
Diversity and Potential Function of Prokaryotic and Eukaryotic Communities from Different Mangrove Sediments

1
Key Laboratory of Estuarine Ecological Security and Environmental Health, School of Environmental Science and Engineering, Xiamen University Tan Kah Kee College, Zhangzhou 363105, China
2
Guangdong Provincial Key Laboratory of Fishery Ecology and Environment, South China Sea Fisheries Research Institute, Chinese Academy of Fisheries Sciences, Guangzhou 510300, China
3
State Key Laboratory of Marine Environmental Science, College of the Environment and Ecology, Xiamen University, Xiamen 361005, China
*
Author to whom correspondence should be addressed.
Sustainability 2022, 14(6), 3333; https://0-doi-org.brum.beds.ac.uk/10.3390/su14063333
Submission received: 18 January 2022
/
Revised: 3 March 2022
/
Accepted: 8 March 2022
/
Published: 12 March 2022
(This article belongs to the Special Issue Future Directions for Soil Remediation and Environmental Management)
Abstract
:Mangrove trees generally play important roles in protecting intertidal ecosystems. The mangrove root-associated sediments provide a repertoire of microbial communities that contribute to pivotal ecological functions in the system. In the present study, we used the high-throughput sequencing and PICRUSt-predicted functional information (based on 16S/18S rDNA profiles) to investigate the bacterial, archaeal, and fungal communities in two mangrove systems, located in the estuary of the Jiulong River (China), with different contaminated conditions and frequencies of human activity. Diverse distribution patterns for microbial communities were observed in six sediment samples collected from the two survey areas, which were found to be related mainly to the substrates in mangrove sediments. The sediments were predominated by relatively higher ratios of heterotrophic bacteria that participated in the degradation of organic matters, including phylum of Chloroflexi, Acidobacteriota, Desulfobacterota, and Proteobacteria. In addition, Crenarchaeota and Ascomycota presented the highest abundances of archaea and fungi, respectively. The relatively high concentrations of calcium, nitrogen, magnesium, and phosphorus in mangrove sediments correlated significantly with the microbial communities. In addition, although the potential functions were similar in the two sites based on COG and KEGG pathways, the abundances of enzymes involved in the degradation processes of cellulose and hemicellulose and the metabolism of nitrogen and sulfur presented distinctions. These results provide insights into the environmental conditions shaping microbial assemblies of the mangrove sediments under the impacts of human activities; for instance, a more abundant amount of calcium was found in urban areas in this study.
1. Introduction
The intertidal ecosystems are generally known as the interacted regions of the ocean, atmosphere, and terrestrial environments and are believed to be the most momentous coastal habitats in view of their biological productivity and economic value [1]. Mangroves are well known as the dominant flora in intertidal zones such as coastal lagoons, coastlines, and estuaries, where the hydrological conditions are relatively complicated [2,3]. Nonetheless, the unique characteristics of mangroves provide them the ability to accommodate the dynamic environments of intertidal regions and, in addition, to protect coastal environments, aquacultures, and living conditions for numerous organisms [4]. Moreover, mangrove sediments and trees along the coastlines have also been planted to efficiently absorb most of the substrates possessed in contaminated soils and wastewater over the last 40 years [5,6,7].
Similar to the plants in terrestrial environments, mangroves also rely on the reciprocal benefits with various microbial assemblies [8]. Compared with sediments of bare intertidal flats, sediments from mangrove ecosystems comprise abundant organic carbon produced by a litter of mangrove plants, root exudates, and sedimentary fragments of phytoplanktons [9], providing adequate ecological niches for the development of microbial communities’ composition and function [10,11]. Active organic matters are secreted by roots of mangrove plants to subsurface sediments, which lead to higher microbial abundances and relevant activities in rhizosphere sediments than those of neighboring bulk sediments [9]. In addition, the microbial communities were found to be distinguishing between the inner and outer regions of mangrove sediments [12]. Therefore, elucidating the interactions or relationships between the mangroves and microbial communities is of great importance in revealing the mechanisms involved in the biological functions of mangrove forests. The collaborations of mangroves and microbial communities in sediments were found to drive the maintenances of biodiversity, community stability, and ecosystem functioning [13]. Microbial communities, either bacteria or fungi, have been reported to function importantly in decomposition processes of organic carbon, as well as other substrates in the sediments of mangroves [14,15,16]. High-throughput sequencing of 16S rDNA and 18S rDNA genes have been applied in studying the prokaryotic and eukaryotic communities, respectively, and their metabolic potentials in sediments of mangroves [17,18]. However, even though several general patterns between the mangroves and related microbial communities were reported, a few studies were conducted to investigate the diversity and functions of microorganisms under different contaminated sediments of mangroves.
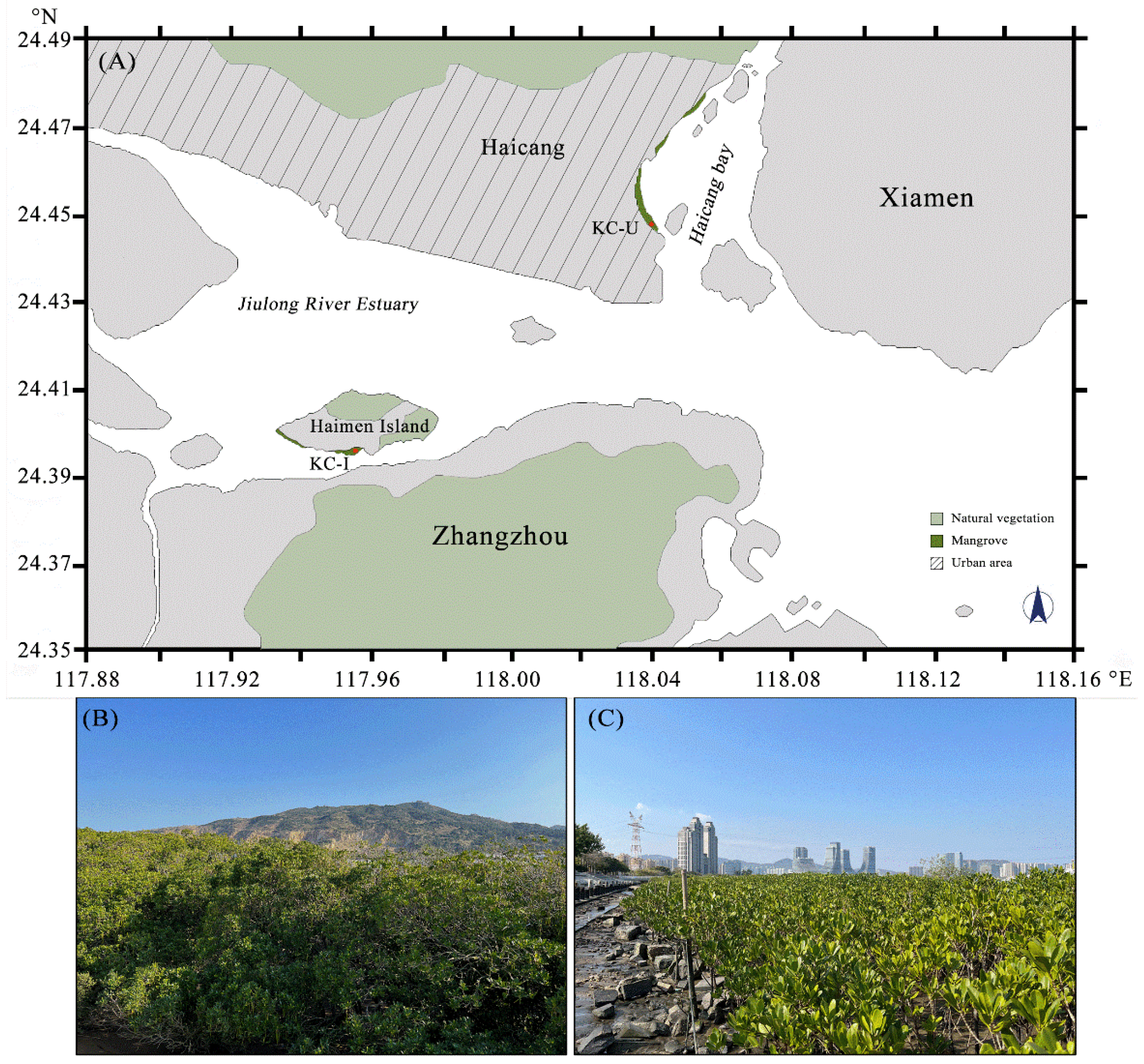
Jiulong River is the second largest river in Fujian Province, China. The estuary of the river presents a complicated hydrological condition owing to the influence of the tide and is a typical wetland where mangroves are the dominant vegetation. Many studies have focused on the environmental conditions and organisms inhabited in this region. However, the diversities and potential functions of prokaryotic and eukaryotic communities in mangrove sediments were seldom investigated. In this study, the mangrove systems, under different contaminated conditions and frequency of human activities, were selected as the survey areas where grew the same mangrove species, Kandelia candel, to ensure consistency. The KC-U site was located in an urban area with relatively high human impact, while the KC-I site was located in Haimen Island in the estuary with aquacultures around. Sequencing of both 16S and 18S rDNA was carried out to reveal the compositions of microbial communities, and how they were influenced by environmental factors in the sediments of mangrove ecosystems. Based on 16S rDNA profiles, we finally predicted the potential functions that were distinct (e.g., degradation activities of cellulose and hemicellulose and functional enzymes involved in nitrogen and sulfur metabolism) between the two areas.
