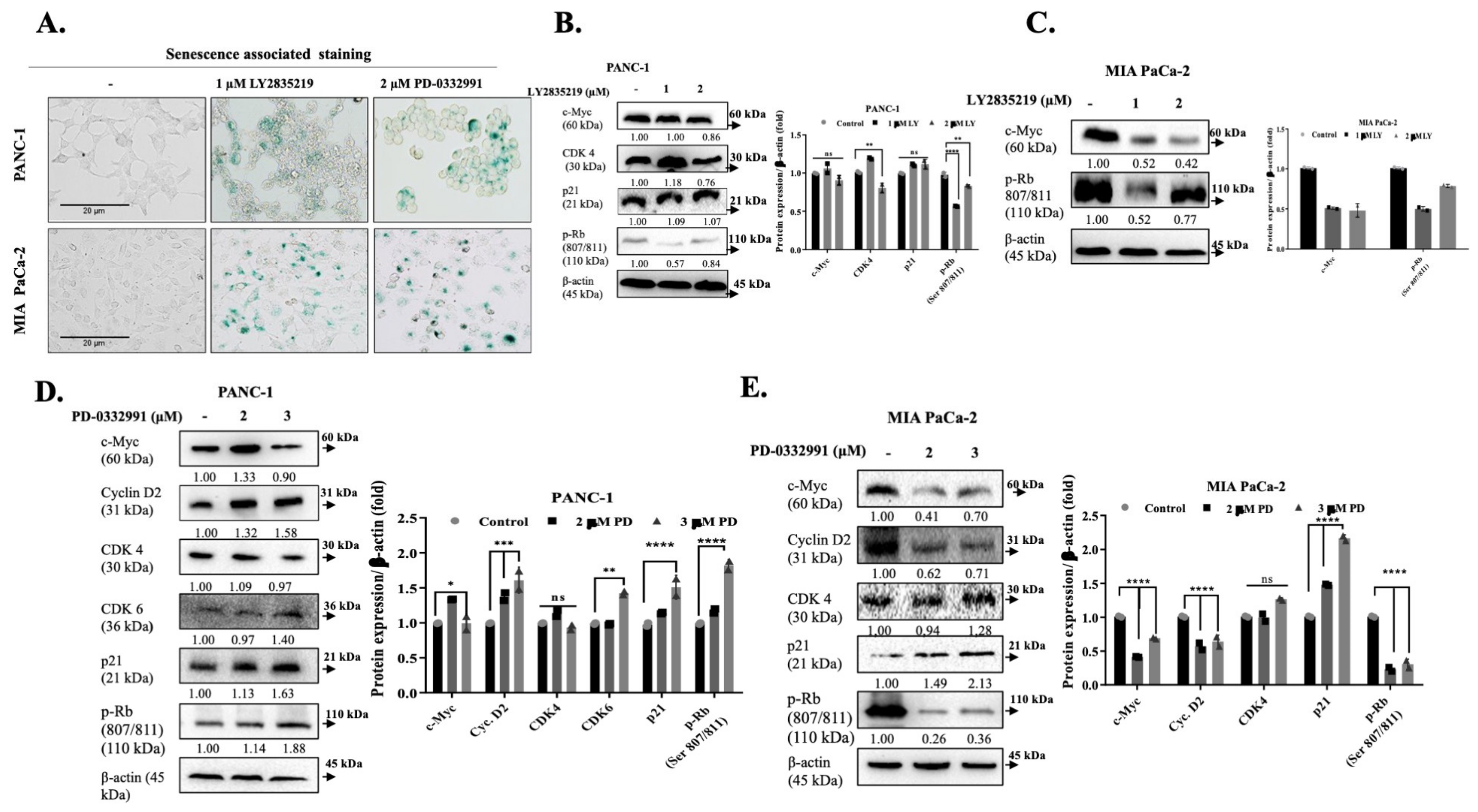
2.2. LY2835219 and PD-0332991 Arrest the Cell Cycle and Led to Senescence
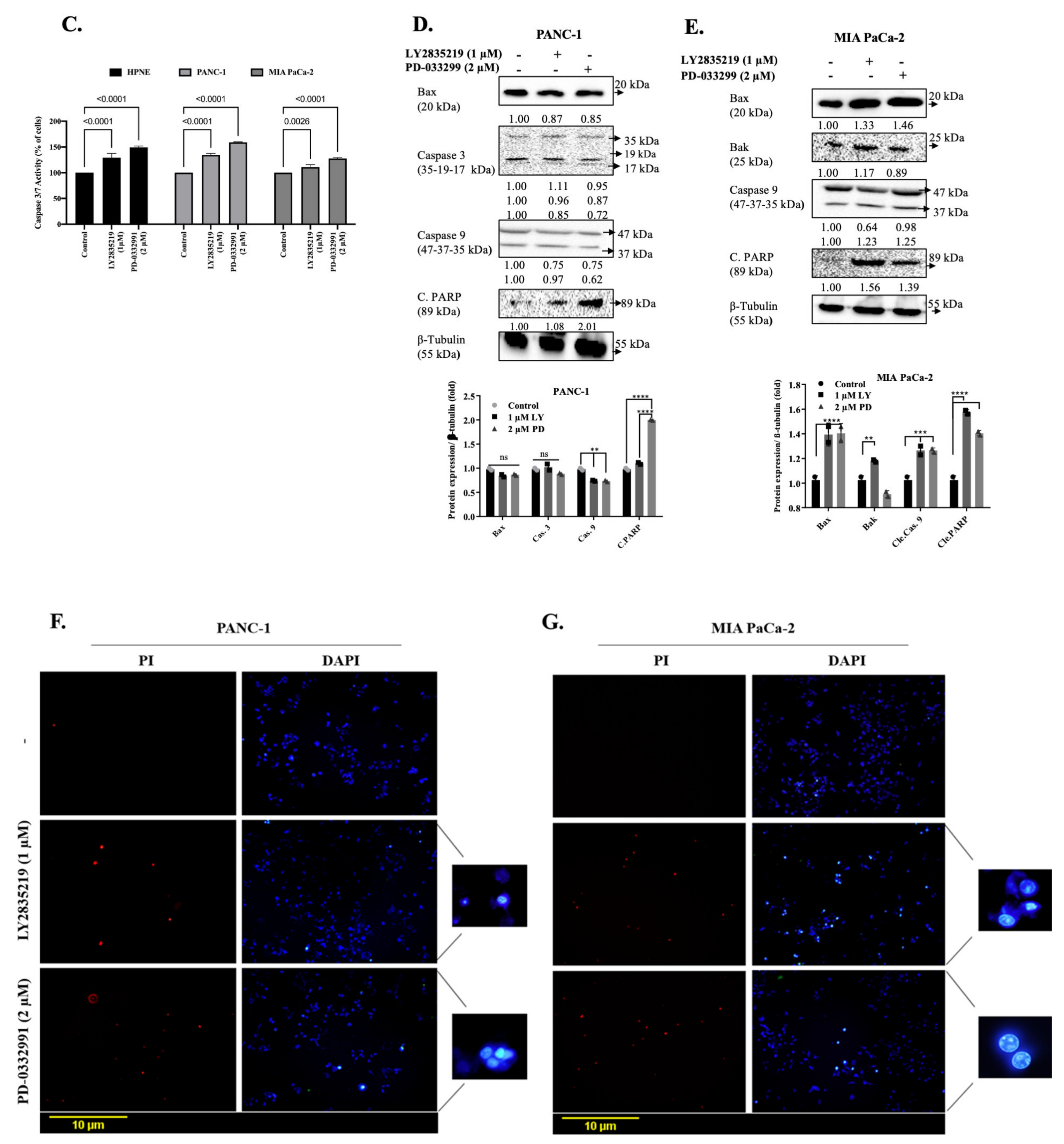
2.3. LY2835219 and PD-0332991 Induced Apoptosis in Time-Dependent Manner But the Cell Death Decision Was Taken at a Later Stage
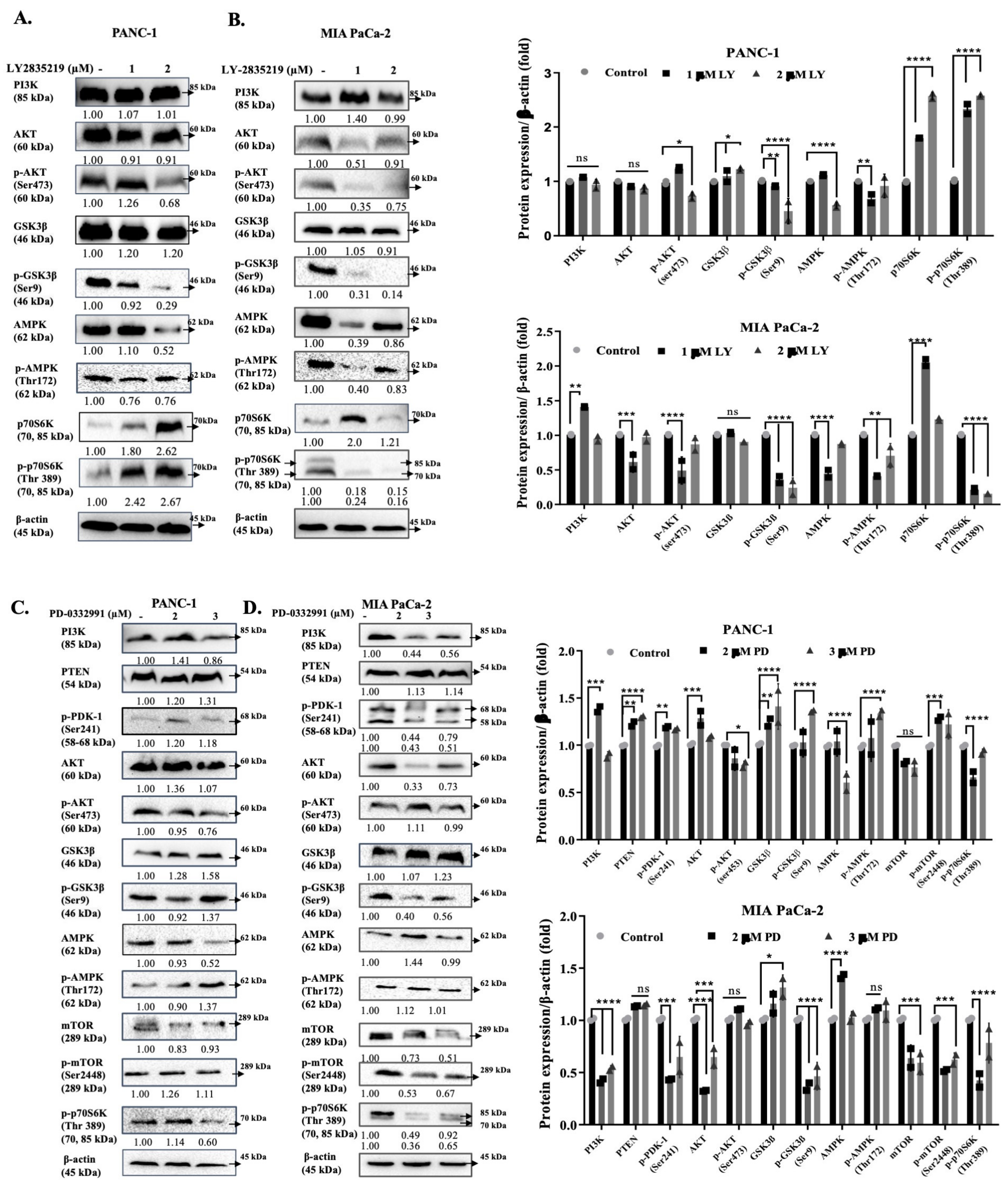
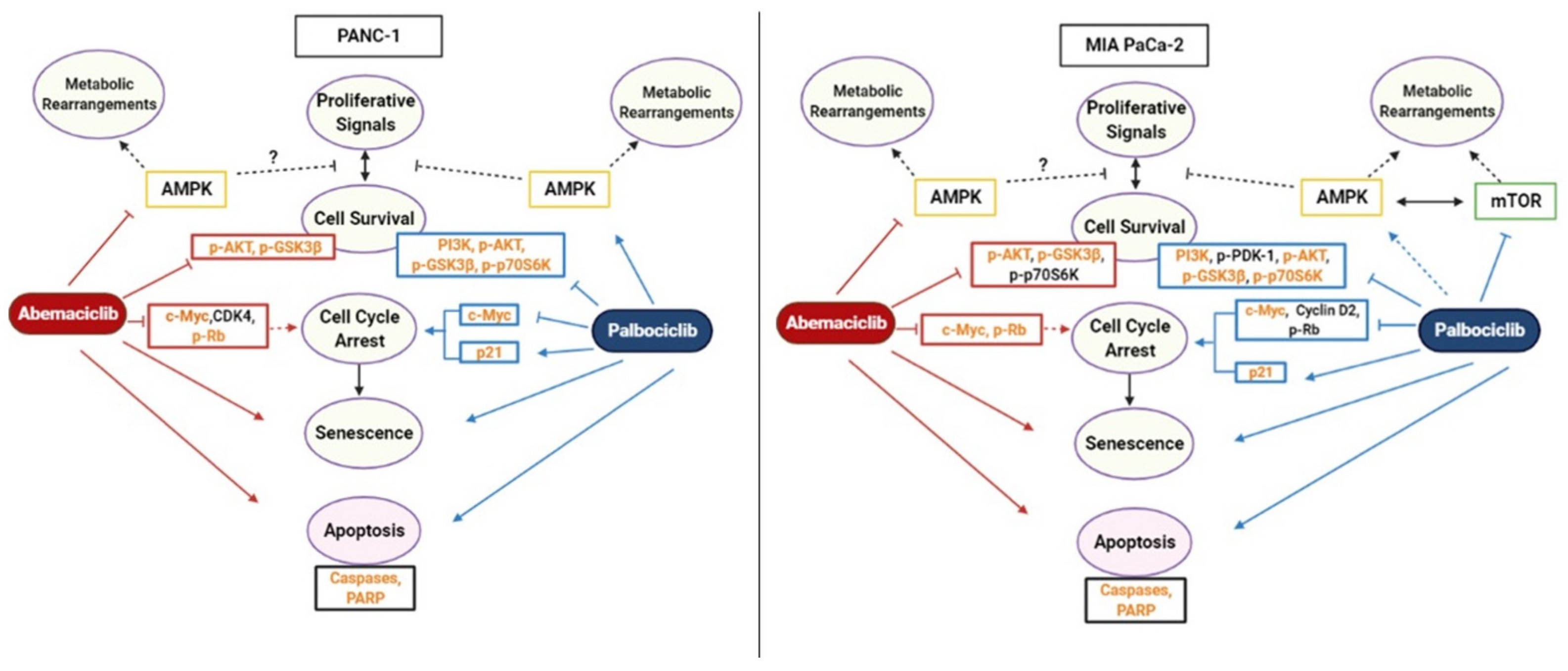
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Reagents
4.2. Cell Viability Assay
4.3. Colony Formation Assay
4.4. Determination of Apoptotic Cell Death and Cell Survival by Fluorescent Microscopy and Caspase 3/7 Activity Assay
4.5. Senescence β-Galactosidase Staining
4.6. Cell Cycle Analysis by Flow Cytometry
4.7. Determination of Cell Death by Annexin V/PI Analysis
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mihaljevic, A.L.; Michalski, C.W.; Friess, H.; Kleeff, J. Molecular mechanism of pancreatic cancer—Understanding proliferation, invasion, and metastasis. Langenbeck’s Arch. Surg. 2010, 395, 295–308. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Gordon-Dseagu, V.L.; Devesa, S.S.; Goggins, M.; Stolzenberg-Solomon, R. Pancreatic cancer incidence trends: Evidence from the Surveillance, Epidemiology and End Results (SEER) population-based data. Int. J. Epidemiol. 2018, 47, 427–439. [Google Scholar] [CrossRef]
- Conway, J.R.; Herrmann, D.; Evans, T.J.; Morton, J.P.; Timpson, P. Combating pancreatic cancer with PI3K pathway inhibitors in the era of personalised medicine. Gut 2019, 68, 742–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Young, R.J.; Waldeck, K.; Martin, C.; Foo, J.H.; Cameron, D.P.; Kirby, L.; Do, H.; Mitchell, C.; Cullinane, C.; Liu, W.; et al. Loss of CDKN2A expression is a frequent event in primary invasive melanoma and correlates with sensitivity to the CDK4/6 inhibitor PD0332991 in melanoma cell lines. Pigment. Cell Melanoma Res. 2014, 27, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Flanders, T.Y.; Pollock, P.M.; Haywardt, N.K.; Gene, C. The CDKN2A (pl6) Gene and Human Cancer. Molecular Med. 1997, 3, 5–20. [Google Scholar] [CrossRef] [Green Version]
