Development and Validation of a UHPLC-ESI-MS/MS Method for Quantification of Oleandrin and Other Cardiac Glycosides and Evaluation of Their Levels in Herbs and Spices from the Belgian Market


Abstract
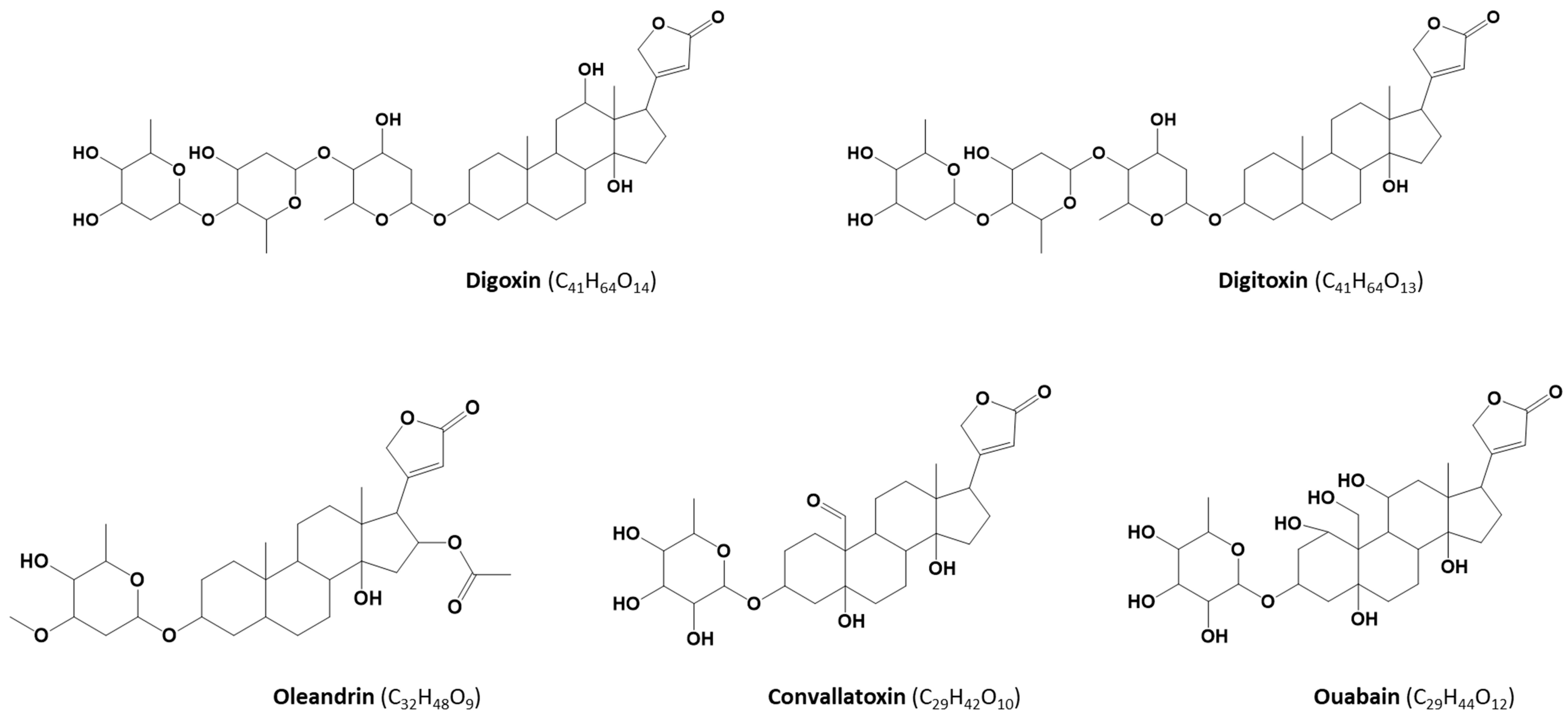
:1. Introduction
2. Results
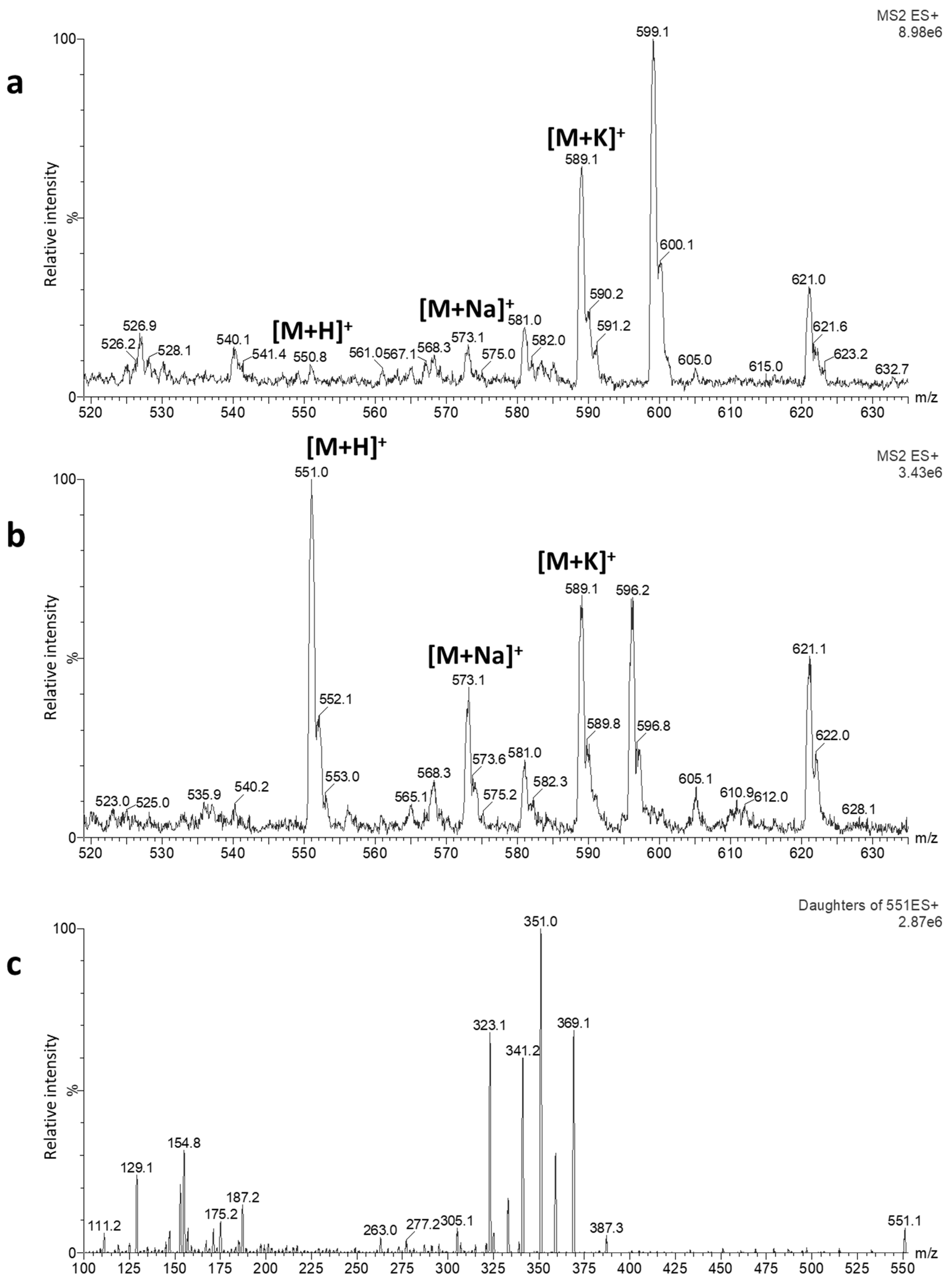
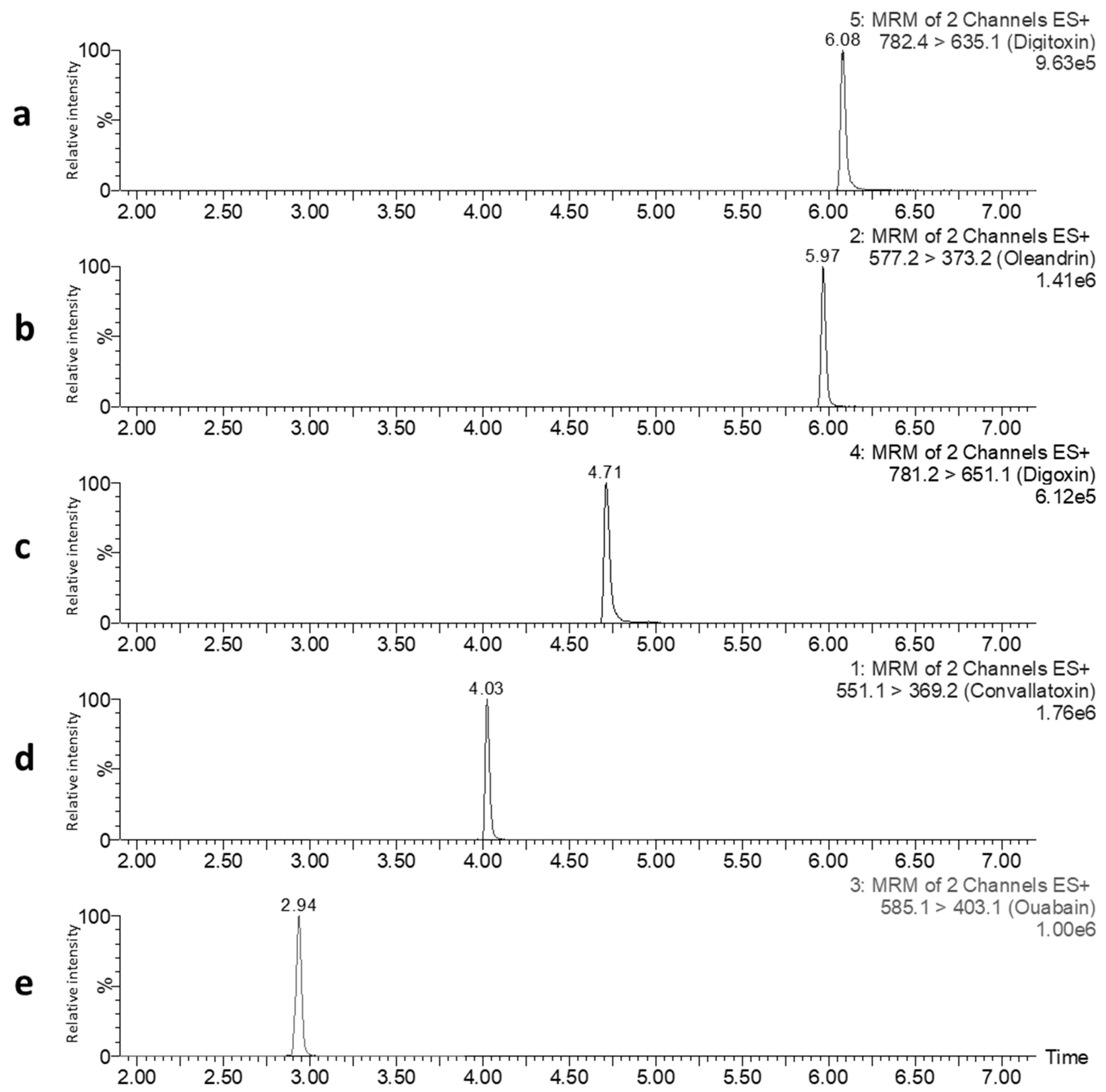
2.1. Optimization of LC-MS/MS Conditions
2.2. Optimization of Sample Preparation
2.3. Method Validation
2.4. Method Application for Analysis of Culinary Herbs
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Standards, Reagents, and Consumables
5.2. UHPLC-MS/MS Conditions
5.3. Samples
5.4. Sample Preparation
5.5. Validation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fozzard, H.A.; Sheets, M.F. Cellular mechanism of action of cardiac glycosides. J. Am. Coll. Cardiol. 1985, 5, 10A–15A. [Google Scholar] [CrossRef] [Green Version]
- Hollman, A. Plants and cardiac glycosides. Br. Heart J. 1985, 54, 258–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero, C.P.; Medarde, M.; San Feliciano, A. A short review on cardiotonic steroids and their aminoguanidine analogues. Molecules 2000, 5, 51–81. [Google Scholar] [CrossRef] [Green Version]
- Kanji, S.; MacLean, R.D. Cardiac glycoside toxicity More than 200 years and counting. Crit. Care Clin. 2012, 28, 527–535. [Google Scholar] [CrossRef]
