Status of Asp29 and Asp40 in the Interaction of Naja atra Cardiotoxins with Lipid Bilayers

Abstract
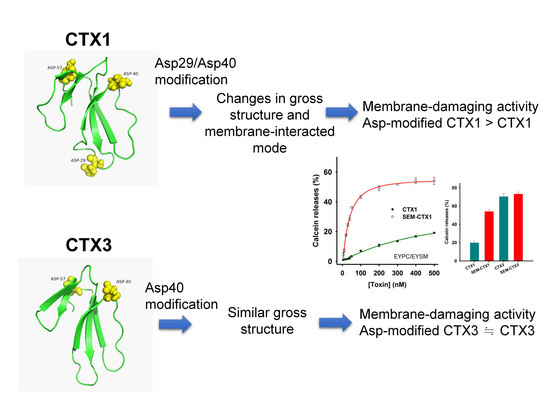
:
1. Introduction
2. Results
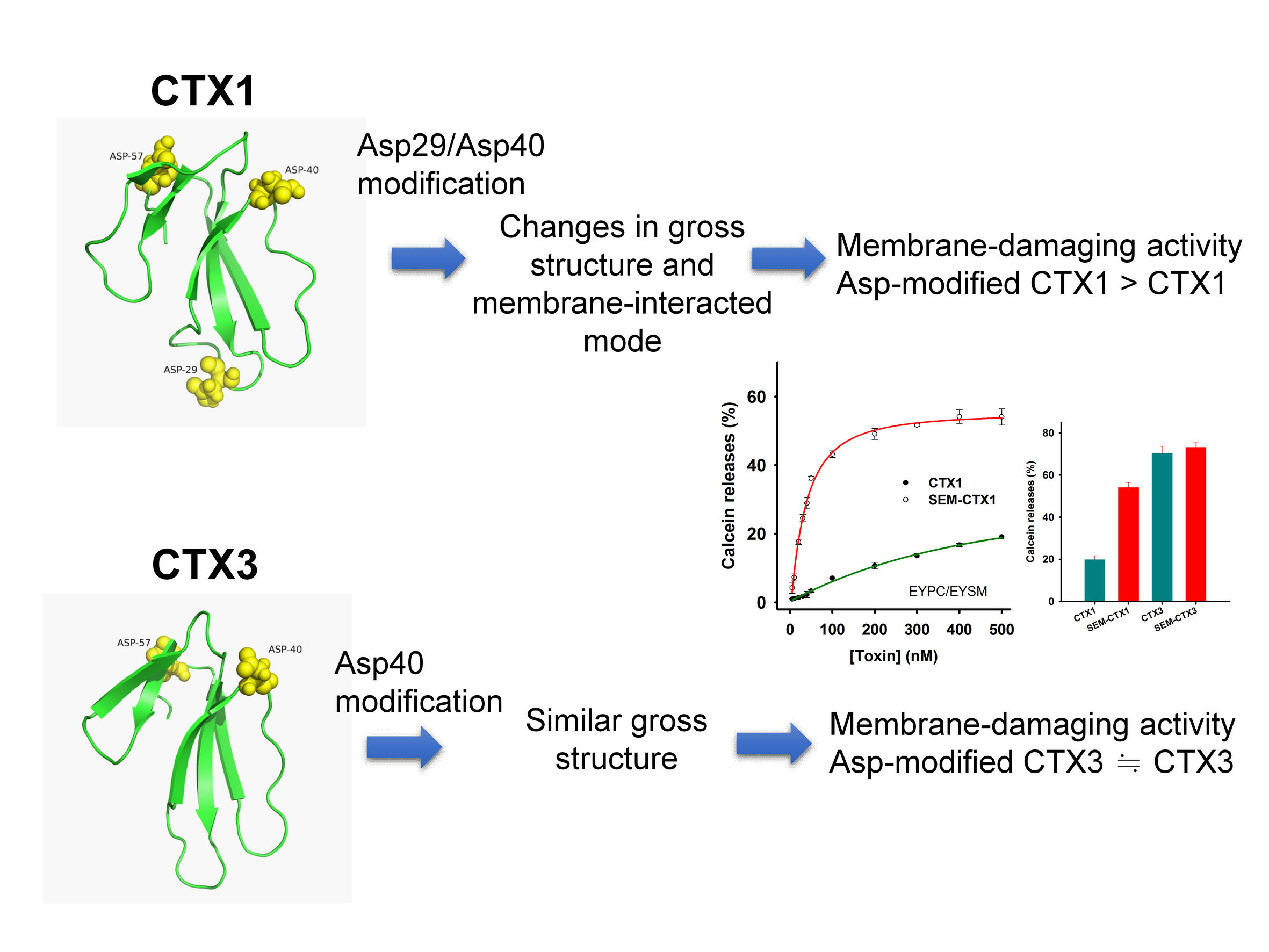
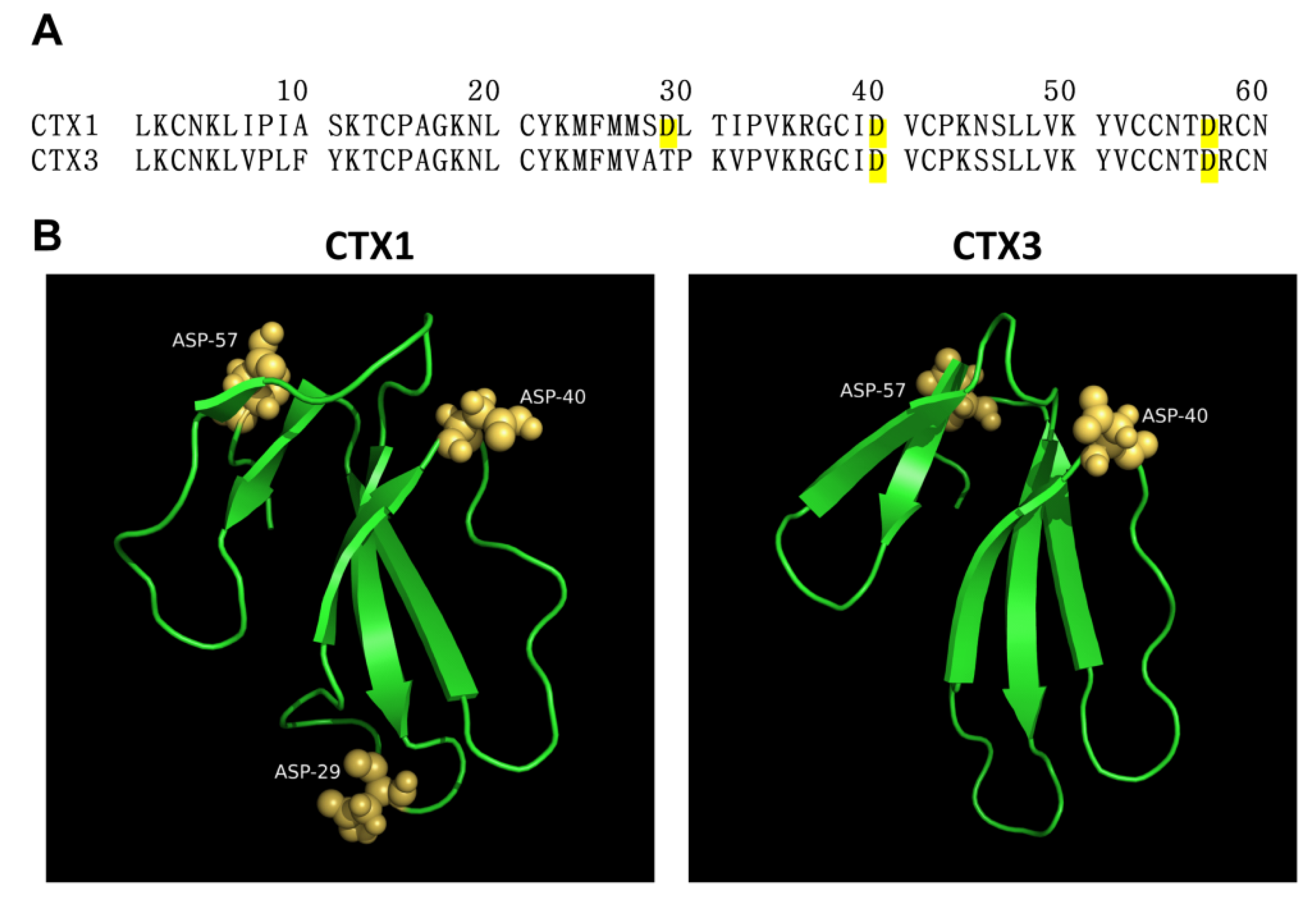
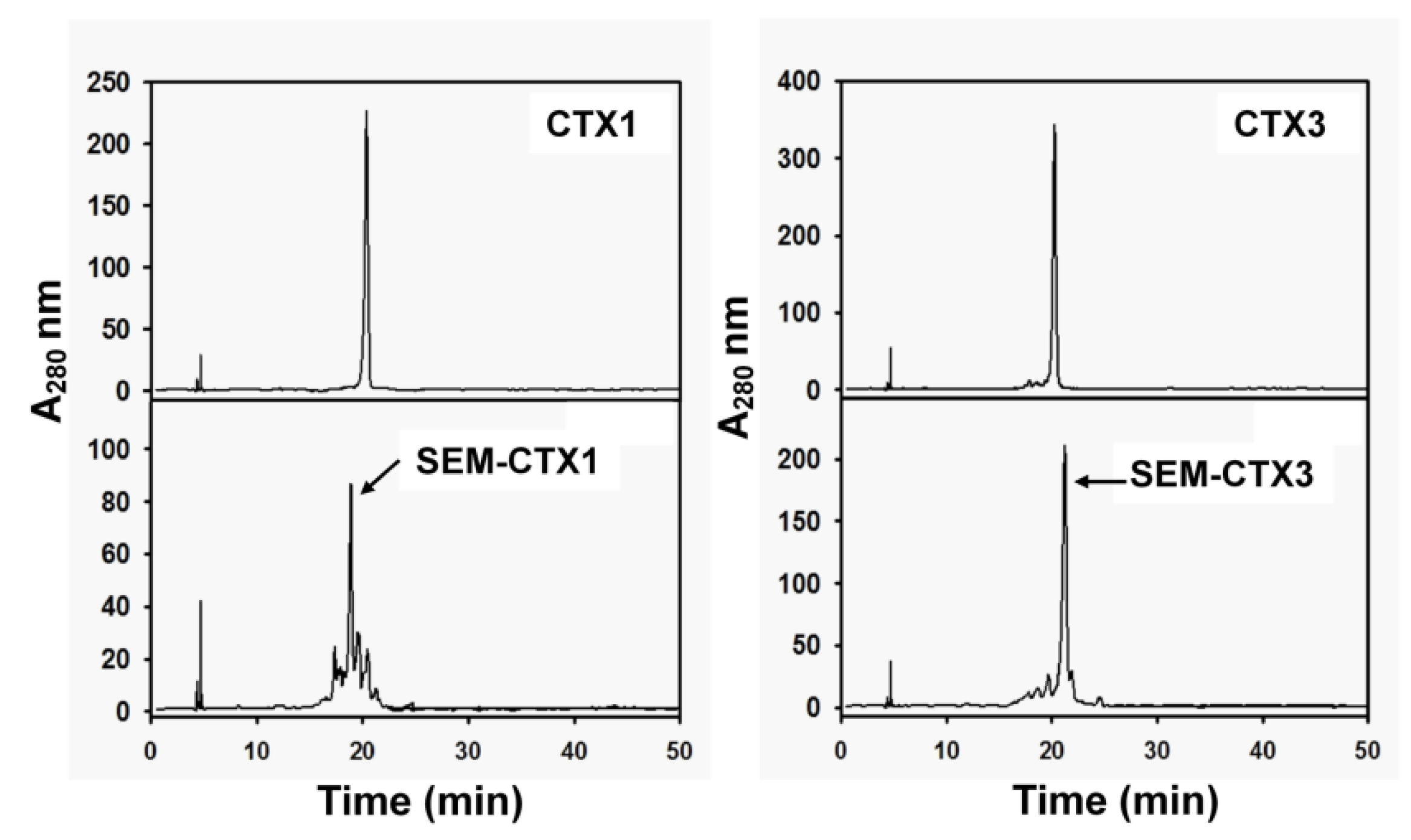
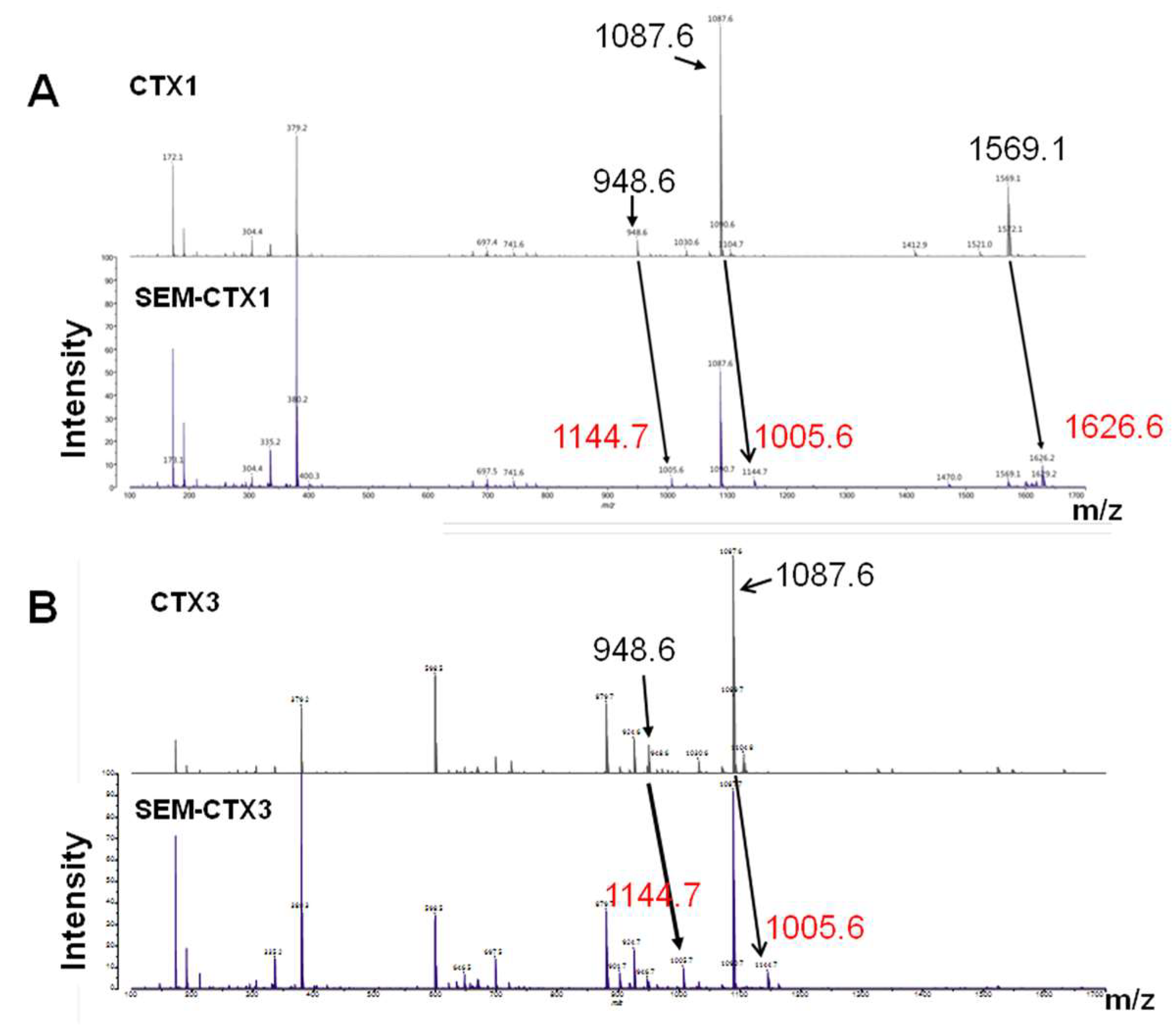
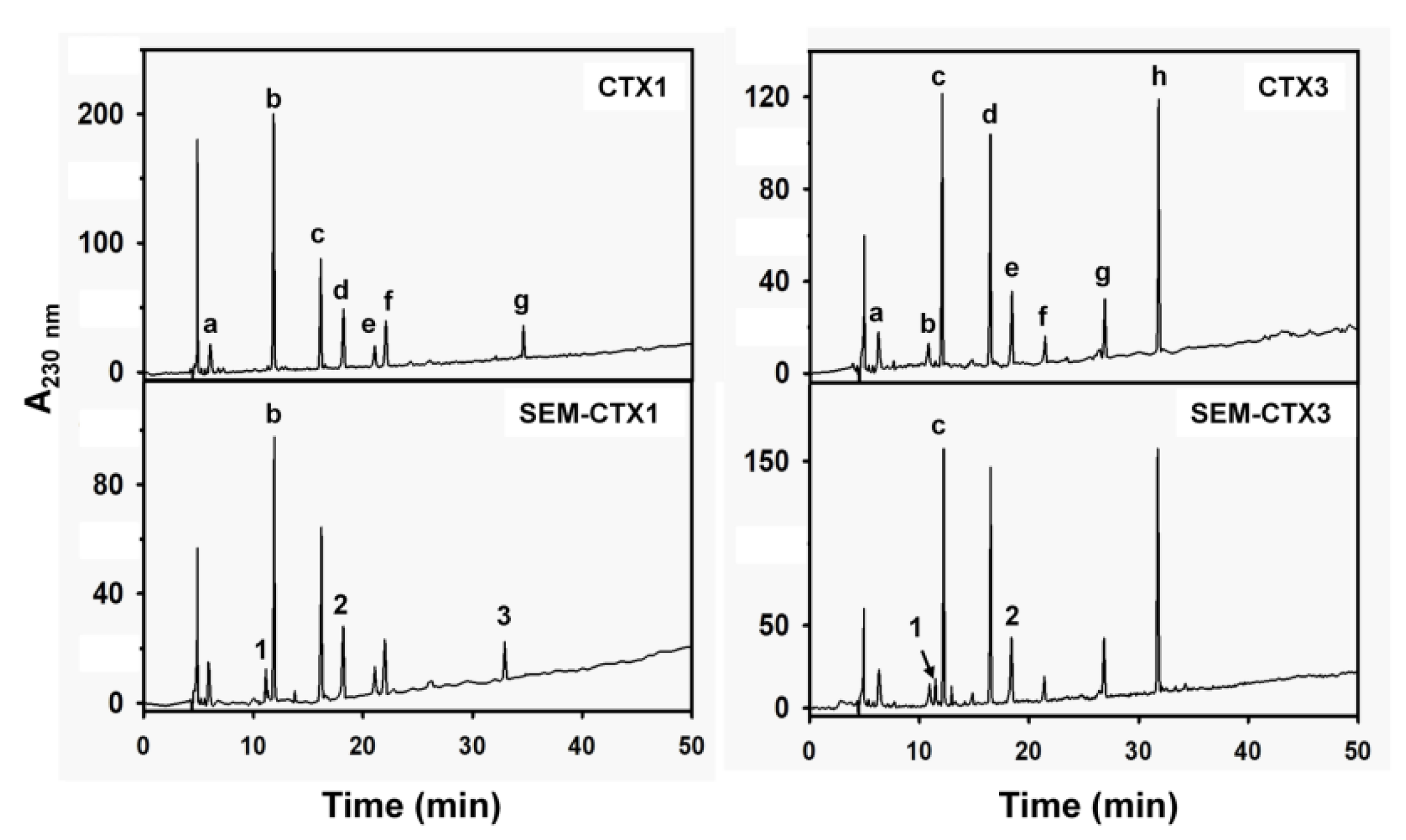
2.1. SEM-CTX1 Contains Modified Asp29 and Asp40
2.2. Asp40 Is Identified to Be Modified in SEM-CTX3
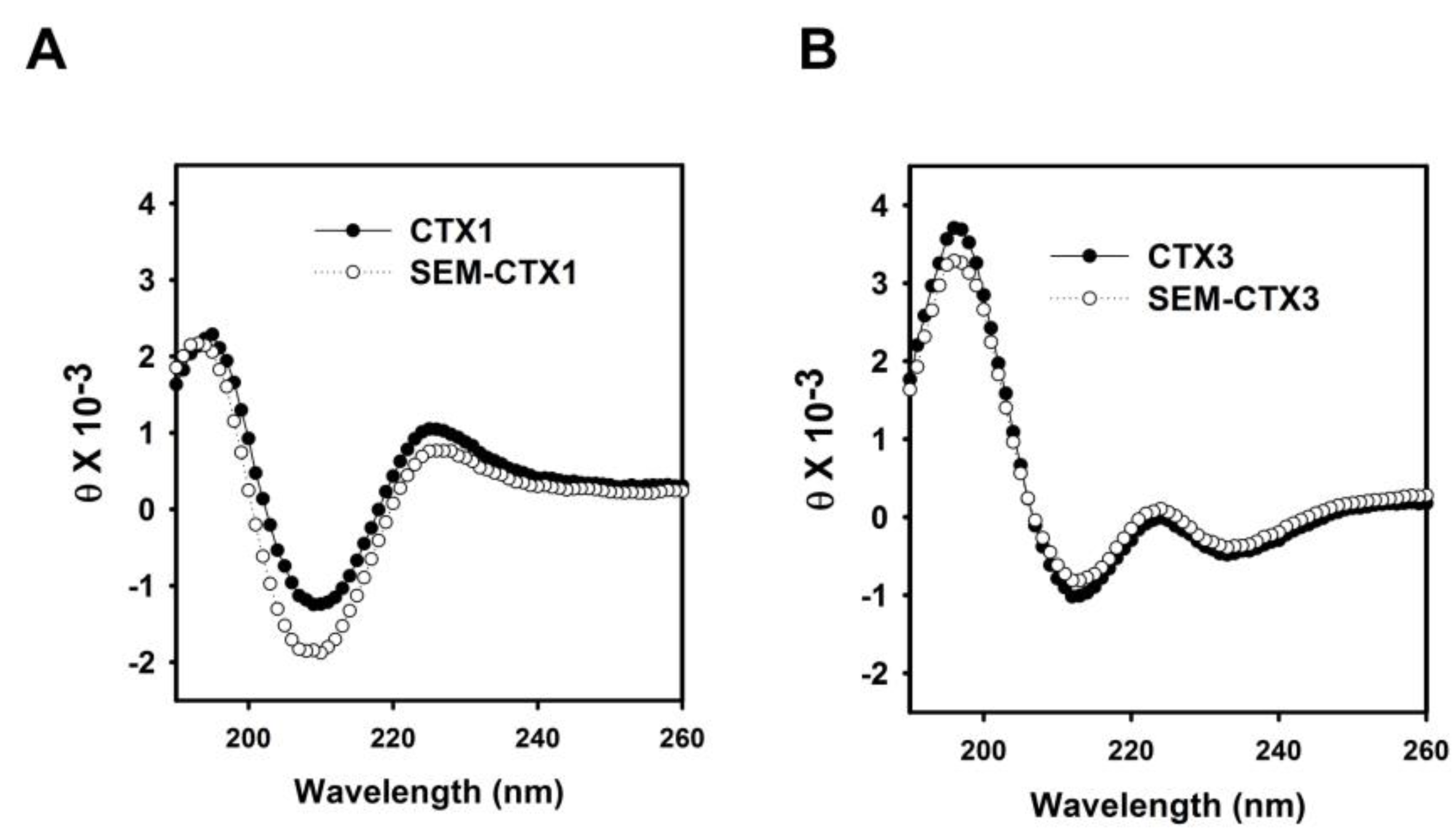
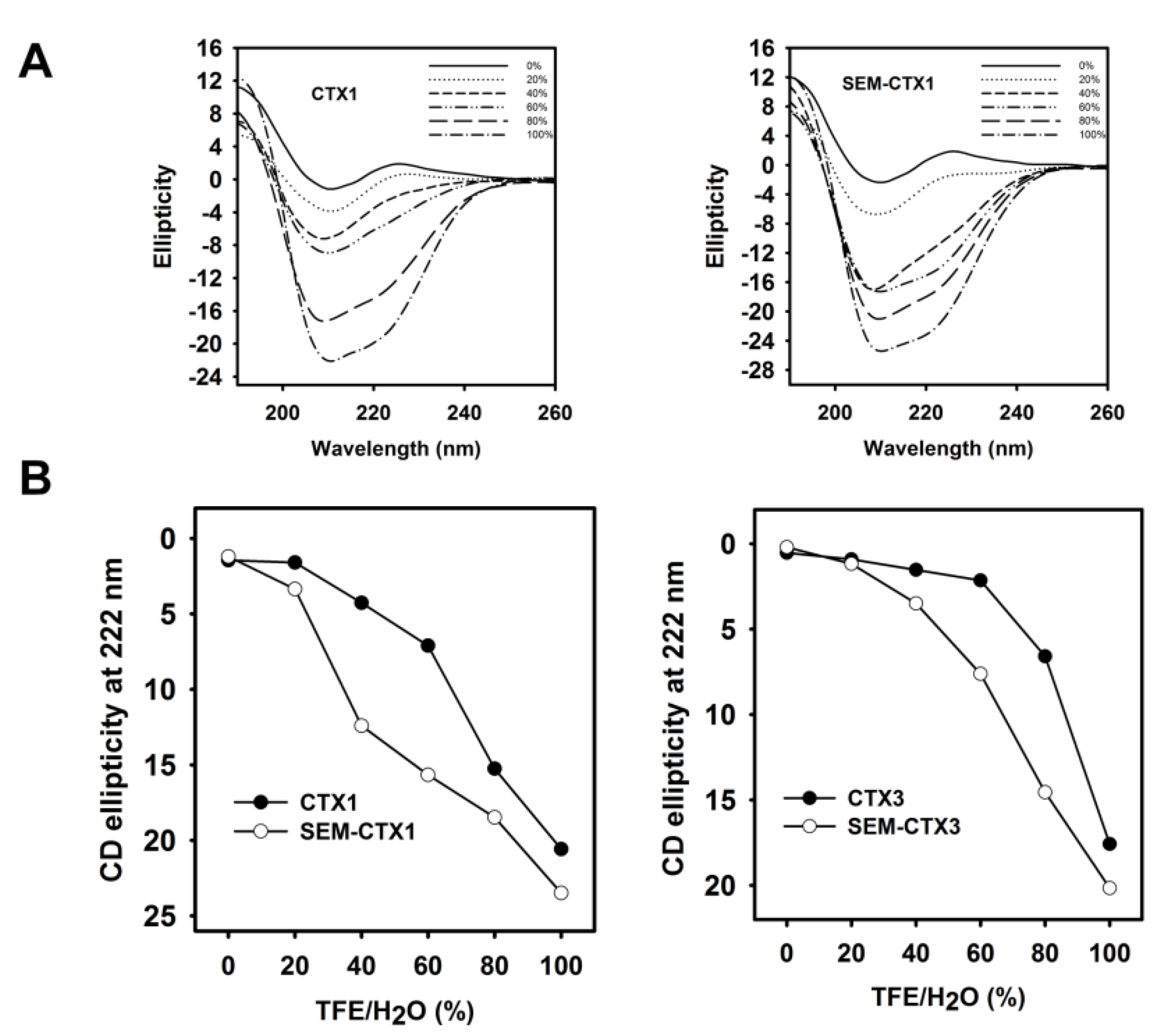
2.3. The Structural Transition of SEM-CTX1 and SEM-CTX3 Response to Trifluoroethanol (TFE)-Induced Effects Are Different from That of Native Toxins
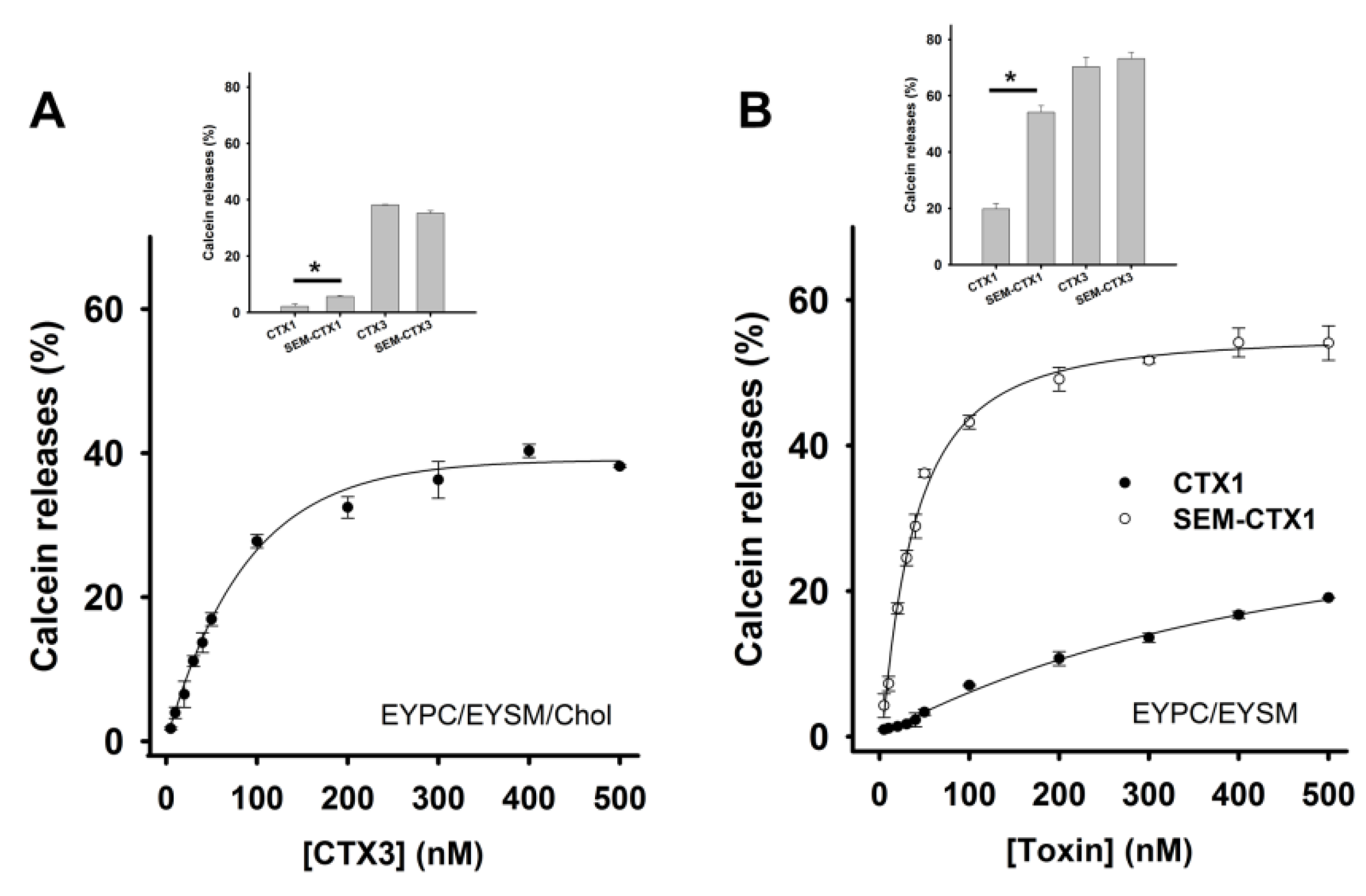
2.4. Modification of Asp Residues Markedly Increases Membrane-Damaging Activity of CTX1
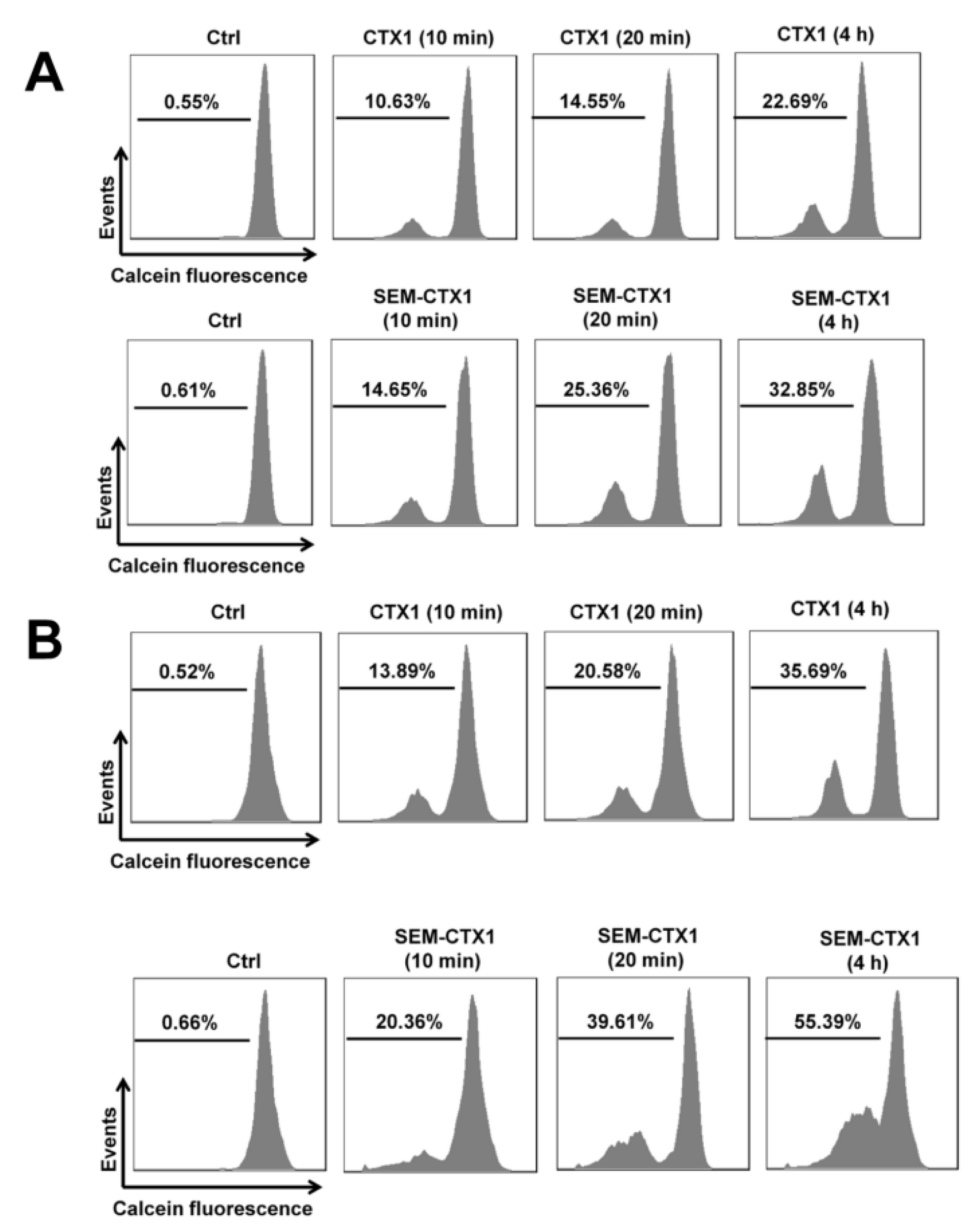
2.5. Modification of Asp Residues Promotes the Ability of CTX1 to Induce Membrane Permeabilization of K562 Cells
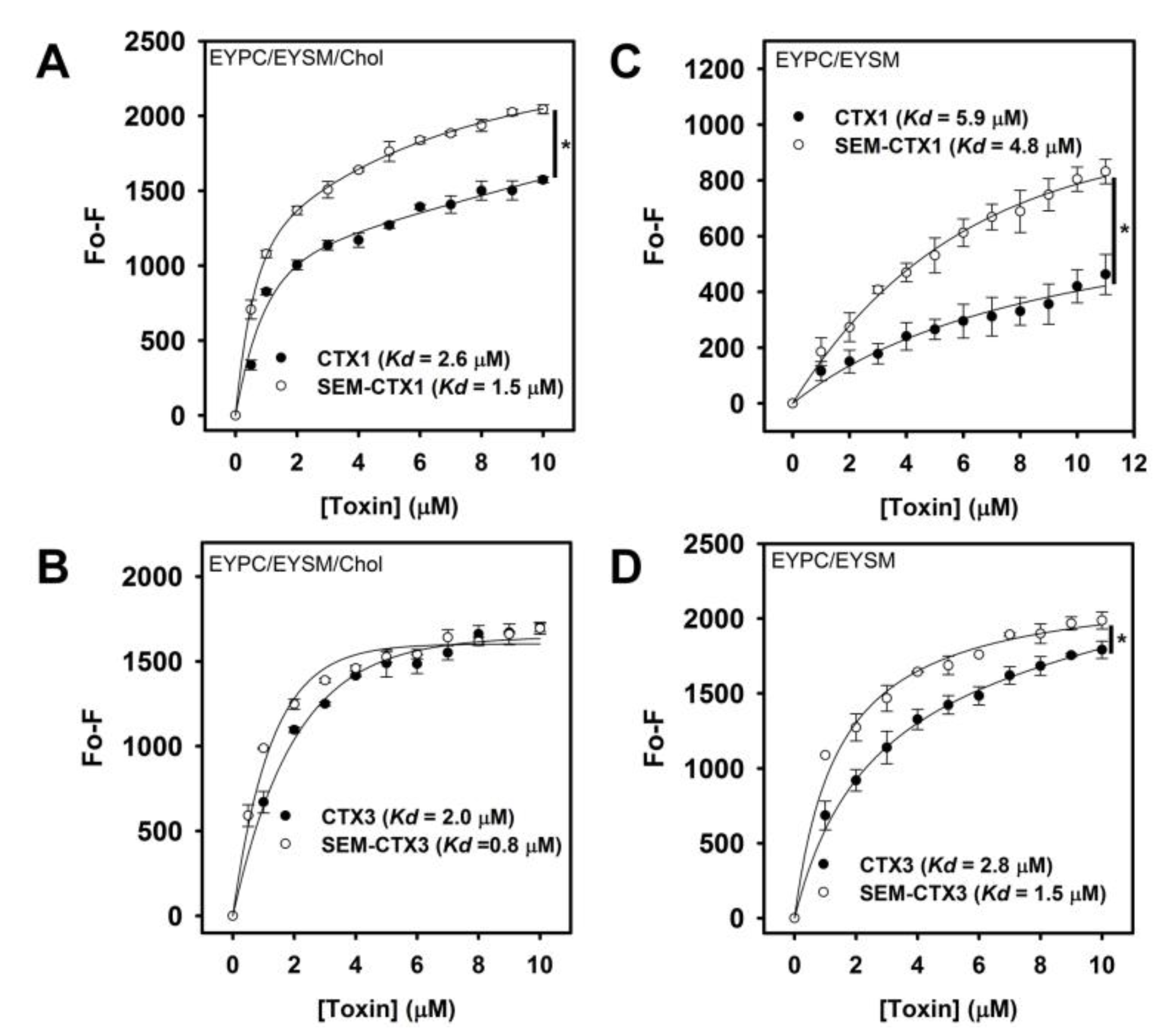