- Regel, I.; Kong, B.; Raulefs, S.; Erkan, M.; Michalski, C.W.; Hartel, M.; Kleeff, J. Energy metabolism and proliferation in pancreatic carcinogenesis. Langenbeck’s Arch. Surg. 2012, 397, 507–512. [Google Scholar] [CrossRef]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T. Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 tumor suppressor gene: Important milestones at the various steps of tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Fajas, L.; Lopez-Mejia, I.C. CDK4, a new metabolic sensor that antagonizes AMPK. Mol. Cell. Oncol. 2018, 5, e1409862. [Google Scholar] [CrossRef]
- Lopez-Mejia, I.C.; Lagarrigue, S.; Giralt, A.; Martinez-Carreres, L.; Zanou, N.; Denechaud, P.D.; Castillo-Armengol, J.; Chavey, C.; Orpinell, M.; Delacuisine, B.; et al. CDK4 Phosphorylates AMPKα2 to Inhibit Its Activity and Repress Fatty Acid Oxidation. Mol. Cell 2017, 68, 336–349.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogarty, S.; Hardie, D.G. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim. Biophys. Acta Proteins Proteom. 2010, 1804, 581–591. [Google Scholar] [CrossRef]
- Hardie, D.G.; Iwadate, Y.; Yumura, S. The AMP-activated protein kinase pathway—New players upstream and downstream. J. Cell Sci. 2004, 117, 5479–5487. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Qian, W.; Li, J.; Jiang, Z.; Cheng, L.; Yan, B.; Cao, J.; Sun, L.; Zhou, C.; Lei, M. Loss of AMPK activation promotes the invasion and metastasis of pancreatic cancer through an HSF1-dependent pathway. Mol. Oncol. 2017, 11, 1475–1492. [Google Scholar] [CrossRef]
- Iriana, S.; Ahmed, S.; Gong, J.; Annamalai, A.A.; Tuli, R.; Hendifar, A.E. Targeting mTOR in pancreatic ductal adenocarcinoma. Front. Oncol. 2016, 6, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Cretella, D.; Ravelli, A.; Fumarola, C.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Alfieri, R.; Biondi, A.; Generali, D.; Bonelli, M.; et al. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef]
- Murthy, D.; Attri, K.S.; Singh, P.K. Phosphoinositide 3-kinase signaling pathway in pancreatic ductal adenocarcinoma progression, pathogenesis, and therapeutics. Front. Physiol. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Bondar, V.M.; Sweeney-Gotsch, B.; Andreeff, M.; Mills, G.B.; McConkey, D.J. Inhibition of the phosphatidylinositol 3′-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol. Cancer Ther. 2002, 1, 989–997. [Google Scholar]