- Morsy, N. Cardiac glycosides in medicinal plants. In Aromatic and Medicinal Plants (Back to Nature); El-Shemy, H.A., Ed.; InTechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Botelho, A.F.M.; Pierezan, F.; Soto-Blanco, B.; Melo, M.M. A review of cardiac glycosides: Structure, toxicokinetics, clinical signs, diagnosis and antineoplastic potential. Toxicon 2019, 158, 63–68. [Google Scholar] [CrossRef]
- Wasfi, I.A.; Zorod, O.; Al katheeri, N.A.; Al Awadhi, A.M. A fatal case of oleandrin poisoning. Forensic. Sci. Int. 2008, 179, e31–e36. [Google Scholar] [CrossRef]
- Gechtman, C.; Guidugli, F.; Marocchi, A.; Masarin, A.; Zoppi, F. Unexpectedly dangerous escargot stew: Oleandrin poisoning through the alimentary chain. J. Anal. Toxicol. 2006, 30, 683–686. [Google Scholar] [CrossRef] [Green Version]
- Hugues, T.; Anoult, M.; Beaub, N.; Yaici, K.; Mélandri, P.; Saoudi, N.; Gibelin, P. Intoxication volontaire au laurier rose; cas clinique et revue de la littérature. A non fatal Nerium oleander self-poisoning; case report and discussion. Ann. Cardiol. Angeiol. 2012, 61, 128–131. [Google Scholar] [CrossRef]
- Papi, L.; Luciani, A.B.; Forni, D.; Guisiani, M. Unexpected double lethal oleander poisoning. Am. J. Forensic. Med. Pathol. 2012, 33, 93–97. [Google Scholar] [CrossRef]
- Bavunoğlu, I.; Balta, M.; Türkmen, Z. Oleander poisoning as an example of self-medication attempt. Balkan. Med. J. 2016, 33, 559–562. [Google Scholar] [CrossRef]