2. Materials and Methods
2.1. Sample Collections
Two different locations, where K. candel is the primary mangrove plant, were selected as the sampling sites in the present study. The sediment samples KC-I were collected from Haimen Island located at the Jiulong River Estuary, Fujian Province, China (KC-I: 117°57′ E, 24°24′ N, Figure 1A). Samples KC-U were gathered from Haicang Bay in the western sea area of Xiamen, Fujian Province, China (KC-U: 118°2′ E, 24°27′ N, Figure 1A). Due to the pivotal driving function of temperature in shaping microbial communities in sediments [19,20,21], the samplings were conducted on 10 and 12 November 2020 (18–24 °C) to reduce the influence of temperature. All samples were collected from the uppermost 10–20 cm of sediment near the roots of K. candel. For each location, three biological replicates were sampled. The sediment samples were then transported to the laboratory and pre-filtered with a 2 mm pore-size sieve (with the purpose of removing debris, stones, large metazoans, and grains), followed by being separated as two groups. One group was air-dried at room temperature for the determination of environmental parameters and the other group was stored at −20 °C until DNA isolation.
2.2. Environmental Parameters Measurement
The physical and chemical parameters of the sediment samples were measured. The concentration analysis of total nitrogen and ammonium (NH4-N) was carried out using the Kjeldahl method in terms of digestion and distillation [22,23]. The determination of total phosphorus was implemented by the molybdate colorimetric measurement using the 721-spectrophotometer. For the total organic carbon (Corg) concentration, the analysis was performed with dichromate oxidation, followed by titration with acidified ferrous ammonium sulfate [24]. Samples were also undertaken to determine the amount of sediment sodium, potassium, calcium, and magnesium by inductively coupled plasma atomic emission (ICP–AES) spectroscopy. In terms of pH, a digital pH meter (pH6+, Thermo Scientific, Waltham, MA, USA) was applied to measure the values.
2.3. DNA Extraction, PCR, and Illumina Sequencing
Extraction of DNA was conducted using the FastDNA SPIN extraction kit for soil (MP Biomedicals, Santa Ana, California, USA). The V3-V4 hypervariable region of prokaryotic 16S rDNA was amplified utilizing the primers 515 F (5′-GTGYCAGCMGCCGCGGTAA-3′) and 806 R (5′-GGACTACNVGGGTWTCTAAT-3′) [25]. The eukaryotic 18S rDNA was amplified using the primers SSU0817 F (5′-TTAGCATGGAATAATRRAATAGGA-3′) and 1196 R (5′-TCTGGACCTGGTGAGTTTCC-3′) [26]. Each DNA sample was individually PCR-amplified. All PCR reactions were carried out in 30 μL reactions with 15 μL of Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM of forward and reverse primers, and about 10 ng template DNA. Thermal cycling was as follows: initial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, elongation at 72 °C for 60 s, and finally, 72 °C for 5 min. Negative PCR controls without template DNA were included for the reactions. Subsequently, all amplicons were sequenced on a single run using the Illumina MiSeq 2x300 bp platform (Illumina, San Diego, CA, USA).
2.4. Sequence Assembly, Clustering, and Annotations
After the separation of raw data to each sample based on the barcodes, both barcodes and primer sequences were removed. Separated raw data were then merged into raw tags with the software FLASH (V1.2.11, https://0-ccb-jhu-edu.brum.beds.ac.uk/software/FLASH/index.shtml, accessed date: 10 April 2021) [27], followed by the filtrations of the raw tags by quality filters processed using QIIME (V1.9.1, http://qiime.org/index.html, accessed date: 14 April 2021) [28]. Filtered tags were then classified into operational taxonomic units (OTUs) at a similarity of 0.97 with the removal of chimera using USEARCH (V7.0 http://drive5.com/uparse/, accessed date: 19 April 2021) [29]. OTUs from all sequencing datasets were annotated accordingly, using the RDP classifier (V11.5, http://rdp.cme.msu.edu/accessed date: 22 April 2021) [30] confronted against the Silva (release 128) [31] by a confidence threshold of 0.7. The OTUs with ambiguous annotation or unclassified results by one of the given taxonomic groups were presented as “others”.
2.5. Predicted Profiles of Functions
On the basis of 16S rDNA data, the potential functional information was reestablished for bacterial and archaeal assemblages from the mangrove sediments using the software PICRUSt [32]. Closed-reference 97% OTU picking was conducted and then normalized by PICRUSt for each OTU. For all samples, estimable gene families were predicted from metagenomic predictions according to the Cluster of Orthologous Groups (COGs) and KEGG Orthology (KO). The nearest sequenced taxon index (NSTI) represents the accuracy of prediction and the lower values generally suggest better accuracies. Predicted KO terms were classified into the second hierarchy of the KEGG pathway. Based on the KEGG identification, the pathways, comprising enzymes that participated in the biological degradation of (hemi-)cellulose and the metabolic processes of nitrogen and sulfur, were then acquired. According to the 18S rDNA data, functional classifications of fungi were analyzed using FUNGuild [33].
2.6. Statistical Analysis
For the analysis of α-diversity, the community diversity parameters (Shannon index) were calculated using the Mothur software (V1.30.2, https://www.mothur.org/wiki/Download_mothur, accessed date: 9 May 2021) [34], and the rarefaction curves were drawn by R (version 4.1.0). A two-tailed Student’s t-test was conducted to test whether the environmental factors of each location were significantly discrepant. The results of α-diversity were subsequently compared using one-way ANOVA and Student’s t-test. For β-diversity analysis, principal component analysis (PCA) was operated in R. The analysis of similarity (ANOSIM) was applied to statistically test for significant variations in assemblages between the two sites. Mantel tests were performed in R using the “vegan” package to obtain the correlations between the environmental factors and the microbial communities (based on Bray–Cutis similarity). For environmental parameters, Euclidean distance matrices were calculated via the R-based dist. function. Partial Mantel tests were also performed between class richness and environmental factors.
3. Results and Discussion
3.1. Overview of the Sediments from the Survey Region
The investigation was carried out in the offshore area, as described above, from 10 November 2020 to 12 November 2020, and the sediment samples from the mangrove forest of K. candel were collected. Based on the subsequent determinations of physical and chemical parameters, the results shown in Table 1 revealed several differences between the sampling sites. The concentrations of magnesium and calcium in KC-I were much lower than that in KC-U. By contrast, the concentrations of nitrogen and phosphorus in KC-I were obviously higher, which may be the result of the aquaculture around the estuary of Jiulong River. Moreover, discrepancies were also observed for the concentrations of organic carbon and pH between the two sampling regions, presenting an inverse relationship. Previous studies reported that the total organic carbon and pH in various natural environments generally have a negative correlation owing to their innate internal relationship [35,36]. The decomposition of a relatively high abundance of organic carbon could reduce the environmental pH by producing organic acids [37]. This was consistent with the adverse concentrations of organic carbon and pH in this study, also indicating that the sediments from Haimen Island were more fertile.
3.2. Diversity and Distribution of Prokaryotes and Eukaryotes
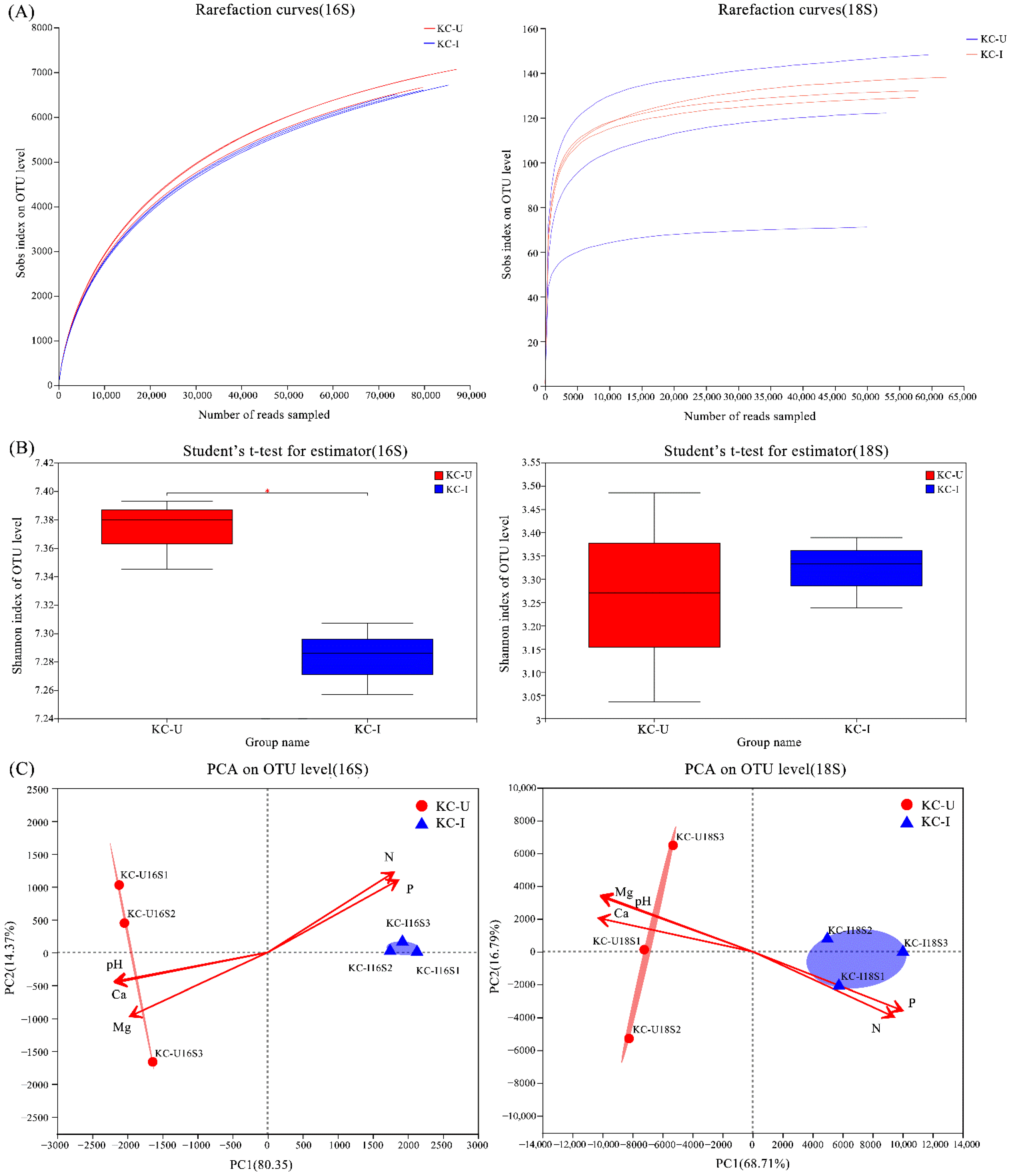
For prokaryotic microbial communities, 16s rDNA sequencing obtained 586,767 high-quality sequences, which were clustered subsequently into a total of 11,460 OTUs. As for eukaryotic microorganisms, 18s rDNA sequencing produced 349,972 high-quality sequences, which were then clustered totally into 300 OTUs. The rarefaction curves were basically saturated for each sample (Figure 2A). According to the results of Mann–Whitney U test, the Shannon index value of prokaryotes samples in KC-U was significantly higher (p ≤ 0.05) than that in KC-I (Figure 2B). In β-diversity analysis, PCA results for prokaryotes and eukaryotes both suggested that the communities shifted across the two areas by partitioning of the sites (Figure 2C). According to the scatter plots of the first two axes, these four variables illustrated two sampling locations with distinct environmental factors at both organismal levels: the KC-U site was associated with the concentration of magnesium, calcium, and pH, while the KC-I station was related to the concentrations of nitrogen and phosphorus.