2.6. Native and Carboxyl Group-Modified CTXs Show Similar Lipid-Binding Capability
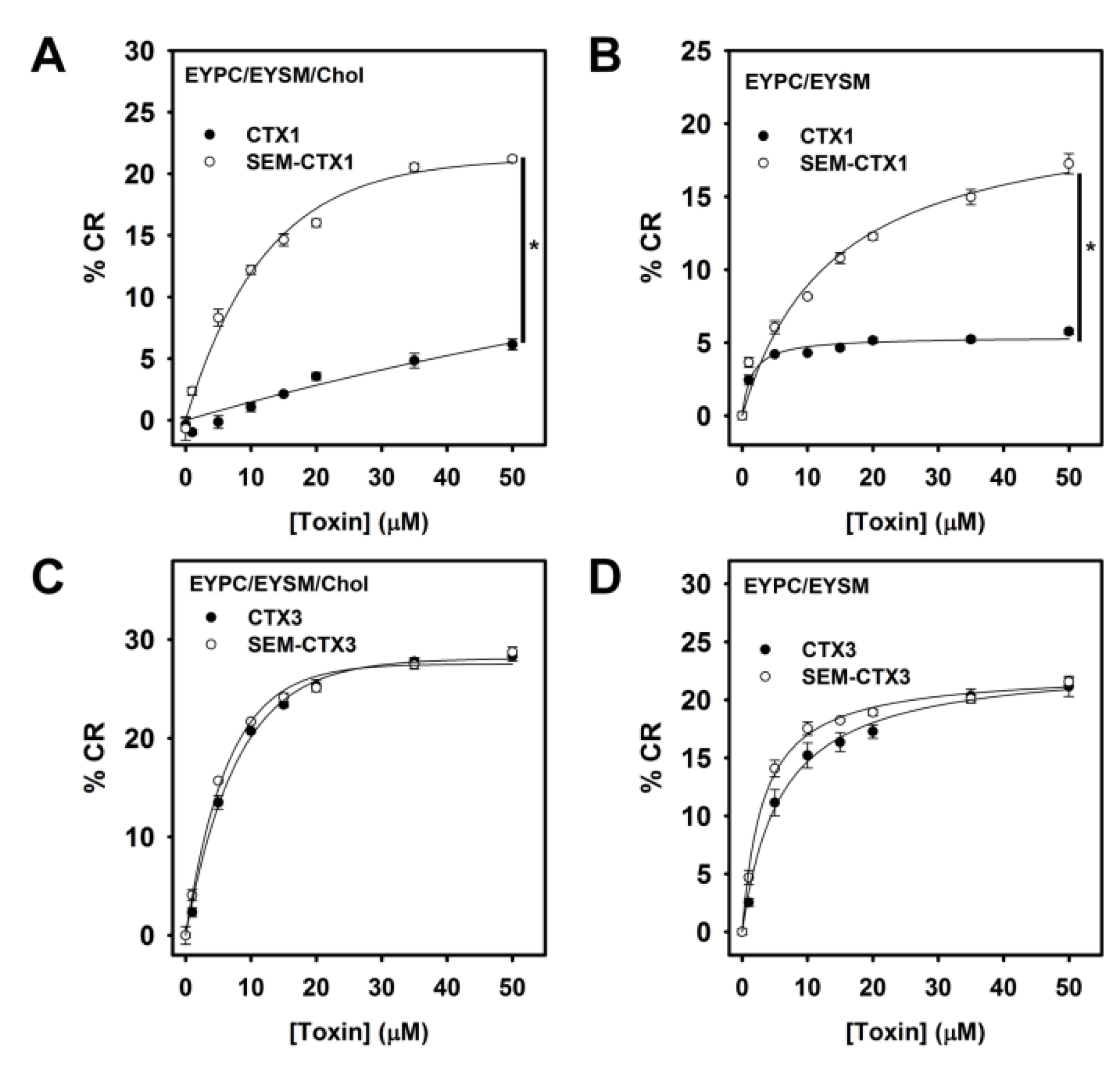
2.7. Native and Carboxyl Group-Modified CTXs Adopt Different Conformation for Interacting with Membrane
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Reagents
5.2. Preparation of SEM-CTX1 and SEM-CTX3
5.3. Separation of Tryptic Peptides
5.4. NanoUPLC-MS/MS Analyses
5.5. Measurement of Circular Dichroism (CD) Spectra
5.6. Membrane Leakage Induced by CTXs
5.7. Lipid-Binding Experiments
5.8. Colorimetric Response of the Phospholipid/PDA Vesicles
5.9. The Membrane Leakage of Calcein-Labeled Cells
5.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dufton, M.J.; Hider, R.C. The structure and pharmacology of Elapid cytotoxins. In Snake Toxins; Harvey, A.L., Ed.; Pergamon Press: New York, NY, USA, 1991; pp. 259–302. [Google Scholar]
- Konshina, A.G.; Krylov, N.A.; Efremov, R.G. Cardiotoxins: Functional role of local conformational changes. J. Chem. Inf. Model 2017, 57, 2799–2810. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, S.; Cheng, X.W.; Inoue, A.; Hu, L.; Piao, L.; Yu, C.; Goto, H.; Xu, W.; Zhao, G.; Lei, Y.; et al. Cathepsin K activity controls cardiotoxin-induced skeletal muscle repair in mice. J. Cachexia Sarcopenia Muscle 2018, 9, 160–175. [Google Scholar] [CrossRef] [PubMed]
- Chiou, J.T.; Shi, Y.J.; Wang, L.J.; Huang, C.H.; Lee, Y.C.; Chang, L.S. Naja atra Cardiotoxin 3 Elicits Autophagy and Apoptosis in U937 Human Leukemia Cells through the Ca2+/PP2A/AMPK Axis. Toxins 2019, 11, 527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.G.; Tjong, S.C.; Wu, P.L.; Kuo, J.H.; Wu, K. Role of heparan sulfates and glycosphingolipids in the pore formation of basic polypeptides of cobra cardiotoxin. Adv. Exp. Med. Biol. 2010, 677, 143–149. [Google Scholar] [PubMed]
- Gorai, B.; Karthikeyan, M.; Sivaramanm, T. Putative membrane lytic sites of P-type and S-type cardiotoxins from snake venoms as probed by all-atom molecular dynamics simulations. J. Mol. Model 2016, 22, 238. [Google Scholar] [CrossRef] [PubMed]