- Smith, T.W. Pharmacokinetics, bioavailability and serum levels of cardiac glycosides. J. Am. Coil. Cordial. 1985, 5, 43A–50A. [Google Scholar] [CrossRef] [Green Version]
- Doherty, J.E.; Kane, J.J. Clinical pharmacology of digitalis glycosides. Annu. Rev. Med. 1975, 26, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Lahrtz, H.g.; Reinold, H.M.; van Zwieten, P.A. Serum concentration and urinary excretion of 3H-ouabain in patients suffering from liver or kidney diseases. Pharmacol. Clinica. 1969, 1, 114–118. [Google Scholar] [CrossRef]
- Lisalo, E. Clinical pharmacokinetics of digoxin. Clin. Pharm. 1977, 2, 1–16. [Google Scholar] [CrossRef]
- Hinderling, P.H.; Garrett, E.R.; Wester, R.C. Pharmacokinetics of b-methyldigoxin in healthy humans I: Intravenous studies. J. Pharm. Sci. 1977, 66, 242–253. [Google Scholar] [CrossRef]
- Baecher, S.; Kroiss, M.; Fassnacht, M.; Vogeser, M. No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS. Clin. Chim. Acta 2014, 431, 87–92. [Google Scholar] [CrossRef]
- Bylda, C.; Thiele, R.; Kobold, U.; Volmer, D.A. Simultaneous quantification of digoxin, digitoxin, and their metabolites in serum using high performance liquid chromatography-tandem mass spectrometry. Drug Test. Anal. 2015, 7, 937–946. [Google Scholar] [CrossRef]
- Frommherz, L.; Köhler, H.; Brinkmann, B.; Lehr, M.; Beike, J. LC-MS assay for quantitative determination of cardio glycoside in human blood samples. Int. J. Legal. Med. 2008, 122, 109–114. [Google Scholar] [CrossRef]