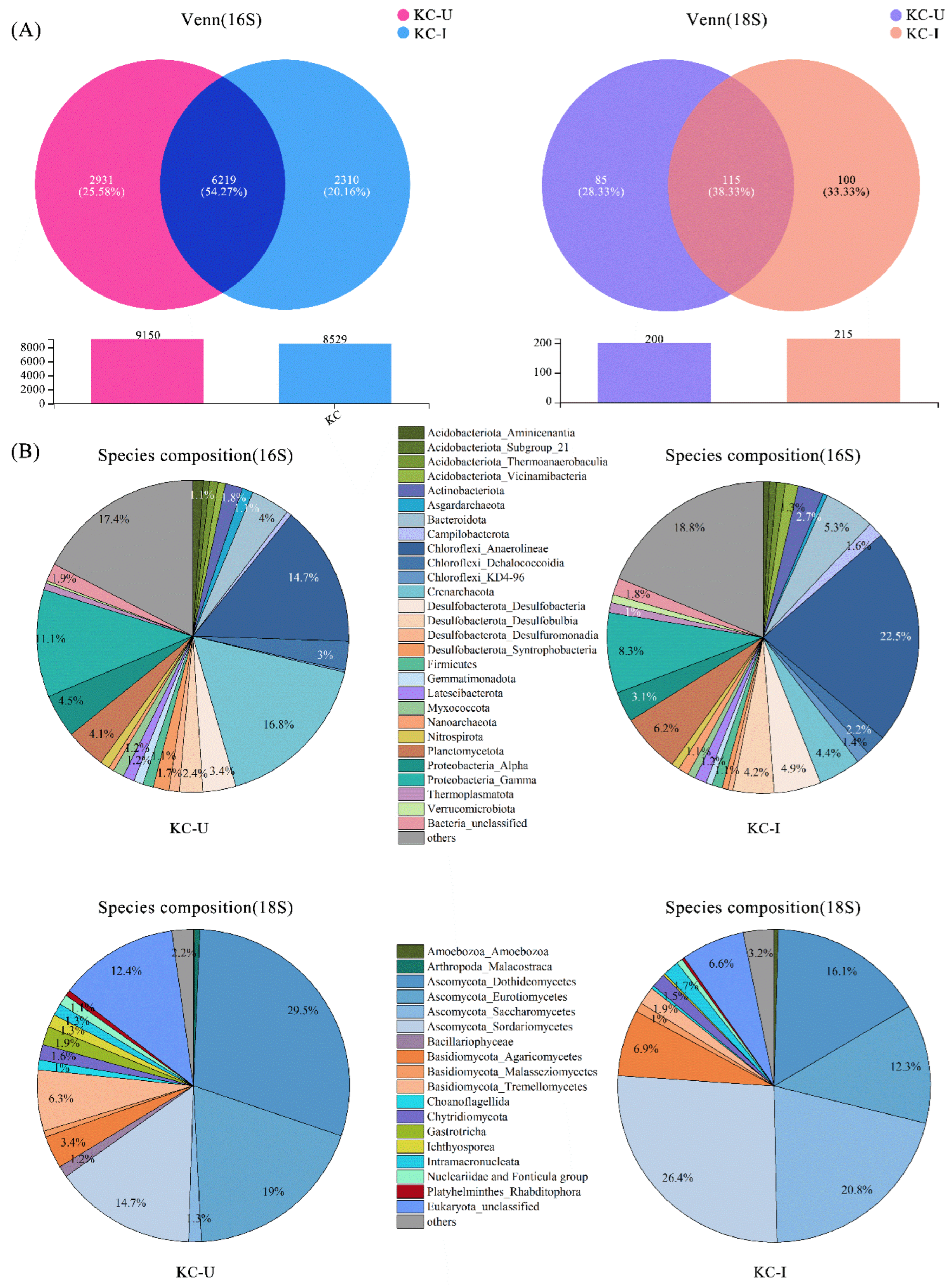
The relative abundances of prokaryotic and eukaryotic classifications in the two locations presented different patterns (Figure 3). The abundance analysis of OTUs at 16S rDNA level showed that a total of 6219 (54.27%) OTUs were detected in both samples, and 2931 (25.58%) and 2310 (20.16%) OTUs were observed exclusively from KC-U and KC-I, respectively (Figure 3A). At the 18S rDNA level, the results showed that a total of 115 (38.33%) OTUs were detected in both samples, and 85 (28.33%) and 100 (33.33%) OTUs existed exclusively in KC-U and KC-I, respectively (Figure 3A).