- El-Masry, O.S.; Al-Sakkaf, K.; Brown, B.L.; Dobson, P.R. Differential crosstalk between the AMPK and PI3K/Akt pathways in breast cancer cells of differing genotypes: Leptin inhibits the effectiveness of AMPK activation. Oncol. Rep. 2015, 34, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Han, F.; Li, C.F.; Cai, Z.; Zhang, X.; Jin, G.; Zahang, W.N.; Xu, C.; Wang, C.-Y.; Morrow, J.; Zhang, S.; et al. The critical role of AMPK in driving Akt activation under stress, tumorigenesis and drug resistance. Nat. Commun. 2018, 9, 4728. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Gong, J.; Luo, X.; Zang, M.; Guo, W.; Wen, R.; Luo, Z. AMPK exerts dual regulatory effects on the PI3K pathway. J. Mol. Signal. 2010, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, W.W.; Kurokawa, M. Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer. Cancer Drug Resist. 2019, 3, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.; Xu, J.; Sun, T. Cyclin-dependent kinases 4/6 inhibitors in breast cancer: Current status, resistance, and combination strategies. J. Cancer 2019, 10, 5504–5517. [Google Scholar] [CrossRef] [PubMed]
- Jingwen, B.; Yaochen, L.; Guojun, Z. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar] [CrossRef]
- Eggersmann, T.K.; Degenhardt, T.; Gluz, O.; Wuerstlein, R.; Harbeck, N. CDK4/6 Inhibitors Expand the Therapeutic Options in Breast Cancer: Palbociclib, Ribociclib and Abemaciclib. BioDrugs 2019, 33, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Franco, J.; Witkiewicz, A.K.; Knudsen, E.S. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget 2014, 5, 6512–6525. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; McAllister, S.S.; Zhao, J.J. CDK4/6 Inhibition in Cancer: Beyond Cell Cycle Arrest. Trends Cell Biol. 2018, 28, 911–925. [Google Scholar] [CrossRef]
- Marra, A.; Curigliano, G. Are all cyclin-dependent kinases 4/6 inhibitors created equal? NPJ Breast Cancer 2019, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Frederick, M.O.; Lowery, C.D.; Shackleford, T.; Renschler, M.; Stephens, J.; Flack, R.; Blosser, W.; Gupta, S.; Stewart, J.; Webster, Y.; et al. Abemaciclib is Active in Preclinical Models of Ewing’s Sarcoma via Multi-pronged Regulation of Cell Cycle, DNA Methylation, and Interferon Pathway Signaling. Clin. Cancer Res. 2018, 12, 151–161. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, F.S.; Chen, Y.L.; Hung, M.H.; Chu, P.Y.; Tsai, M.H.; Chen, L.J.; Hsiao, Y.J.; Shih, C.T.; Chang, M.J.; Chao, T.I.; et al. Palbociclib induces activation of AMPK and inhibits hepatocellular carcinoma in a CDK4/6-independent manner. Mol. Oncol. 