- Grabowski, T.; Świerczewska, A.; Borucka, B.; Sawicka, R.; Sasinowska-Motyl, M.; Gumułka, S.W.; Zahariev, Y.; Mitova, A.; Zhilkova, K. Rapid chromatographic/mass spectrometric method for digoxin quantification in human plasma. Pharm. Chem. J. 2009, 43, 710–715. [Google Scholar] [CrossRef]
- Guan, F.; Ishii, A.; Seno, H.; Watanabe-Suzuki, K.; Kumazawa, T.; Suzuki, O. Identification and quantification of cardiac glycosides in blood and urine samples by HPLC/MS/MS. Anal. Chem. 1999, 71, 4034–4043. [Google Scholar] [CrossRef]
- Mitamura, K.; Horikawa, A.; Yamane, Y.; Ikeda, Y.; Fujii, Y.; Shimada, K. Determination of digoxin in human serum using stable isotope dilution liquid chromatography/electrospray ionization-tandem mass spectrometry. Biol. Pharm. Bull. 2007, 30, 1653–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, J.X.; Yan, H.; Shen, M.; Shen, B.H.; Liu, W. Determination of oleandrin in blood and liver samples by LC-MS/MS. Fa Yi Xue Za Zhi 2018, 34, 585–589. [Google Scholar] [CrossRef]
- Gosetti, F.; Nebbia, C.; Ceci, L.; Carelli, G.; Marengo, E. UHPLC-MS/MS determination of oleandrin in blood and tissues of dairy cattle poisoned by oleander (Nerium oleander). Anal. Methods 2019, 11, 5562–5567. [Google Scholar] [CrossRef]
- Dasgupta, A.; Datta, P. Rapid detection of oleander poisoning using digoxin immunoassays: Comparison of five assays. Ther. Drug Monit. 