Development and Application of Extraction Methods for LC-MS Quantification of Microcystins in Liver Tissue


 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
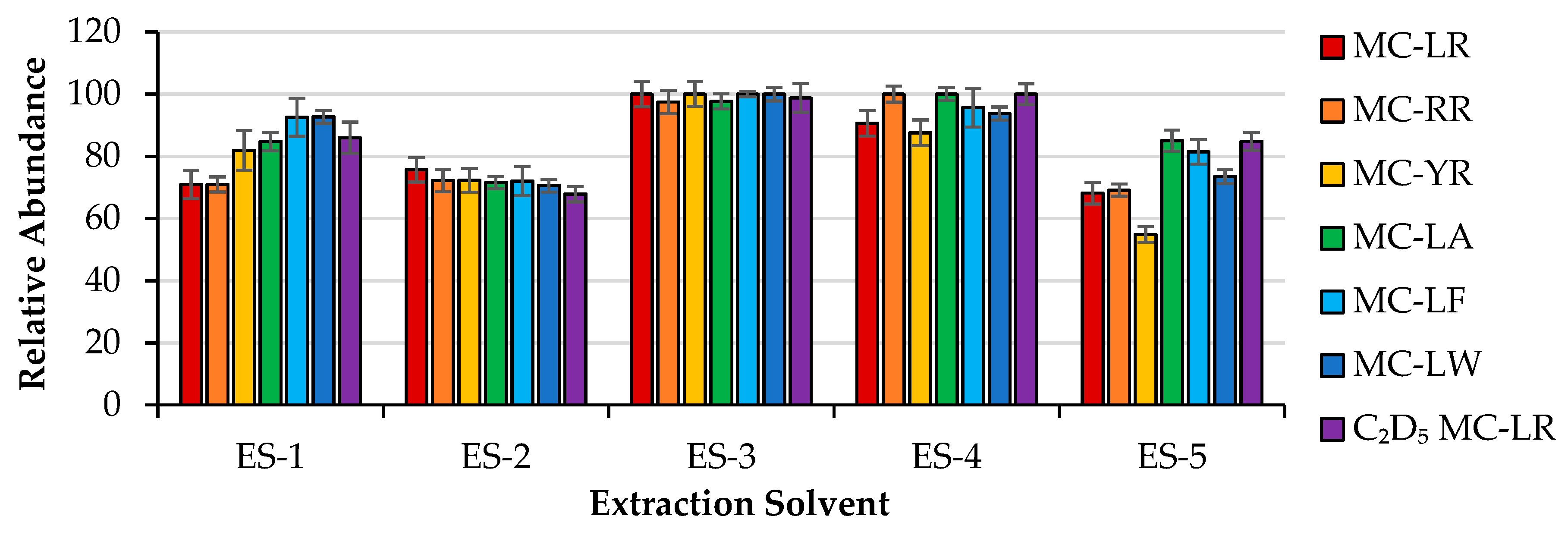
2.1. Extraction Optimization
2.2. Recovery Experiments
2.3. Calibration Curves and LOQs
2.4. Quantification of MC-LR and MC-LR-Cys in Livers of Gavaged Mice
3. Conclusion
4. Materials and Methods
4.1. Reagents
4.2. Materials and Instruments
4.3. Mouse Liver Collection
4.4. Sample Preparation for Optimization and Recovery Experiments
4.5. Sample Preparation for Calibration Curves and Livers of Mice Gavaged with MC-LR
4.6. Liquid Chromatography and Mass Spectrometry
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carmichael, W.W.; Jones, C.L.A.; Mahmood, N.A.; Theiss, W.C. Algal toxins and water-based diseases. CRC Crit. Rev. Environ. Contr. 1985, 15, 275–313. [Google Scholar] [CrossRef]
- Francis, G. Poisonous Australian lake. Nature 1878, 18, 11–12. [Google Scholar] [CrossRef] [Green Version]
- Bishop, C.T.; Anet, E.F.; Gorham, P.R. Isolation and identification of the fast-death factor in Microcystis aeruginosa NRC-1. Can. J. Biochem. Physiol. 1959, 37, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, P.; Anderson, D.M.; Kaoru, Y.; White, A.W. The economic effects of harmful algal blooms in the United States: Estimates, assessment issues, and information needs. Estuaries 2002, 25, 819–837. [Google Scholar] [CrossRef]
- Oudra, B.; Loudiki, M.; Vasconcelos, V.; Sabour, B.; Sbiyyaa, B.; Oufdou, K.; Mezrioul, N. Detection and quantification of microcystins from cyanobacteria strains isolated from reservoirs and ponds in Morocco. Environ. Toxicol. 2002, 17, 32–39. [Google Scholar] [CrossRef]
- Sinang, S.C.; Poh, K.B.; Shamsudin, S.; Sinden, A. Preliminary assessment of cyanobacteria diversity and toxic potential in ten freshwater lakes in Selangor, Malaysia. Bull. Environ. Contam. Toxicol. 2015, 95, 542–547. [Google Scholar] [CrossRef]
- Pouria, S.; de Andrade, A.; Barbosa, J.; Cavalcanti, R.L.; Barreto, V.T.; Ward, C.J.; Preiser, W.; Poon, G.K.; Neild, G.H.; Codd, G.A. Fatal microcystin intoxication in haemodialysis unit in Caruaru, Brazil. Lancet 1998, 352, 21–26. [Google Scholar] [CrossRef]
- Ernst, B.; Hitzfeld, B.; Dietrich, D. Presence of Planktothrix sp. and cyanobacterial toxins in Lake Ammersee, Germany and their impact on whitefish (Coregonus lavaretus L.). Environ. Toxicol. 2001, 16, 483–488. [Google Scholar] [CrossRef]
- Jochimsen, E.M.; Carmichael, W.W.; An, J.S.; Cardo, D.M.; Cookson, S.T.; Holmes, C.E.; Antunes, M.B.; de Melo Filho, D.A.; Lyra, T.M.; Barreto, V.S.; et al. Liver failure and death after exposure to microcystins at a hemodialysis center in Brazil. N. Engl. J. Med. 1998, 338, 873–878. [Google Scholar] [CrossRef]