2017, 11, 1035–1049. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Wang, Y.; Li, S.; Wang, Y.; Tan, P.; He, J.; Chen, Y.; Deng, H.; Huang, W.; Lin, X.; et al. AMPK/TSC2/mTOR pathway regulates replicative senescence of human vascular smooth muscle cells. Exp. Ther. Med. 2018, 16, 4853–4858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [Green Version]
- Law, M.E.; Corsino, P.E.; Narayan, S.; Law, B.K. Cyclin-dependent kinase inhibitors as anticancer therapeutics. Mol. Pharmacol. 2015, 88, 846–852. [Google Scholar] [CrossRef] [Green Version]
- Musgrove, E.A.; Caldon, C.E.; Barraclough, J.; Stone, A.; Sutherland, R.L. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer 2011, 11, 558–572. [Google Scholar] [CrossRef] [PubMed]
- Dhir, T.; Schultz, C.W.; Jain, A.; Brown, S.Z.; Haber, A.; Goetz, A.; Xi, C.; Su, G.H.; Xu, L.; Posey, J., 3rd; et al. Abemaciclib is effective against pancreatic cancer cells and synergizes with HuR and YAP1 inhibition. Mol. Cancer Res. 2019, 17, 2029–2041. [Google Scholar] [CrossRef] [Green Version]
- Bonelli, M.; La Monica, S.; Fumarola, C.; Alfieri, R. Multiple effects of CDK4/6 inhibition in cancer: From cell cycle arrest to immunomodulation. Biochem. Pharmacol. 2019, 170, 113676. [Google Scholar] [CrossRef] [PubMed]
- Skowron, M.A.; Vermeulen, M.; Winkelhausen, A.; Becker, T.K.; Bremmer, F.; Petzsch, P.; Schönberger, S.; Calaminus, G.; Köhrer, K.; Albers, P.; et al. CDK4/6 inhibition presents as a therapeutic option for paediatric and adult germ cell tumours and induces cell cycle arrest and apoptosis via canonical and non-canonical mechanisms. Br. J. Cancer 2020, 123, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Tu, C.; Cheng, X.; Xu, Z.; Wang, X.; Shen, J.; Chai, K.; Chen, W. Inflammatory and Senescent Phenotype of Pancreatic Stellate Cells Induced by Sqstm1 Downregulation Facilitates Pancreatic Cancer Progression. Int. J. Biol. Sci. 2019, 15, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 24, 503–511. [Google Scholar] [CrossRef]
- Duong, H.Q.; Hwang, J.S.; Kim, H.J.; Seong, Y.S.; Bae, I. BML-275, an AMPK inhibitor, induces DNA damage, G2/M arrest and apoptosis in human pancreatic cancer cells. Int. J. Oncol. 2012, 41, 2227–2236. [Google Scholar] [CrossRef] [Green Version]