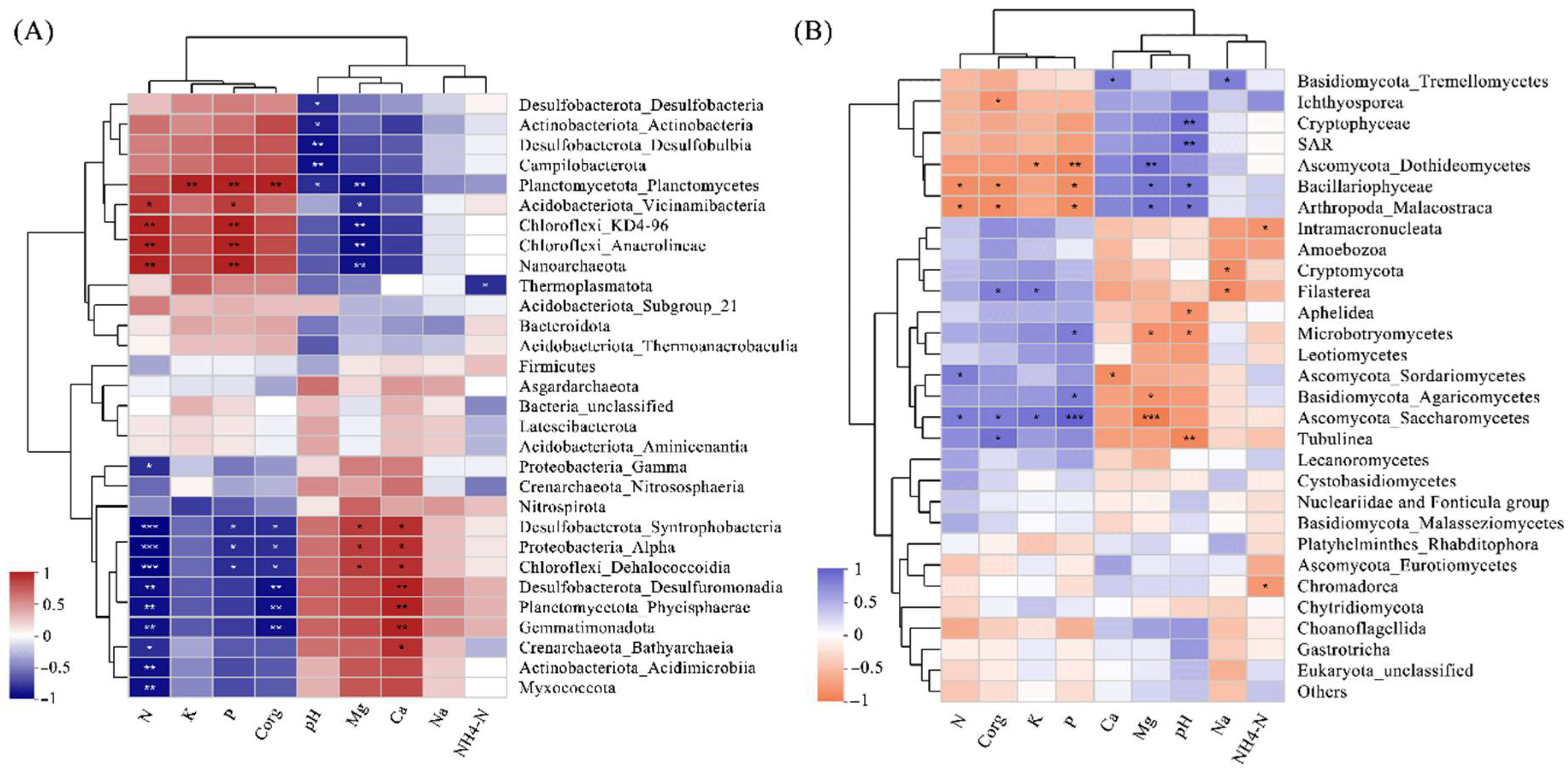
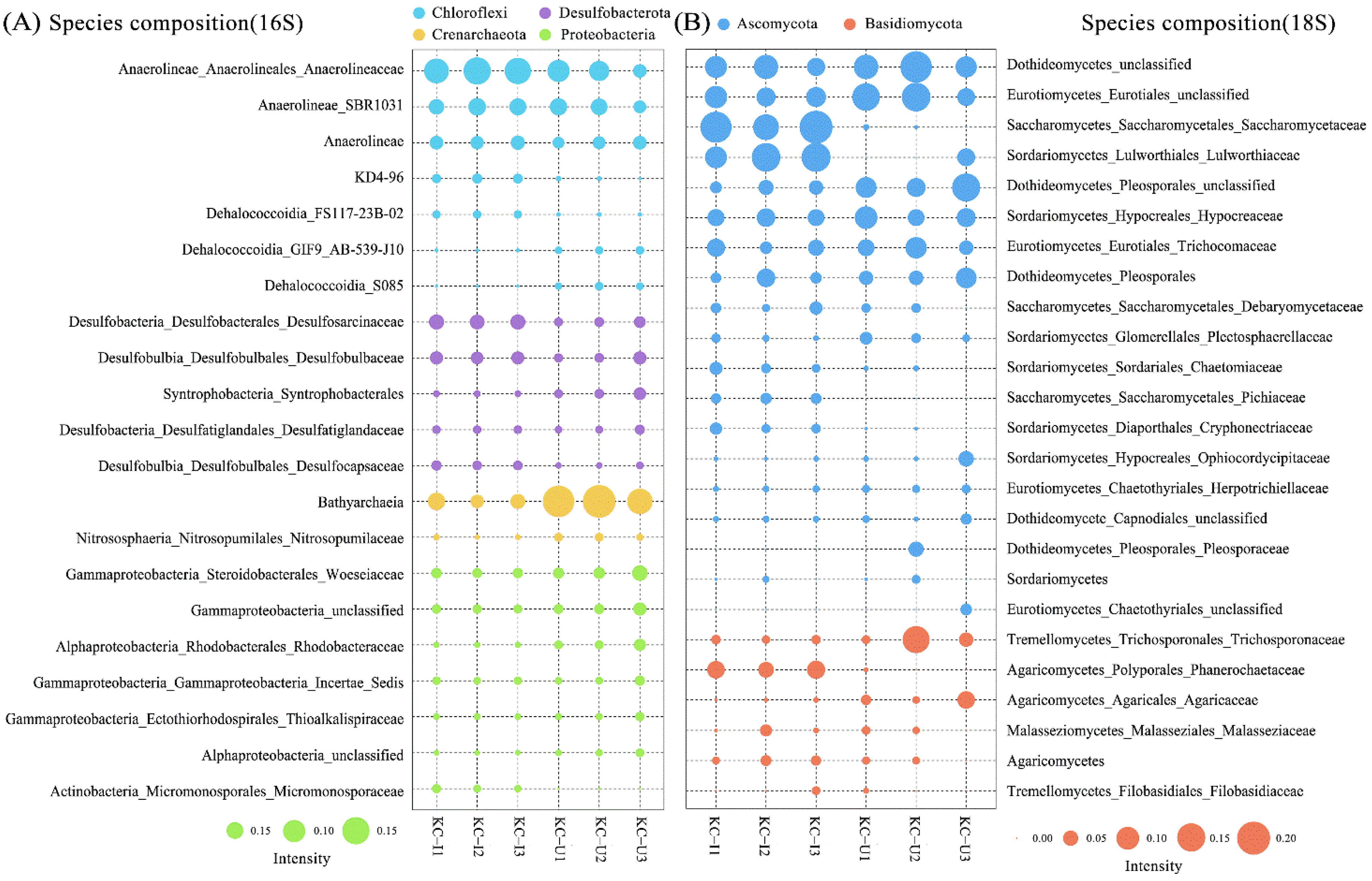
Among the prokaryotic communities, bacteria accounted for 85.5% of the total sequences, comprising 79.3% and 91.7% in KC-U and KC-I, respectively. The account of archaea in KC-U (20.6%) was more than in KC-I (8.3%). The diversities of bacteria communities were similar between the KC-U and KC-I sites (Figure 3B). At the taxonomic order of phylum, the most abundant group from the two locations was Chloroflexi, of which the Anaerolineae was the dominant class. Chloroflexi was also detected in other studies focused on the mangrove root-associated microbial communities, while it usually presented relatively low abundances [16,38,39,40]. The class Anaerolineae, as high abundant anaerobic bacteria in mangrove sediments, functioned in the biological reactions of reducing dissimilatory nitrate to ammonium under facultative hypoxia environments and made contributions to the generation of ammonia nitrogen [41]. Moreover, the heterotrophic bacteria Anaerolineae was reported to occupy higher proportions in the upper soil zones of mangroves and participate in the degradation of organic compounds [9]. In this study, the relative abundance of Anaerolineae was positively correlated with organic carbon, nitrogen, and phosphorus, indicating a potential promotion of Anaerolineae by these substrates, especially in the KC-I site, with higher concentrations of nitrogen and phosphorus discharged from nearby aquaculture (Figure 4A). Moreover, Acidobacteriota, Desulfobacterota, and Proteobacteria also exhibited high abundances in the investigated areas, which was consistent with previous studies [39,42]. These results suggested that Chloroflexi, Acidobacteriota, Desulfobacterota, and Proteobacteria might constitute the core prokaryotic communities of mangrove sediments. On the other hand, the Bacteroidota was identified in both regions. Generally, Bacteroidota was considered as the specialists during the degradation of macromolecules in the mangrove sediments, such as complex polysaccharides and proteins, to acquire carbon, amino acids, and sulfur [43]. In terms of archaea, Crenarchaeota was observed in both sites, consistent with the fact that Crenarchaeota were usually found to predominate among the archaeal communities in subsurface sediments of mangroves [17]. In addition, the majority of obtained Crenarchaeota belonged to Bathyarchaeota class, a worldwide distributed taxon in anoxic sediments [44]. The abundance of Bathyarchaeota was suggested to be related to the total organic carbon [45,46]. However, in this study, Bathyarchaeota was detected to show higher abundances from KC-U (16.8%) than that from KC-I (4.4%), in which the total organic carbon was relatively lower. Further investigations might be required to elucidate this feature. Moreover, several other archaea such as Euryarchaeota and Asgardaeota were found in this study. These groups function possibly in transporting and assimilating peptides in mangrove sediments, but they were observed as the minor phylum in both sites. Further analysis at the classification level of family was focused on several dominant phyla mentioned above. In Chloroflexi, the family Anerolineaceae presented the highest abundances, followed by SBR1031, both of which belonged to the order Anaerolineae. Unlike this, diverse subgroups at the family level showed similar abundances in both phyla of Desulfobacterota and Proteobacteria. Moreover, only the family Nitrosopumilaceae from Crenarchaeota was identified with a relatively lower content, while no detailed classification information was obtained for Bathyarchaeia (Figure 5A).
As for the eukaryotes in mangrove sediments, fungi communities were primarily focused. Fungi accounted for 82.78% of total sequences based on the 18S rDNA sequencing, comprising 77.0% and 88.5% in KC-U and KC-I, respectively. The composition of eukaryotic communities differed obviously between the two sites (Figure 3B). At the level of phylum, Ascomycota was the dominant taxonomic group in both sites, while the class compositions of Ascomycota were different. Among the subdivided class, Dothideomycetes and Eurotiomycetes exhibited a higher proportion of eukaryotic communities in KC-U, while the Sordariomycetes were the dominant class in KC-I. Moreover, the Saccharomycetes present higher abundance in KC-I (20.8%) than that in KC-U (1.3%). Previous investigations reported that the groups Dothideomycetes and Sordariomycetes were detected to be highly abundant in the mangrove leaves and soils [15,39,47]. Saccharomycetes was detected mainly in the rhizosphere soil compartment [39] or as the dominant group (99.5%) in polycyclic aromatic hydrocarbons (PAHs) contaminated mangrove forest [48]. Members from the Saccharomycetes class grew frequently in anthropogenically polluted sites [49] and could enhance the bioavailability and degradability of PAHs [50]. Therefore, the more abundant Saccharomycetes detected from KC-I suggested a possible higher PAHs pollution in mangrove sediments from Haimen Island. In addition, microbial assembly in Ascomycota showed a high diversity of compositions at the family level, as shown in Figure 5B. The family Saccaromycetaceae and Lulworthiaceae, which presented higher content in KC-I, were the dominant subclusters in the order of Saccharomycetes and Sordariomycetes, respectively. For Dothideomycetes, almost no family group with high abundances was observed at the family level. In the phylum Basidiomycota, the class with the highest abundances was Agaricomycetes from the KC-I site, while for the KC-U site, the dominant class was Tremellomycetes. On the other hand, the multiformity of communities in Basidiomycota was relatively lower than that in Ascomycota. Detection of the phyla described above was consistent with the previous studies on mangrove ecosystems, indicating potential usages of these fungi from mangrove sediments for bioremediation [51,52].