- Chien, K.Y.; Chiang, C.M.; Hseu, Y.C.; Vyas, A.A.; Rule, G.S.; Wu, W. Two distinct types of cardiotoxin as revealed by the structure and activity relationship of their interaction with zwitterionic phospholipid dispersions. J. Biol. Chem. 1994, 269, 14473–14483. [Google Scholar] [PubMed]
- Efremov, R.G.; Volynsky, P.E.; Nolde, D.E.; Dubovskii, P.V.; Arseniev, A.S. Interaction of cardiotoxins with membranes: A molecular modeling study. Biophys. J. 2002, 83, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.C.; Hsu, J.H.; Sheu, Y.C.; Chiang, C.M.; Wu, W.G.; Fann, W.; Tsao, P.H. Effect of D57N mutation on membrane activity and molecular unfolding of cobra cardiotoxin. Biophys. J. 1998, 75, 2382–2388. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; Baldwin, R.L. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry 1997, 36, 8413–8421. [Google Scholar] [CrossRef] [PubMed]
- Roccatano, D.; Colombo, G.; Fioroni, M.; Mark, A.E. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: A molecular dynamics study. Proc. Natl. Acad. Sci. USA 2002, 99, 12179–12184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, Y.L.; Wang, J.J.; Chang, L.S. Effect of cholesterol on membrane-damaging activity of Naja nigricollis toxin γ toward phospholipid vesicles. Toxicon 2009, 54, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Gurr, M.I.; Harwood, T.L.; Frayn, K.N. Lipid Biochemistry, 5th ed.; Blackwell Science: Oxford, UK, 2002. [Google Scholar]
- Chiou, Y.L.; Kao, P.H.; Liu, W.H.; Lin, S.R.; Chang, L.S. Roles of lysine residues and N-terminal alpha-amino group in membrane-damaging activity of Taiwan cobra cardiotoxin 3 toward anionic and zwitterionic phospholipid vesicles. Toxicon 2010, 55, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.; Golding, C.A.; Van Veen, M.; O’shea, P. The use of fluoresceinphosphatidylethanolamine (FPE) as a real-time probe for peptide-membrane interactions. Mol. Membr. Biol. 1995, 12, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Kao, P.H.; Lin, S.R.; Wu, M.J.; Chang, L.S. Membrane-bound conformation and phospholipid