2004, 26, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Galey, F.D.; Holstege, D.M.; Plumlee, K.H.; Tor, E.; Johnson, B.; Anderson, M.L.; Blanchard, P.C.; Brown, F. Diagnosis of oleander poisoning in livestock. J. Vet. Diagn. Investig. 1996, 8, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Hamada, K.; Iwamoto, A.; Miyazaki, S.; Yamanaka, N.; Guruge, K.S. Determination of bovine blood oleandrin by high-performance liquid chromatography and postcolumn derivatization. J. Chromatogr. Sci. 2002, 40, 515–518. [Google Scholar] [CrossRef] [Green Version]
- Namera, A.; Yashiki, M.; Okada, K.; Iwasaki, Y.; Kojima, T. Rapid quantitative analysis of oleandrin in human blood by high-performance liquid chromatography. Nihon. Hoizaku. Zasshi. 1997, 51, 315–318. [Google Scholar]
- Praveen, U.S.; Gowtham, M.D.; Yogaraje-Gowda, C.V.; Nayak, V.G.; Mohan, B.M. Detection of residues of cardenolides of Nerium oleander by high-performance thin-layer chromatography in autopsy samples. Int. J. Med. Toxicol. Forensic. Med. 2012, 2, 135–142. [Google Scholar]
- Anastassiades, M.; Lehotay, S.J.; Stajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partiotioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [Green Version]
- Commission Decision of 12 August 2002 implementing Council Directive 96/23/EC concerning the performance of analytical methods and the interpretation of the results (2002/657/EC). OJ 2002, L221, 8–36. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32002D0657&from=EN (accessed on 16 January 2020).