- Falconer, I.; Langley, B.J.; Runnegar, M. Liver pathology in mice in poisoning by the blue-green alga Microcystis aeruginosa. Aust. J. Biol. Sci. 1980, 34, 179–188. [Google Scholar] [CrossRef] [Green Version]
- Nishiwaki-Matsushima, R.; Ohta, T.; Nishiwaki, S.; Suganuma, M.; Kohyama, K.; Ishikawa, T.; Carmichael, W.W.; Fujiki, H. Liver tumor promotion by the cyanobacterial cyclic peptide toxin microcystin-LR. J. Cancer Res. Clin. 1992, 118, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, W.W. Algal Toxins. In Advances in Botanical Research; Callow, J.A., Ed.; Academic Press: Cambridge, MA, USA, 1986; Volume 12, pp. 47–101. [Google Scholar]
- Carmichael, W.W.; Beasley, V.; Bunner, D.L.; Eloff, J.N.; Falconer, I.; Gorham, P.; Harada, K.-I.; Krishnamurthy, T.; Min-Juan, Y.; Moore, R.E.; et al. Naming of cyclic heptapeptide toxins of cyanobacteria (blue-green algae). Toxicon 1988, 26, 971–973. [Google Scholar] [CrossRef]
- Meriluoto, J.; Spoof, L.; Codd, G.A. Handbook of cyanobacterial monitoring and cyanotoxin analysis. Anal. Bioanal. Chem. 2018, 410, 1405–1406. [Google Scholar] [CrossRef]
- MacKintosh, R.W.; Dalby, K.N.; Campbell, D.G.; Cohen, P.T.; Cohen, P.; MacKintosh, C. The cyanobacterial toxin microcystin binds covalently to cysteine-273 on protein phosphatase 1. FEBS Lett. 1995, 371, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKintosh, C.; Beattie, K.A.; Klumpp, S.; Cohen, P.; Codd, G.A. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 1990, 264, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, R.; Yoshizawa, S.; Watanabe, M.F.; Harada, K.; Furusawa, M.; Carmichael, W.W.; Fujiki, H. In vitro and in vivo effects of protein phosphatase inhibitors, microcystins and nodularin, on mouse skin and fibroblasts. Biochem. Biophys. Res. Commun. 1990, 171, 867–874. [Google Scholar] [CrossRef]
- Yoshizawa, S.; Matsushima, R.; Watanabe, M.F.; Harada, K.-I.; Ichihara, A.; Carmichael, W.W.; Fujiki, H. Inhibition of protein phosphatases by microcystins and nodularin associated with hepatotoxicity. J. Cancer Res. Clin. 1990, 116, 609–614. [Google Scholar] [CrossRef]
- Runnegar, M.; Berndt, N.; Kaplowitz, N. Microcystin uptake and inhibition of protein phosphatases: Effects of chemoprotectants and self-inhibition in relation to known hepatic transporters. Toxicol. Appl. Pharmacol. 1995, 134, 264–272. [Google Scholar] [CrossRef]
- Ito, E.; Takai, A.; Kondo, F.; Masui, H.; Imanishi, S.; Harada, K.-I. Comparison of protein phosphatase inhibitory activity and apparent toxicity of microcystins and related compounds. Toxicon 2002, 40, 1017–1025. [Google Scholar] [CrossRef]