3.3. Environmental Factors Shaping the Microbiome Communities
A Mantel test was conducted to ascertain the influence of each environmental parameter on constructing the microbiome communities in the selected locations of mangrove forest. Among the different factors, calcium exhibited the relatively highest impact on the communities at the eukaryotic and prokaryotic levels (Table 2). Calcium is generally required to maintain cell structure, motility, and cell division, and plays an important role in the permeability of ions, sugar, and amino acids [53,54]. The high correlation values between calcium and microbiome communities were found in samples of mangroves sediments from the urban area [55]. Coincidentally, the KC-U site was also located in the urban area and presented high correlations with calcium (Figure 2C). These results suggest the possibility that the microbiome communities of mangrove sediments might be influenced indirectly by human activities, which resulted in more discharge of calcium near the urban area. Moreover, nitrogen, magnesium, and phosphorus also presented less significant correlations, indicating that they may have potential contributions to shaping the composition of microbial communities. Generally, these macronutrients are of high relevance to the activity and growth of microorganisms [14,56].
For prokaryotic groups, numbers of prokaryotic communities were significantly influenced by nitrogen, especially the taxonomic classes of Syntrophobacteria, alpha-proteobacteria, and Dehalococcoidia (Figure 4A). Additionally, phosphorus, organic carbon, magnesium, and calcium showed either positive or negative correlations with several groups, such as Desulfuromonadia, Phycisphaerae, Gemmatimonadota, etc. It is worth mentioning that pH only presented a negative correlation with several classes from prokaryotic communities. In terms of eukaryotic communities, Dothideomycetes and Saccharomycetes presented significant correlations with magnesium and phosphorus (Figure 4B) but with opposite trends. According to the abundances of Dothideomycetes and Saccharomycetes, shown in Figure 3B, the reversed relationship indicated their diverse requirements of the magnesium and phosphorus in mangrove sediments. Another class of Ascomycota, Sordariomycetes, which differed from Dothideomycetes and Saccharomycetes, presented a low correlation with the environmental factors.
3.4. Microbial Functional Prediction
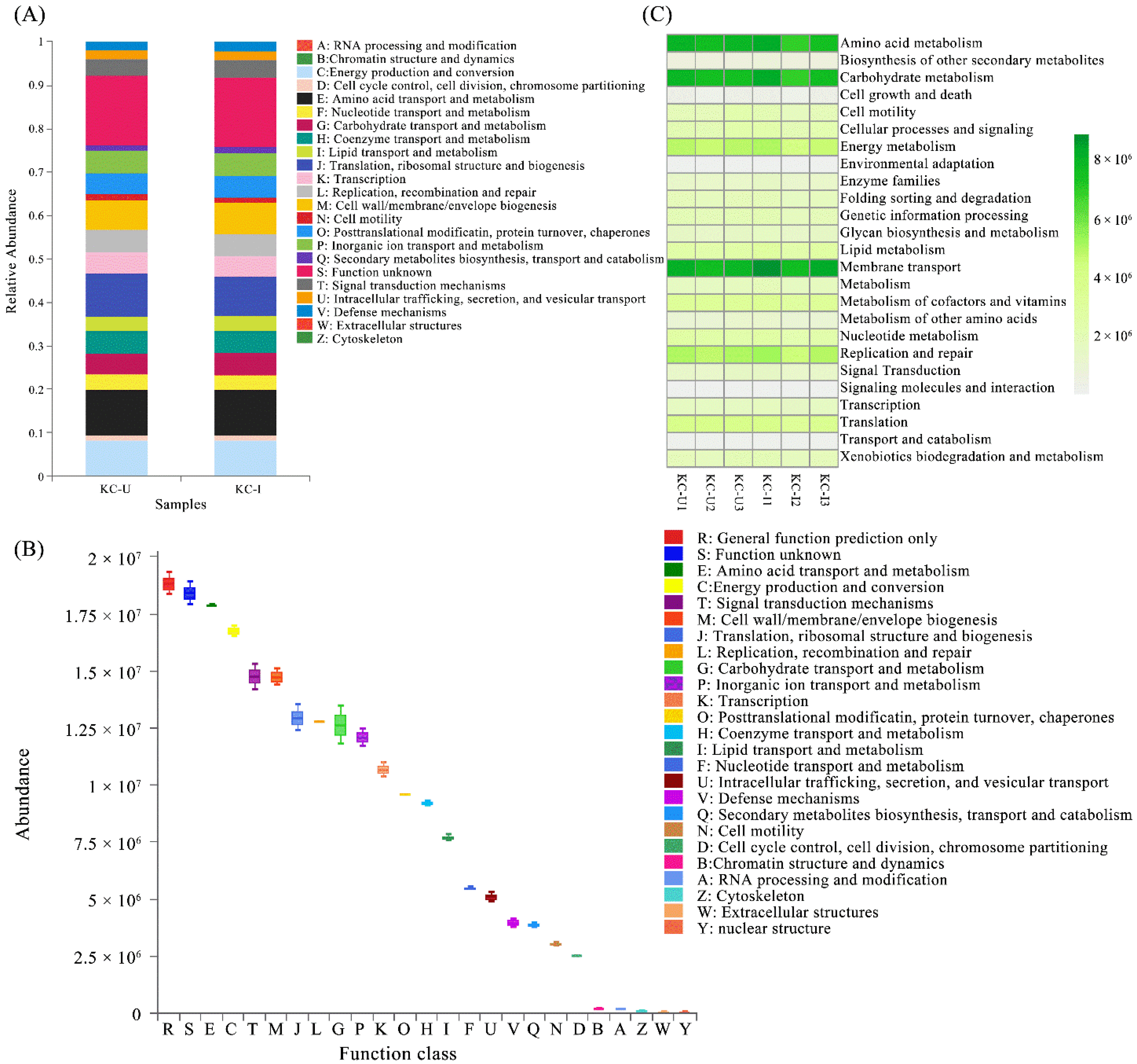
PICRUSt and FUNGuild software programs were utilized to predict the functional profiles of microorganisms from the two sites based on the clusters of orthologs groups database. The PICRUSt-based NSTI values of the sediments were higher than 0.03 (NSTI = 0.14 ± 0.01), and therefore, the prediction of microbiome functions and pivotal genes suggest a general indicator of the trends in community functions on the prokaryotic level. PICRUSt assignment of predicted metagenomics contents at the level of COGs indicated a similar functional pattern between KC-U and KC-I sites (Figure 6A). Based on the integration of OTUs from both sites, the main prokaryotic community functions in the mangrove sediments were found to participate mainly in amino acid transport and metabolism, energy production and conversion, and signal transduction and mechanisms (Figure 6B). The results of FUNGuild analysis assigned all the OTUs to a total of 7 trophic modes and 19 guilds, of which the dominant trophic mode was Saprotroph, with a percentage of 48.23%, followed by Pathotroph–Saprotroph–Symbiotroph (2.29%) and Pathotroph (1.99%) (Supplementary Table S1). The high abundance of fungal groups belonging to Saprotroph was expectable in view of the growth conditions in mangrove ecosystems. Moreover, the compositions of fungal trophic modes were similar to the situation reported, and almost all guilds showed no significant differences between the two sites [57].