- Mulder, P.P.J.; De Nijs, M.; Castellari, M.; Hortos, M.; MacDonald, S.; Crews, C.; Hajslova, J.; Stranska, M. Occurrence of tropane alkaloids in food. EFSA Support. Publ. 2016, EN-1140. [Google Scholar] [CrossRef] [Green Version]
- Mulder, P.P.J.; López, P.; Castellari, M.; Bodi, D.; Ronczka, S.; Preiss-Weigert, A.; These, A. Occurrence of pyrrolizidine alkaloids in animal- and plant-derived food: Results of a survey across Europe. Food Addit. Contam. Part A 2018, 35, 118–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picron, J.-F.; Herman, M.; Van Hoeck, E.; Goscinny, S. Analytical strategies for the determination of pyrrolizidine alkaloids in plant based food and examination of the transfer rate during the infusion process. Food Chem. 2018, 266, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Kaltner, F.; Rychlik, M.; Gareis, M.; Gottschalk, C. Occurrence and risk assessment of pyrrolizidine alkaloids in spices and culinary herbs from various geographical origins. Toxins 2020, 12, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Edler, L.; Grasl-Kraupp, B.; et al. Statement on the risks for human health related to the presence of pyrrolizidine alkaloids in honey, tea, herbal infusions and food supplements. EFSA J. 2017, 15, 4908–4942. [Google Scholar] [CrossRef] [Green Version]
- Arcella, D.; Altieri, A.; Horváth, Z. Scientific report on human acute exposure assessment to tropane alkaloids. EFSA J. 2018, 16, 5160–5189. [Google Scholar] [CrossRef] [Green Version]
- Sugergat, H.; Unger, K.K.; Emmert, J.; Wendt, J.; Mandel, F. Determination of digoxin in human serum by LC-MS with on-line sample preparation. Appl. Notes Agil. Technol. 2001, 15, 5988-364EN. [Google Scholar]
- Nozaki, K.; Tarui, A.; Osaka, I. Elimination technique for alkali metal ion adducts from an electrospray ionization process using an on-line ion suppressor. Anal. Sci. 2010, 26, 715–718. [Google Scholar] [CrossRef] [Green Version]
- Gozalpour, E.; Greupink, R.; Bilos, A.; Verweij, V.; van den Heuvel, J.J.M.W.; Masereeuw, R.; Russel, F.G.M.; Koenderink, J.B. Convallatoxin: A new P-glycoprotein substrate. Eur. J. Pharmacol. 2014, 744, 18–27. [Google Scholar] [CrossRef]
- Ravi, B.G.; Guardian, M.G.E.; Dickman, R.; Wang, Z.Q. Profiling and structural analysis of cardenolides in two species of Digitalis using liquid chromatography coupled with high-resolution mass spectrometry. J. Chromatogr. A 2020, in press. [Google Scholar] [CrossRef]
- Carlier, J.; Guitton, J.; Romeuf, L.; Bévalot, F.; Boyer, B.; Fanton, L.; Gaillard, Y. Screening approach by ultra-high performance liquid chromatography-tandem mass spectrometry for the blood quantification of thirty-four toxic principles of plant origin. Application to forensic toxicology. J. Chromatogr. B 2015, 975, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Kanno, S.; Watanabe, K.; Yamagishi, I.; Hirano, S.; Minakata, K.; Gonmori, K.; Suzuki, O. Simultaneous analysis of cardiac glycosides in blood and urine by thermoresponsive LC-MS-MS. Anal. Bioanal. Chem. 2011, 399, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- SANTE/12682/2019. Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed; European Commission: Brussels, Belgium, 2020; p. 49. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Analyte | Precursor Ion (m/z) | Cone Voltage (V) | Product Ions (m/z) | Collision Energy (eV) |
|---|---|---|---|---|
| Oleandrin | 577.2 [M + H]+ | 30 | 373.21 433.1 | 15 10 |