- Kondo, F.; Matsumoto, H.; Yamada, S.; Ishikawa, N.; Ito, E.; Nagata, S.; Ueno, Y.; Suzuki, M.; Harada, K. Detection and identification of metabolites of microcystins formed in vivo in mouse and rat livers. Chem. Res. Toxicol. 1996, 9, 1355–1359. [Google Scholar] [CrossRef]
- Guo, X.; Xie, P.; Chen, J.; Tuo, X.; Deng, X.; Li, S.; Yu, D.; Zeng, C. Simultaneous quantitative determination of microcystin-LR and its glutathione metabolites in rat liver by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2014, 963, 54–61. [Google Scholar] [CrossRef]
- Kondo, F.; Ikai, Y.; Oka, H.; Okumura, M.; Ishikawa, N.; Harada, K.; Matsuura, K.; Murata, H.; Suzuki, M. Formation, characterization, and toxicity of the glutathione and cysteine conjugates of toxic heptapeptide microcystins. Chem. Res. Toxicol. 1992, 5, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Lad, A.; Su, R.C.; Breidenbach, J.D.; Sanchez, N.K.; Khalaf, F.K.; Zhang, S.; Kleinhenz, A.L.; Dube, P.; Mohammed, C.J.; Crawford, E.L.; et al. Chronic low dose oral exposure to microcystin-LR exacerbates hepatic injury in a murine model of non-alcoholic fatty liver disease. Toxins 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, S.; Kaya, K. Quail spleen is enlarged by microcystin RR as a blue-green algal hepatotoxin. Nat. Toxins 1993, 1, 283–285. [Google Scholar] [CrossRef]
- Fawell, J.K.; Mitchell, R.E.; Everett, D.J.; Hill, R.E. The toxicity of cyanobacterial toxins in the mouse: I microcystin-LR. Hum. Exp. Toxicol. 1999, 18, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, P.; Tang, R.; Zhang, X.; Li, L.; Li, D. In vivo studies on the toxic effects of microcystins on mitochondrial electron transport chain and ion regulation in liver and heart of rabbit. Comp. Biochem. Physica C 2008, 148, 204–210. [Google Scholar] [CrossRef] [PubMed]
- McLellan, N.L.; Manderville, R.A. Toxic mechanisms of microcystins in mammals. Toxicol. Res. 2017, 6, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Sedan, D.; Giannuzzi, L.; Rosso, L.; Marra, C.A.; Andrinolo, D. Biomarkers of prolonged exposure to microcystin-LR in mice. Toxicon 2013, 68, 9–17. [Google Scholar] [CrossRef]
- Cantor, G.H.; Beckonert, O.; Bollard, M.E.; Keun, H.C.; Ebbels, T.M.; Antti, H.; Wijsman, J.A.; Bible, R.H.; Breau, A.P.; Cockerell, G.L.; et al. Integrated histopathological and urinary metabonomic investigation of the pathogenesis of microcystin-LR toxicosis. Vet. Pathol. 2013, 50, 159–171. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Human Microcystin (MC) ELISA Kit. Available online: https://www.mybiosource.com/human-elisa-kits/microcystin-mc/9300557 (accessed on 17 December 2019).