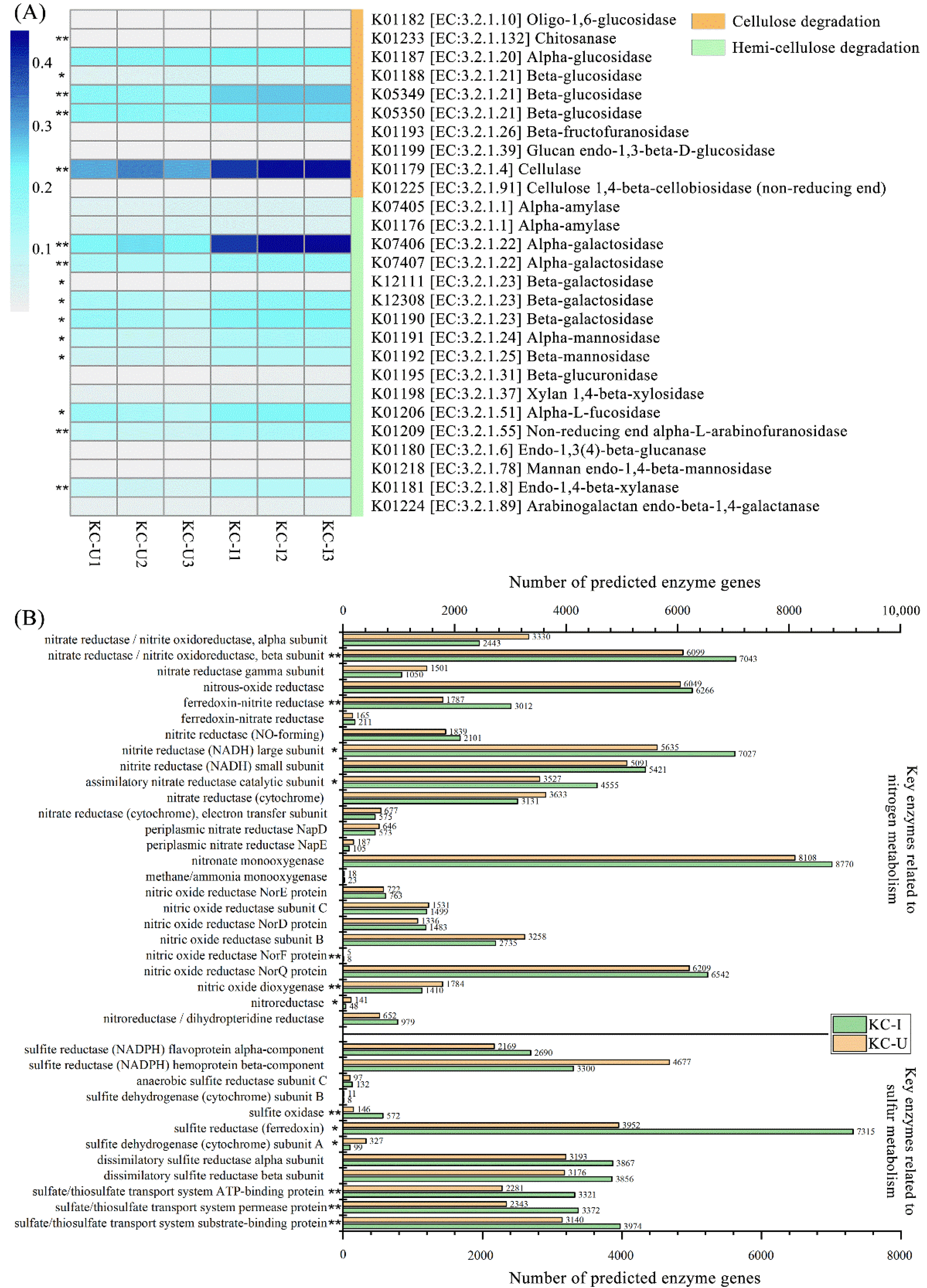
In addition, the results of KEGG pathways also suggested that the metabolic processes related to the prokaryotic community functions were similar between the two sites (Figure 6C). In terms of the decomposition of (hemi-)cellulose, which is generally abundant in the sediments of mangrove wetland, many enzymes involved in the degradation of them were detected (Figure 7A). The abundances of these enzymes presented higher abundances in KC-I than that in KC-U, especially the enzymes such as beta-glucosidase (EC:3.2.1.21), cellulose (EC:3.2.1.4), α-galactosidase (EC:3.2.1.22), and β-galactosidase (EC:3.2.1.23). The more plentiful enzymes in KC-I samples might suggest a more active degradation of (hemi-)celluloses in the KC-I site, even if the two sites exhibited a similar pattern of carbohydrate metabolism based on KEGG pathways.
Furthermore, the analyses of key enzymes in the metabolism of nitrogen and sulfur were carried out (Figure 7B). The most abundant enzymes related to nitrogen metabolism were the nitrate reductase, nitrous-oxide reductase, nitrite reductase (NADH) subunit, and nitronate monooxygenase. The first three enzymes are involved generally in denitrification or dissimilatory nitrate reduction, coupled to the pathways of oxidizing organic compounds. In this study, the abundances of these enzymes exhibited higher levels in KC-I, which might be related to the relatively higher concentration of organic carbon in KC-I than that in KC-U. Nitronate monooxygenase is flavin mononucleotide (FMN)-dependent that oxidizes (anionic) alkyl nitronates with dioxygen and, in the case of the enzyme from Neurospora crassa, (neutral) nitroalkanes to the corresponding carbonyl compounds and nitrite [58]. A previous study revealed that the high abundance of nitronate monooxygenase could effectively promote the nitrification of NH4-N and reduce the overlying water and porewater in the sediments [59]. However, less attention has been paid to this enzyme in mangrove sediments. On the other hand, less than 30 genes annotated as ammonia monooxygenase was observed in both sites. The low abundance of ammonia monooxygenase might be caused by the periodic anaerobic condition of the mangrove sediments, due to the strict aerobic requirement of ammonia oxidization [60]. In addition, most of the enzymes related to sulfur metabolism were identified to participate in the processes of sulfur reduction, which occurs constantly at low dissolved oxygen conditions (Figure 7B). The number of oxidation genes, such as sulfite oxidase, could be reduced by the low dissolved oxygen conditions in the mangrove sediments, which are conducive to inhibiting the production of black and smelly compounds including FeS and H2 S [59].
4. Conclusions
This study revealed the main prokaryotic and eukaryotic groups participating in the degradation of organic compounds and absorbing most of the substrates in mangrove sediments under different contaminated conditions and frequencies of human activities. In terms of the communities from bacteria, the phyla Chloroflexi, Acidobacteriota, Desulfobacterota, and Proteobacteria exhibited high abundances in the mangrove sediments. At the class level of these phyla, the heterotrophic bacteria Anaerolineae were the dominant groups in the mangrove sediments from the KC-I site under conditions of aquaculture and less human impact. In addition, the Crenarchaeota and Ascomycota presented the highest abundances of archaea and fungi, respectively, participating in diverse metabolic processes in the mangrove sediments with different environmental conditions. Among the environmental factors, calcium had the highest impact on microbial communities, which might be influenced by human activities in the urban area. The substrates, including nitrogen, magnesium, and phosphorus in mangrove sediments, were also observed to have potential contributions to shaping the compositions of microbial communities. Furthermore, results indicated that the predictive potential functions utilizing COG and KEGG pathways were similar in the two sites. However, the enzymes of interest were identified that predominated differentially in the two regions, which might be related to the relatively higher concentration of organic carbon in KC-I sites. Results in the present study indicated that the diversities and potential roles of microbial communities in mangrove sediments experience different conditions and effects of human activities. The microbial communities played positive roles in accommodating contaminated intertidal ecosystems. Nonetheless, more investigations are needed to investigate the spatial and temporal compositions of the sediments’ communities in broader, more extensive regions.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/su14063333/s1, Table S1: Abundances of predicted functional classifications of fungal communities.