| Digoxin | 781.2 [M + H]+ | 25 | 651.1 391.1 | 10 15 |
| Digitoxin | 782.4 [M + NH4]+ | 30 | 635.1 375.1 | 10 20 |
| Convallatoxin | 551.1 [M + H]+ | 20 | 369.2 351.2 | 10 20 |
| Ouabain | 585.1 [M + H]+ | 30 | 403.1 385.1 | 15 20 |
| Digoxin-d3 2 | 784.2 [M + H]+ | 25 | 654.1 394.2 | 10 15 |
| Analyte | Reporting Limit (ng/g) | Linear Range (ng/g) | R2 | Concentration Level | Recovery ± SD1 | Repeatability (RSDr) (%) | Reproducibility (RSDwR) (%) | Measurement Uncertainty |
|---|---|---|---|---|---|---|---|---|
| (ng/g) | (%) | (%) | ||||||
| Oleandrin | 2.5 | 2.5–200 | 0.9993 | 5 | 82 ± 7 | 7 | 8 | 17 |
| 25 | 86 ± 6 | 6 | 7 | 14 | ||||
| 100 | 80 ± 6 | 6 | 7 | 15 | ||||
| Mean: 83 ± 6 | Mean: 6 | Mean: 8 | ||||||
| Digoxin | 10 | 10–400 | 0.9995 | 10 | 121 ± 11 | 11 | 11 | 21 |
| 50 | 114 ± 7 | 7 | 7 | 13 | ||||
| 200 | 111 ± 10 | 6 | 10 | 19 | ||||
| Mean: 115 ± 10 | Mean: 9 | Mean: 9 | ||||||
| Digitoxin | 20 | 20–400 | 0.9995 | 20 | 86 ± 10 | 14 | 14 | 27 |
| 100 | 96 ± 11 | 9 | 12 | 24 | ||||
| 400 | 95 ± 13 | 7 | 15 | 31 | ||||
| Mean: 92 ± 12 | Mean: 10 | Mean: 14 | ||||||
| Convallatoxin | 10 | 10–400 | 0.9994 | 10 | 52 ± 7 | 7 | 14 | 28 |
| 50 | 55 ± 9 | 12 | 17 | 34 | ||||
| 200 | 57 ± 7 | 10 | 12 | 25 | ||||
| Mean: 55 ± 7 | Mean: 9 | Mean: 14 |
| Analyte | Reporting Limit (ng/mL) | Linear Range (ng/mL) | R2 | Concentration Level | Recovery ± SD1 | Repeatability (RSDr) (%) | Reproducibility (RSDwR) (%) | Measurement Uncertainty |
|---|---|---|---|---|---|---|---|---|
| (ng/mL) | (%) | (%) | ||||||
| Oleandrin | 0.1 | 0.1–20 | 0.9987 | 0.5 | 80 ± 13 | 3 | 18 | 36 |
| 2.5 | 84 ± 9 | 3 | 13 | 26 | ||||
| 10 | 87 ± 5 | 2 | 7 | 13 | ||||
| Mean: 84 ± 10 | Mean: 3 | Mean: 13 | ||||||
| Digoxin | 1 | 1–200 | 0.9993 | 5 | 93 ± 10 | 4 | 12 | 24 |
| 25 | 92 ± 6 | 2 | 7 | 14 | ||||
| 100 | 92 ± 4 | 3 | 5 | 9 | ||||
| Mean: 92 ± 7 | Mean: 3 | Mean: 8 | ||||||
| Digitoxin | 1 | 1–200 | 0.9975 | 5 | 86 ± 14 | 7 | 19 | 37 |
| 25 | 95 ± 10 | 6 | 12 | 24 | ||||
| 100 | 88 ± 5 | 3 | 5 | 11 | ||||
| Mean: 89 ± 11 | Mean: 5 | Mean: 12 | ||||||
| Convallatoxin | 1 | 1–200 | 0.9993 | 5 | 91 ± 15 | 4 | 19 | 36 |
| 25 | 94 ± 7 | 2 | 8 | 26 | ||||
| 100 | 96 ± 3 | 1 | 3 | 16 | ||||
| Mean: 94 ± 9 | Mean: 3 | Mean: 10 | ||||||
| Ouabain | 1 | 1–200 | 0.9994 | 5 | 87 ± 14 | 4 | 18 | 37 |
| 25 | 89 ± 11 | 6 | 13 | 17 | ||||
| 100 | 93 ± 7 | 4 | 8 | 7 | ||||
| Mean: 90 ± 11 | Mean: 5 | Mean: 13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malysheva, S.V.; Mulder, P.P.J.; Masquelier, J. Development and Validation of a UHPLC-ESI-MS/MS Method for Quantification of Oleandrin and Other Cardiac Glycosides and Evaluation of Their Levels in Herbs and Spices from the Belgian Market. Toxins 2020, 12, 243. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040243
Malysheva SV, Mulder PPJ, Masquelier J. Development and Validation of a UHPLC-ESI-MS/MS Method for Quantification of Oleandrin and Other Cardiac Glycosides and Evaluation of Their Levels in Herbs and Spices from the Belgian Market. Toxins. 2020; 12(4):243. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040243
Chicago/Turabian StyleMalysheva, Svetlana V., Patrick P. J. Mulder, and Julien Masquelier. 2020. "Development and Validation of a UHPLC-ESI-MS/MS Method for Quantification of Oleandrin and Other Cardiac Glycosides and Evaluation of Their Levels in Herbs and Spices from the Belgian Market" Toxins 12, no. 4: 243. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040243