components modulate membrane-damaging activity of Taiwan cobra cardiotoxins. Toxicon 2009, 53, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Kao, P.H.; Lin, S.R.; Chang, L.S. Differential binding to phospholipid bilayers modulates membrane-damaging activity of Naja naja atra cardiotoxins. Toxicon 2009, 54, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Forouhar, F.; Huang, W.N.; Liu, J.H.; Chien, K.Y.; Wu, W.G.; Hsiao, C.D. Structural basis of membrane-induced cardiotoxin A3 oligomerization. J. Biol. Chem. 2003, 278, 21980–21988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.J.; Huang, C.H.; Lee, Y.C.; Wang, L.J.; Chiou, J.T.; Chang, L.S. Naja atra cardiotoxins enhance the protease activity of chymotrypsin. Int. J. Biol. Macromol. 2019, 136, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.S.; Chou, Y.C.; Lin, S.R.; Wu, B.N.; Lin, J.; Hong, E.; Sun, Y.J.; Hsiao, C.D. A novel neurotoxin, cobrotoxin b, from Naja naja atra (Taiwan cobra) venom: Purification, characterization, and gene organization. J. Biochem. 1997, 122, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.S.; Lu, C.Y. Direct in situ labeling of target drugs with a fluorophore probe to improve MALDI-MS detection sensitivity in micro-liter plasma. Sci. Rep. 2019, 9, 10787. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Chen, Y.J.; Kao, P.H.; Chang, L.S. Bovine serum albumin with glycated carboxyl groups shows membrane-perturbing activities. Arch. Biochem. Biophys. 2014, 564, 43–51. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tryptic Peptides | Positions (#) | Predicted Mass (Da) | Measured Mass (Da) |
|---|---|---|---|
| CTX1 | |||
| MFMMSDLTIPVKR | 24–36 (Asp29) | 1569 | 1569.1 |
| GC(a)IDVC(a)PK | 37–44 (Asp40) | 950 | 948.6 |
| YVC(a)C(a)NTDR | 51–58 (Asp57) | 1089 | 1087.6 |
| SEM-CTX1 | |||
| MFMMSD(b)LTIPVKR | 24–36 (Asp29) | 1626 | 1626.6 |
| GC(a)ID(b)VC(a)PK | 37–44 (Asp40) | 1007 | 1005.6 |
| YVC(a)C(a)NTD(b)R | 51–58 (Asp57) | 1146 | 1144.7 |
| CTX3 | |||
| GC(a)IDVC(a)PK | 37–44 (Asp40) | 950 | 948.6 |
| YVC(a)C(a)NTDR | 51–58 (Asp57) | 1089 | 1087.6 |