Author Contributions
Conceptualization, Y.Z. and H.G.; software, Y.Z. and S.Z.; validation, S.Z. and C.L.; formal analysis, Y.Z., H.G. and C.L.; writing—original draft preparation, Y.Z. and H.G.; writing—review and editing, Y.Z., H.G., S.Z. and C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially supported by research grants from the National Natural Science Foundation of China (Project No. 41806124), the Fund of Guangdong Provincial Key Laboratory of Fishery Ecology and Environment (FEEL-2020-10), and Guangdong Basic and Applied Basic Research Foundation (Grant Number 2020 A1515010331).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The raw data presented in this study are openly available in the NCBI Sequence Read Archive (SRA) under accession number PRJNA797991.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ortega-Morales, B.O.; Chan-Bacab, M.J.; De la Rosa-García, S.D.C.; Camacho-Chab, J.C. Valuable processes and products from marine intertidal microbial communities. Curr. Opin. Biotechnol. 2010, 21, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Donato, D.C.; Kauffman, J.B.; Murdiyarso, D.; Kurnianto, S.; Stidham, M.; Kanninen, M. Mangroves among the most carbon-rich forests in the tropics. Nat. Geosci. 2011, 4, 293–297. [Google Scholar] [CrossRef]
- Maher, D.T.; Santos, I.R.; Golsby-Smith, L.; Gleeson, J.; Eyre, B.D. Groundwater-derived dissolved inorganic and organic carbon exports from a mangrove tidal creek: The missing mangrove carbon sink? Limnol. Oceanogr. 2013, 58, 475–488. [Google Scholar] [CrossRef]
- Giri, C.; Ochieng, E.; Tieszen, L.L.; Zhu, Z.; Singh, A.; Loveland, T.; Masek, J.; Duke, N. Status and distribution of mangrove forests of the world using earth observation satellite data. Glob. Ecol. Biogeogr. 2011, 20, 154–159. [Google Scholar] [CrossRef]
- Amaral, V.; Penha-Lopes, G.; Paula, J. Effects of vegetation and sewage load on mangrove crab condition using experimental mesocosms. Estuar. Coast. Shelf Sci. 2009, 84, 300–304. [Google Scholar] [CrossRef]
- Ouyang, X.; Guo, F. Paradigms of mangroves in treatment of anthropogenic wastewater pollution. Sci. Total Environ. 2016, 544, 971–979. [Google Scholar] [CrossRef]
- Ouyang, X.; Guo, F. Intuitionistic fuzzy analytical hierarchical processes for selecting the paradigms of mangroves in municipal wastewater treatment. Chemosphere 2018, 197, 634–642. [Google Scholar] [CrossRef]
- De Paula, N.M.; da Silva, K.; Brugnari, T.; Haminiuk, C.W.I.; Maciel, G.M. Biotechnological potential of fungi from a mangrove ecosystem: Enzymes, salt tolerance and decolorization of a real textile effluent. Microbiol. Res. 2022, 254, 126899. [Google Scholar] [CrossRef]
- Zhu, P.; Wang, Y.; Shi, T.; Zhang, X.; Huang, G.; Gong, J. Intertidal zonation affects diversity and functional potentials of bacteria in surface sediments: A case study of the Golden Bay mangrove, China. Appl. Soil Ecol. 2018, 130, 159–168. [Google Scholar] [CrossRef]
- Bouillon, S.; Moens, T.; Overmeer, I.; Koedam, N.; Dehairs, F. Resource utilization patterns of epifauna from mangrove forests with contrasting inputs of local versus imported organic matter. Mar. Ecol. Prog. Ser. 2004, 278, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, K.; Dhal, N.K. Potential microbial diversity in mangrove ecosystems: A review. Indian J. Mar. Sci. 2009, 38, 249–256. [Google Scholar] [CrossRef]
- Jiang, X.-T.; Peng, X.; Deng, G.-H.; Sheng, H.-F.; Wang, Y.; Zhou, H.-W.; Tam, N.F.-Y. Illumina sequencing of 16S rRNA tag revealed spatial variations of bacterial communities in a mangrove wetland. Microb. Ecol. 2013, 66, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Wardle, D.A.; Bardgett, R.D.; Klironomos, J.N.; Setälä, H.; van der Putten, W.H.; Wall, D.H. Ecological linkages between aboveground and belowground biota. Science 2004, 304, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Purahong, W.; Kapturska, D.; Pecyna, M.J.; Jariyavidyanont, K.; Kaunzner, J.; Juncheed, K.; Uengwetwanit, T.; Rudloff, R.; Schulz, E.; Hofrichter, M.; et al. Effects of forest management practices in temperate beech forests on bacterial and fungal communities involved in leaf litter degradation. Microb. Ecol. 2015, 69, 905–913. [Google Scholar] [CrossRef]
- Yao, H.; Sun, X.; He, C.; Maitra, P.; Li, X.-C.; Guo, L.-D. Phyllosphere epiphytic and endophytic fungal community and network structures differ in a tropical mangrove ecosystem. Microbiome 2019, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.-J.; Pan, J.; Duan, C.-H.; Wang, Y.-M.; Liu, Y.; Sun, J.; Zhou, H.-C.; Song, X.; Li, M. Prokaryotic diversity in mangrove sediments across Southeastern China fundamentally differs from that in other biomes. Msystems 2019, 4, e00442-19. [Google Scholar] [CrossRef] [Green Version]
- Fiard, M.; Cuny, P.; Sylvi, L.; Hubas, C.; Jezequel, R.; Lamy, D.; Walcker, R.; El Houssainy, A.; Heimburger-Boavida, L.-E.; Robinet, T.; et al. Mangrove microbiota along the urban-to-rural gradient of the Cayenne estuary (French Guiana, South America): Drivers and potential bioindicators. Sci. Total Environ. 2022, 807, 150667. [Google Scholar] [CrossRef]
- Zainal Abidin, D.H.; Mohd Nor, S.A.; Lavoué, S.; Rahim, M.A.; Jamaludin, N.A.; Mohammed Akib, N.A. DNA-based taxonomy of a mangrove-associated community of fishes in Southeast Asia. Sci. Rep. 2021, 11, 17800. [Google Scholar] [CrossRef]
- Cheung, M.K.; Wong, C.K.; Chu, K.H.; Kwan, H.S. Community structure, dynamics and interactions of bacteria, archaea and fungi in subtropical coastal wetland sediments. Sci. Rep. 2018, 8, 14397. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Fang, A.; Yu, X.; Zhang, K.; He, Z.; Wang, C.; Peng, Y.; Xiao, F.; Yang, T.; Zhang, W.; et al. Microbially-driven sulfur cycling microbial communities in different mangrove sediments. Chemosphere 2021, 273, 128597. [Google Scholar] [CrossRef]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremner, J.M.; Breitenbeck, G.A. A simple method for determination of ammonium in semimicro-kjeldahl analysis of soils and plant materials using a block digester. Commun. Soil Sci. Plant Anal. 1983, 14, 905–913. [Google Scholar] [CrossRef]
- Bremner, J.M.; Tabatabai, M.A. Use of an ammonia electrode for determination of ammonium in Kjeldahl analysis of soils. Commun. Soil Sci. Plant Anal. 1972, 3, 159–165. [Google Scholar] [CrossRef]
- Walkley, A.; Black, I.A. An examination of the degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci. 1934, 37, 29–38. [Google Scholar] [CrossRef]
- Saenz, J.S.; Roldan, F.; Junca, H.; Arbeli, Z. Effect of the extraction and purification of soil DNA and pooling of PCR amplification products on the description of bacterial and archaeal communities. J. Appl. Microbiol. 2019, 126, 1454–1467. [Google Scholar] [CrossRef] [PubMed]
- Rousk, J.; Baath, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Schloss, P.D.; Gevers, D.; Westcott, S.L. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE 2011, 6, e27310. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Zhang, C. Investigating spatially varying relationships between total organic carbon contents and pH values in European agricultural soil using geographically weighted regression. Sci. Total Environ. 2021, 752, 141977. [Google Scholar] [CrossRef]
- Reisser, M.; Purves, R.S.; Schmidt, M.W.I.; Abiven, S. Pyrogenic carbon in soils: A literature-based inventory and a global estimation of its content in soil organic carbon and stocks. Front. Earth Sci. 2016, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Andersson, S.; Nilsson, S.I. Influence of pH and temperature on microbial activity, substrate availability of soil-solution bacteria and leaching of dissolved organic carbon in a mor humus. Soil Biol. Biochem. 2001, 33, 1181–1191. [Google Scholar] [CrossRef]
- Ghosh, A.; Bhadury, P. Investigating monsoon and post-monsoon variabilities of bacterioplankton communities in a mangrove ecosystem. Environ. Sci. Pollut. Res. Int. 2018, 25, 5722–5739. [Google Scholar] [CrossRef]
- Purahong, W.; Sadubsarn, D.; Tanunchai, B.; Wahdan, S.F.M.; Sansupa, C.; Noll, M.; Wu, Y.-T.; Buscot, F. First insights into the microbiome of a mangrove tree reveal significant differences in taxonomic and functional composition among plant and soil compartments. Microorganisms 2019, 7, 585. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, W.; Yu, X.; Hu, R.; Luo, Z.; Liu, X.; Zheng, X.; Xiao, F.; Peng, Y.; He, Q.; Tian, Y.; et al. Diversity, function and assembly of mangrove root-associated microbial communities at a continuous fine-scale. NPJ Biofilms Microbiomes 2020, 6, 52. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, Y.; Liu, J.; Yang, X.; Ren, Y.; Miao, H.; Pan, Y.; Lv, J.; Yan, G.; Ding, L.; et al. Exploring the mechanisms of organic matter degradation and methane emission during sewage sludge composting with added vesuvianite: Insights into the prediction of microbial metabolic function and enzymatic activity. Bioresour. Technol. 2019, 286, 121397. [Google Scholar] [CrossRef] [PubMed]
- Tongununui, P.; Kuriya, Y.; Murata, M.; Sawada, H.; Araki, M.; Nomura, M.; Morioka, K.; Ichie, T.; Ikejima, K.; Adachi, K. Mangrove crab intestine and habitat sediment microbiomes cooperatively work on carbon and nitrogen cycling. PLoS ONE 2021, 16, e0261654. [Google Scholar] [CrossRef]
- Sackett, J.D.; Kruger, B.R.; Becraft, E.D.; Jarett, J.K.; Stepanauskas, R.; Woyke, T.; Moser, D.P.; Bruno, V. Four draft single-cell genome sequences of novel, nearly identical Kiritimatiellaeota strains isolated from the continental deep subsurface. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Pan, J.; Wang, F.; Gu, J.-D.; Li, M. Bathyarchaeota: Globally distributed metabolic generalists in anoxic environments. FEMS Microbiol. Rev. 2018, 42, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Liang, Q.; Niu, M.; Wang, F. High occurrence of Bathyarchaeota (MCG) in the deep-sea sediments of South China Sea quantified using newly designed PCR primers. Environ. Microbiol. Rep. 2017, 9, 374–382. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, R.; Wang, H.; Gong, L.; Man, B.; Xu, Y. Distribution of bathyarchaeota communities across different terrestrial settings and their potential ecological functions. Sci. Rep. 2017, 7, 45028. [Google Scholar] [CrossRef]
- Chi, W.-C.; Chen, W.; He, C.-C.; Guo, S.-Y.; Cha, H.-J.; Tsang, L.M.; Ho, T.W.; Pang, K.-L. highly diverse fungal community associated with leaves of the mangrove plant Acanthus ilicifolius var. xiamenensis revealed by isolation and metabarcoding analyses. PeerJ 2019, 7, e7293. [Google Scholar] [CrossRef] [Green Version]
- Balu, S.; Bhunia, S.; Gachhui, R.; Mukherjee, J. Assessment of polycyclic aromatic hydrocarbon contamination in the Sundarbans, the world’s largest tidal mangrove forest and indigenous microbial mixed biofilm-based removal of the contaminants. Environ. Pollut. 2020, 266, 115270. [Google Scholar] [CrossRef]
- Aranda, E. Promising approaches towards biotransformation of polycyclic aromatic hydrocarbons with Ascomycota fungi. Curr. Opin. Biotechnol. 2016, 38, 1–8. [Google Scholar] [CrossRef]
- Zafra, G.; Absalón, Á.E.; Cuevas, M.D.C.; Cortés-Espinosa, D.V. Isolation and selection of a highly tolerant microbial consortium with potential for PAH biodegradation from heavy crude oil-contaminated soils. Water Air Soil Pollut. 2014, 225, 1–8. [Google Scholar] [CrossRef]
- Verma, A.K.; Raghukumar, C.; Verma, P.; Shouche, Y.S.; Naik, C.G. Four marine-derived fungi for bioremediation of raw textile mill effluents. Biodegradation 2010, 21, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Nazareth, S.W. Diversity of fungi from mangrove sediments of Goa, India, obtained by metagenomic analysis using Illumina sequencing. 3 Biotech 2019, 9, 164. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, D.C. Calcium signalling in bacteria. Mol. Microbiol. 2004, 54, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Pitt, D.; Ugalde, U.O. Calcium in fungi. Plant Cell Environ. 1984, 7, 467–475. [Google Scholar] [CrossRef]
- Mendes, L.W.; Taketani, R.G.; Navarrete, A.A.; Tsai, S.M. Shifts in phylogenetic diversity of archaeal communities in mangrove sediments at different sites and depths in southeastern Brazil. Res. Microbiol. 2012, 163, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Gadd, G.M. Metals, minerals and microbes: Geomicrobiology and bioremediation. Microbiology 2010, 156, 609–643. [Google Scholar] [CrossRef]
- Cao, M.; Cui, L.; Sun, H.; Zhang, X.; Zheng, X.; Jiang, J. Effects of spartina alterniflora invasion on soil microbial community structure and ecological functions. Microorganisms 2021, 9, 138. [Google Scholar] [CrossRef]
- Francis, K.; Russell, B.; Gadda, G. Involvement of a flavosemiquinone in the enzymatic oxidation of nitroalkanes catalyzed by 2-nitropropane dioxygenase. J. Biol. Chem. 2005, 280, 5195–5204. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Li, X.; Li, G.; Tang, W.; Li, C.; Li, J.; Zhao, C.; Du, C.; Liang, X.; Li, W.; et al. New insights into restoring microbial communities by side-stream supersaturated oxygenation to improve the resilience of rivers affected by combined sewer overflows. Sci. Total Environ. 2021, 782, 146903. [Google Scholar] [CrossRef]
- Herbert, R.A. Nitrogen cycling in coastal marine ecosystems. FEMS Microbiol. Rev. 1999, 23, 563–590. [Google Scholar] [CrossRef]
Figure 1.