- Rencuzogulları, O.; Yerlikaya, P.O.; Gürkan, A.Ç.; Arısan, E.D.; Telci, D. Palbociclib, a selective CDK4/6 inhibitor, restricts cell survival and epithelial-mesenchymal transition in Panc-1 and MiaPaCa-2 pancreatic cancer cells. J. Cell. Biochem. 2020, 121, 508–523. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, L.; Zhao, S.; Dicker, D.T.; El-Deiry, W.S. The CDK4/6 inhibitor palbociclib synergizes with irinotecan to promote colorectal cancer cell death under hypoxia. Cell Cycle 2017, 16, 1193–1200. [Google Scholar] [CrossRef]
- Liu, C.Y.; Lau, K.Y.; Hsu, C.C.; Chen, L.J.; Lee, C.H.; Huang, T.T.; Chen, Y.T.; Huang, C.T.; Lin, P.H.; Tseng, L.M. Combination of palbociclib with enzalutamide shows in vitro activity in RB proficient and androgen receptor positive triple negative breast cancer cells. PLoS ONE 2017. [Google Scholar] [CrossRef] [Green Version]
- Bollard, J.; Miguela, V.; De Galarreta, M.R.; Venkatesh, A.; Bian, C.B.; Roberto, M.P.; Tovar, V.; Sia, D.; Molina-Sánchez, P.; Nguyen, C.B.; et al. Palbociclib (PD-0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2016, 66, 1286–1296. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.S.; Karasic, T.B.; DeMichele, A.; Vaughn, D.J.; O’Hara, M.; Perini, R.; Zhang, P.; Lal, P.; Feldman, M.; O’Dwyer, P.J. Palbociclib (PD0332991)—A selective and potent cyclin-dependent kinase inhibitor: A review of pharmacodynamics and clinical development. JAMA Oncol. 2016, 2, 253–260. [Google Scholar] [CrossRef]
- Li, P.; Zhang, X.; Gu, L.; Zhou, J.; Deng, D. P16 methylation increases the sensitivity of cancer cells to the CDK4/6 inhibitor palbociclib. PLoS ONE 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- García-Reyes, B.; Kretz, A.L.; Ruff, J.P.; von Karstedt, S.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; Lemke, J. The emerging role of cyclin-dependent kinases (CDKs) in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2018, 19, 3219. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.S.; Kelly, K.; Paz-Ares, L.G.; Garrido, P.; Jalal, S.; Mahadevan, D.; Gutierrez, M.; Provencio, M.; Schaefer, E.; Shaheen, M.; et al. Abemaciclib in Combination with Single-Agent Options in Patients with Stage IV Non–Small Cell Lung Cancer: A Phase Ib Study. Clin. Cancer Res. 2018, 24, 5543–5551. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Modrak, D.E.; Leon, E.; Goldenberg, D.M.; Gold, D.V. Ceramide regulates gemcitabine-induced senescence and apoptosis in human pancreatic cancer cell lines. Mol. Cancer Res. 2009, 7, 890–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Senescence, cellular senescence, and cancer. Ann. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular Senescence in Cancer and Senescence. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, C.A.; Vargas, L.; Martinez, V.; Bravo, S.; Brown, N.E. Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp. Cell Res. 2017, 360, 390–396. [Google Scholar] [CrossRef]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naz, S.; Sowers, A.; Choudri, R.; Wissler, M.; Gamson, J.; Mathias, A.; Cook, J.A.; Mitchell, J.B. Abemaciclib, a selective CDK4/6 inhibitor, enhances the radiosensitivity of non–small cell lung cancer in vitro and in vivo. Clin. Cancer Res. 2018, 24, 3994–4005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knudsen, E.S.; Kumarasamy, V.; Ruiz, A.; Sivinski, J.; Chung, S.; Grant, A.; Vail, P.; Chauhan, S.S.; Jie, T.; Riall, T.S.; et al. Cell cycle plasticity driven by MTOR signaling: Integral resistance to CDK4/6 inhibition in patient-derived models of pancreatic cancer. Oncogene 2019, 38, 3355–3370. [Google Scholar] [CrossRef]
- Hay, N. Interplay between FOXO, TOR, and Akt. Biochim. Biophys. Acta BBA Bioenerg. 2011, 1813, 1965–1970. [Google Scholar] [CrossRef] [Green Version]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 2–9. [Google Scholar] [CrossRef]
- Perugini, R.A.; McDade, T.P.; Vittimberga, F.J.; Callery, M.P. Pancreatic cancer cell proliferation phosphatidylinositol 3-kinase dependent. J. Surg. Res. 2000, 90, 39–44. [Google Scholar] [CrossRef]
- Orgován, Z.; Keserű, G.M. Small molecule inhibitors of RAS proteins with oncogenic mutations. Cancer Metastasis Rev. 2020, 39, 1107–1126. [Google Scholar] [CrossRef]
- Georgescu, M.-M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, H.Y.; Elpek, G.; Stephanie, A.V.; Zimmerman, M.; Gerald, C.; Chu, H.Y.; Fletcher-Sananikone, E.; Zhang, H.; Liu, Y.; Wang, W.; et al. Pten a major tumor supressor in PDAC. Cancer Discov. 2011, 1, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Michaloglou, C.; Crafter, C.; Siersbaek, R.; Delpuech, O.; Curwen, J.O.; Carnevalli, L.S.; Staniszewska, A.D.; Polanska, U.M.; Cheraghchi-Bashi, A.; Lawson, M.; et al. Combined inhibition of mtor and CDK4/6 is required for optimal blockade of e2f function and long-term growth inhibition in estrogen receptors—Positive breast cancer. Mol. Cancer Ther. 2018, 17, 908–920. [Google Scholar] [CrossRef] [Green Version]