| SEM-CTX3 | |||
| GC(a)ID(b)VC(a)PK | 37–44 (Asp40) | 1007 | 1005.6 |
| YVC(a)C(a)NTD(b)R | 51–58 (Asp57) | 1146 | 1144.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, G.-L.; Shi, Y.-J.; Huang, C.-H.; Lee, Y.-C.; Wang, L.-J.; Chiou, J.-T.; Lu, C.-Y.; Chang, L.-S. Status of Asp29 and Asp40 in the Interaction of Naja atra Cardiotoxins with Lipid Bilayers. Toxins 2020, 12, 262. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040262
Wu G-L, Shi Y-J, Huang C-H, Lee Y-C, Wang L-J, Chiou J-T, Lu C-Y, Chang L-S. Status of Asp29 and Asp40 in the Interaction of Naja atra Cardiotoxins with Lipid Bilayers. Toxins. 2020; 12(4):262. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040262
Chicago/Turabian StyleWu, Guan-Lin, Yi-Jun Shi, Chia-Hui Huang, Yuan-Chin Lee, Liang-Jun Wang, Jing-Ting Chiou, Chi-Yu Lu, and Long-Sen Chang. 2020. "Status of Asp29 and Asp40 in the Interaction of Naja atra Cardiotoxins with Lipid Bilayers" Toxins 12, no. 4: 262. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040262