(A) The locations of sampling sites. KC-U site: 118°2′ E, 24°27′ N; KC-I site: 117°57′ E, 24°24′ N. Diagonals and light green represented regions with relative more urban human activities and more natural vegetation, respectively. Mangrove forests dominated by K. candel were labeled with dark green; (B) photo of KC-I site and (C) photo of KC-U site.
Figure 1.
(A) The locations of sampling sites. KC-U site: 118°2′ E, 24°27′ N; KC-I site: 117°57′ E, 24°24′ N. Diagonals and light green represented regions with relative more urban human activities and more natural vegetation, respectively. Mangrove forests dominated by K. candel were labeled with dark green; (B) photo of KC-I site and (C) photo of KC-U site.

Figure 2.
Alpha and beta diversity analyses of prokaryotic and eukaryotic compositions between the two sites: (A) rarefaction curves of OTUs at 97% similarities; (B) average values of Shannon index of OTUs; (C) PCA for prokaryote and eukaryote communities and significantly related environmental factors for sorting of sites.
Figure 2.
Alpha and beta diversity analyses of prokaryotic and eukaryotic compositions between the two sites: (A) rarefaction curves of OTUs at 97% similarities; (B) average values of Shannon index of OTUs; (C) PCA for prokaryote and eukaryote communities and significantly related environmental factors for sorting of sites.

Figure 3.
(A) Venn diagrams illustrating the abundances of OTUs between the two sites; (B) community compositions of prokaryotic and eukaryotic groups. Shown are the taxa with the relative abundances higher than 1% on average and the format of taxa name phylum_class.
Figure 3.
(A) Venn diagrams illustrating the abundances of OTUs between the two sites; (B) community compositions of prokaryotic and eukaryotic groups. Shown are the taxa with the relative abundances higher than 1% on average and the format of taxa name phylum_class.

Figure 4.
Spearman’s correlations between environmental factors and relative abundances of (A) prokaryotic and (B) eukaryotic groups. Asterisks and double/triple asterisks represent p < 0.05 and p < 0.01, respectively.
Figure 4.
Spearman’s correlations between environmental factors and relative abundances of (A) prokaryotic and (B) eukaryotic groups. Asterisks and double/triple asterisks represent p < 0.05 and p < 0.01, respectively.

Figure 5.
Microbial assemblies of (A) prokaryotic and (B) eukaryotic groups at the family level. Subgroups were named in the format of class_order_family. Only class or class_order was shown when no detailed annotation was obtained.
Figure 5.
Microbial assemblies of (A) prokaryotic and (B) eukaryotic groups at the family level. Subgroups were named in the format of class_order_family. Only class or class_order was shown when no detailed annotation was obtained.

Figure 6.
(A) Comparisons of the compositions of potential COG function classes of prokaryotic community between two sites; (B) the abundances of potential COG function classes of prokaryotic communities from all samples; (C) representative KEGG pathways based on the OTUs abundance in all samples.
Figure 6.
(A) Comparisons of the compositions of potential COG function classes of prokaryotic community between two sites; (B) the abundances of potential COG function classes of prokaryotic communities from all samples; (C) representative KEGG pathways based on the OTUs abundance in all samples.

Figure 7.
(A) Comparison of enzymes involved potentially in the degradation of cellulose and hemicellulose between KC-I and KC-U sites; (B) the abundances of predicted genes functioned in the metabolisms of nitrogen and sulfur in the two sites. t-test comparison statistics are shown: * p < 0.05, ** p < 0.01.
Figure 7.
(A) Comparison of enzymes involved potentially in the degradation of cellulose and hemicellulose between KC-I and KC-U sites; (B) the abundances of predicted genes functioned in the metabolisms of nitrogen and sulfur in the two sites. t-test comparison statistics are shown: * p < 0.05, ** p < 0.01.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Sampling site locations and environmental parameters.
| KC-U | KC-I | |||||
|---|---|---|---|---|---|---|
| Date | 10 November 2020 | 12 November 2020 | ||||
| Location | 118°2′ E, 24°27′ N | 117°57′ E, 24°24′ N | ||||
| S1 | S2 | S3 | S1 | S2 | S3 | |
| Na (g/kg) | 17.03 | 19.57 | 18.14 | 16.29 | 17.18 | 18.37 |
| Mg (g/kg) ** | 16.66 | 17.94 | 18.23 | 11.37 | 12.4 | 10.22 |
| K (g/kg) | 28.22 | 27.32 | 25.98 | 29.71 | 27.43 | 28.59 |
| Ca (g/kg) ** | 14.14 | 15.35 | 14.75 | 4.1 | 3.92 | 4.39 |
| P (mg/kg) ** | 529.14 | 479.6 | 455.87 | 705.87 | 693.09 | 788.34 |
| N (mg/kg) ** | 783.47 | 747.25 | 699.71 | 939.45 | 1009.8 | 996.62 |
| NH4-N (g/kg) | 15.67 | 14.53 | 16.85 | 14.06 | 15.68 | 15.97 |
| Corg (g/kg) * | 15.3 | 14.56 | 11.63 | 19.64 | 17.98 | 17.27 |
| pH ** | 8.25 | 8.15 | 8.24 | 6.19 | 6.36 | 6.2 |
t-test comparison statistics are shown: * p < 0.05, ** p < 0.01.
Table 2.
The correlations between assemblages and environmental factors based on the Mantel test.
| Na | Mg | K | Ca | P | N | NH4-N | Corg | pH | |
|---|---|---|---|---|---|---|---|---|---|
| 16S rDNA Level | 0.354 | 0.436 * | −0.164 | 0.768 ** | 0.393 | 0.529 * | −0.114 | 0.282 | 0.225 |
| 18S rDNA Level | 0.079 | 0.654 | −0.218 | 0.914 ** | 0.657 * | 0.786 * | −0.254 | 0.454 | 0.526 |
The significances are tested based on 999 permutations: * p < 0.05 and ** p < 0.01.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, Y.; Gui, H.; Zhang, S.; Li, C. Diversity and Potential Function of Prokaryotic and Eukaryotic Communities from Different Mangrove Sediments. Sustainability 2022, 14, 3333. https://0-doi-org.brum.beds.ac.uk/10.3390/su14063333
AMA Style
Zhang Y, Gui H, Zhang S, Li C. Diversity and Potential Function of Prokaryotic and Eukaryotic Communities from Different Mangrove Sediments. Sustainability. 2022; 14(6):3333. https://0-doi-org.brum.beds.ac.uk/10.3390/su14063333
Chicago/Turabian StyleZhang, Yong, Hongjie Gui, Shufei Zhang, and Changxu Li. 2022. "Diversity and Potential Function of Prokaryotic and Eukaryotic Communities from Different Mangrove Sediments" Sustainability 14, no. 6: 3333. https://0-doi-org.brum.beds.ac.uk/10.3390/su14063333
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.