- Litchfield, L.M.; Boehnke, K.; Brahmachary, M.; Mur, C.; Bi, C.; Stephens, J.R.; Sauder, J.M.; Gutiérrez, S.M.; McNulty, A.M.; Ye, X.S.; et al. Combined inhibition of PIM and CDK4/6 suppresses both mTOR signaling and Rb phosphorylation and potentiates PI3K inhibition in cancer cells. Oncotarget 2020, 11, 1478–1492. [Google Scholar] [CrossRef]
- Kang, J.I.; Hong, J.Y.; Lee, H.J.; Bae, S.Y.; Jung, C.; Park, H.J.; Lee, S.K. Anti-Tumor Activity of Yuanhuacine by Regulating AMPK/mTOR Signaling Pathway and Actin Cytoskeleton Organization in Non-Small Cell Lung Cancer Cells. PLoS ONE 2015, 10, e0144368. [Google Scholar] [CrossRef] [Green Version]
- Romero-Pozuelo, J.; Figlia, G.; Kaya, O.; Martin-Villalba, A.; Teleman, A.A. Cdk4 and Cdk6 Couple the Cell-Cycle Machinery to Cell Growth via mTORC1. Cell Rep. 2020, 31, 107504. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Guo, X.; Li, W.; Zhang, H. Activation of Wnt/β-catenin signalling via GSK3 inhibitors direct differentiation of human adipose stem cells into functional hepatocytes. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Korur, S.; Huber, R.M.; Sivasankaran, B.; Petrich, M.; Jr, P.M.; Hemmings, B.A.; Merlo, A.; Lino, M.M. GSK3β Regulates Differentiation and Growth Arrest in Glioblastoma. PLoS ONE 2009, 4, e7443. [Google Scholar] [CrossRef]
- Tanaka, Y.; Momose, S.; Tabayashi, T.; Sawada, K.; Yamashita, T.; Higashi, M.; Sagawa, M.; Tokuhira, M.; Rosenwald, A.; Kizaki, M.; et al. Abemaciclib, a CDK4/6 inhibitor, exerts preclinical activity against aggressive germinal center-derived B-cell lymphomas. Cancer Sci. 2020, 111, 749–759. [Google Scholar] [CrossRef] [Green Version]
- Torres-Guzmán, R.; Calsina, B.; Hermoso, A.; Baquero, C.; Alvarez, B.; Amat, J.; McNulty, A.M.; Gong, X.; Boehnke, K.; Du, J.; et al. Preclinical characterization of abemaciclib in hormone receptor positive breast cancer. Oncotarget 2017, 8, 69493–69507. [Google Scholar] [CrossRef] [Green Version]
- Laroche-Clary, A.; Chaire, V.; Algeo, M.P.; Derieppe, M.A.; Loarer, F.L.; Italiano, A. Combined targeting of MDM2 and CDK4 is synergistic in dedifferentiated liposarcomas. J. Hematol. Oncol. 2017, 10, 123. [Google Scholar] [CrossRef] [Green Version]
- Hino, H.; Iriyama, N.; Kokuba, H.; Kazama, H.; Moriya, S.; Takano, N.; Hiramoto, M.; Aizawa, S.; Miyazawa, K. Abemaciclib induces atypical cell death in cancer cells characterized by formation of cytoplasmic vacuoles derived from lysosomes. Cancer Sci. 2020, 111, 2132–2145. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Liu, X.; Dagda, R.K.; Zhang, Y. How AMPK and PKA interplay to regulate mitochondrial function and survival in models of ischemia and diabetes. Oxid. Med. Cell. Longev. 2017, 2017, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a parp-1 and nuclear factor-κB-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arisan, E.D.; Rencuzogullari, O.; Coban, M.; Sevgin, B.; Obakan-Yerlikaya, P.; Çoker-Gürkan, A.; Palavan-Unsal, N. The role of the PI3K/AKT/mTOR signaling axis in the decision of the celastrol-induced cell death mechanism related to the lipid regulatory pathway in prostate cancer cells. Phytochem. Lett. 2020, 39, 73–83. [Google Scholar] [CrossRef]
- Yuedi, D.; Houbao, L.; Pinxiang, L.; Hui, W.; Min, T.; Dexiang, Z. KLF2 induces the senescence of pancreatic cancer cells by cooperating with FOXO4 to upregulate p21. Exp. Cell Res. 2020, 388, 111784. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sevgin, B.; Coban, M.N.; Rencuzogullari, Ö.; Coker-Gurkan, A.; Obakan-Yerlikaya, P.; Uysal Onganer, P.; Arisan, E.D. AMPK Is the Crucial Target for the CDK4/6 Inhibitors Mediated Therapeutic Responses in PANC-1 and MIA PaCa-2 Pancreatic Cancer Cell Lines. Stresses 2021, 1, 48-68. https://0-doi-org.brum.beds.ac.uk/10.3390/stresses1010005
Sevgin B, Coban MN, Rencuzogullari Ö, Coker-Gurkan A, Obakan-Yerlikaya P, Uysal Onganer P, Arisan ED. AMPK Is the Crucial Target for the CDK4/6 Inhibitors Mediated Therapeutic Responses in PANC-1 and MIA PaCa-2 Pancreatic Cancer Cell Lines. Stresses. 2021; 1(1):48-68. https://0-doi-org.brum.beds.ac.uk/10.3390/stresses1010005
Chicago/Turabian StyleSevgin, Bortecine, Merve Nur Coban, Özge Rencuzogullari, Ajda Coker-Gurkan, Pinar Obakan-Yerlikaya, Pinar Uysal Onganer, and Elif Damla Arisan. 2021. "AMPK Is the Crucial Target for the CDK4/6 Inhibitors Mediated Therapeutic Responses in PANC-1 and MIA PaCa-2 Pancreatic Cancer Cell Lines" Stresses 1, no. 1: 48-68. https://0-doi-org.brum.beds.ac.uk/10.3390/stresses1010005