- Ito, E.; Kondo, F.; Harada, K.-I. First report on the distribution of orally administered microcystin-LR in mouse tissue using an immunostaining method. Toxicon 2000, 38, 37–48. [Google Scholar] [CrossRef]
- Greer, B.; Maul, R.; Campbell, K.; Elliott, C.T. Detection of freshwater cyanotoxins and measurement of masked microcystins in tilapia from Southeast Asian aquaculture farms. Anal. Bioanal. Chem. 2017, 409, 4057–4069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanan, T.; Lazorchak, J. EPA Current Research on Cyanotoxins in Fish Tissue; U.S. Environmental Protection Agency: Washington, DC, USA, 2017. [Google Scholar]
- Moreno, I.M.; Molina, R.; Jos, A.; Picó, Y.; Cameán, A.M. Determination of microcystins in fish by solvent extraction and liquid chromatography. J. Chromatogr. A 2005, 1080, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Boyer, G.L. Standardization of microcystin extraction from fish tissues: A novel internal standard as a surrogate for polar and non-polar variants. Toxicon 2009, 53, 238–245. [Google Scholar] [CrossRef]
- Altaner, S.; Puddick, J.; Fessard, V.; Feurstein, D.; Zemskov, I.; Wittmann, V.; Dietrich, R.D. Simultaneous detection of 14 microcystin congeners from tissue samples using UPLC-ESI-MS/MS and two different deuterated synthetic microcystins as internal standards. Toxins 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Krężel, A.; Maret, W. The biological inorganic chemistry of zinc ions. Arch. Biochem. Biophys. 2016, 611, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Heroes, E.; Rip, J.; Beullens, M.; Van Meervelt, L.; De Gendt, S.; Bollen, M. Metals in the active site of native protein phosphatase-1. J. Inorg. Biochem. 2015, 149, 1–5. [Google Scholar] [CrossRef]
- Xiong, Y.; Luo, D.J.; Wang, X.L.; Qiu, M.; Yang, Y.; Yan, X.; Wang, J.Z.; Ye, Q.F.; Liu, R. Zinc binds to and directly inhibits protein phosphatase 2A in vitro. Neurosci. Bull. 2015, 31, 331–337. [Google Scholar] [CrossRef] [Green Version]
- Palagama, D.S.W.; Baliu-Rodriguez, D.; Lad, A.; Levison, B.S.; Kennedy, D.J.; Haller, S.T.; Westrick, J.; Hensley, K.; Isailovic, D. Development and applications of solid-phase extraction and liquid chromatography-mass spectrometry methods for quantification of microcystins in urine, plasma, and serum. J. Chromatogr. A 2018, 1573, 66–77. [Google Scholar] [CrossRef]
- Palagama, D.S.W.; West, R.E., III; Isailovic, D. Improved solid-phase extraction protocol and sensitive quantification of six microcystins in water using an HPLC-orbitrap mass spectrometry system. Anal. Methods 2017, 9, 2021–2030. [Google Scholar] [CrossRef]
- Bioanalytical Method Validation Guidance for Industry; U.S. Food and Drug Administration: Washington, DC, USA, 2018.
- Furey, A.; Moriarty, M.; Bane, V.; Kinsella, B.; Lehane, M. Ion suppression; a critical review on causes, evaluation, prevention and applications. Talanta 2013, 115, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Xie, P.; Chen, J.; Zhang, D.; Liang, G. Development and validation of a liquid chromatography–tandem mass spectrometry assay for the simultaneous quantitation of microcystin-RR and its metabolites in fish liver. J. Chromatogr. A 2010, 1217, 1455–1462. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
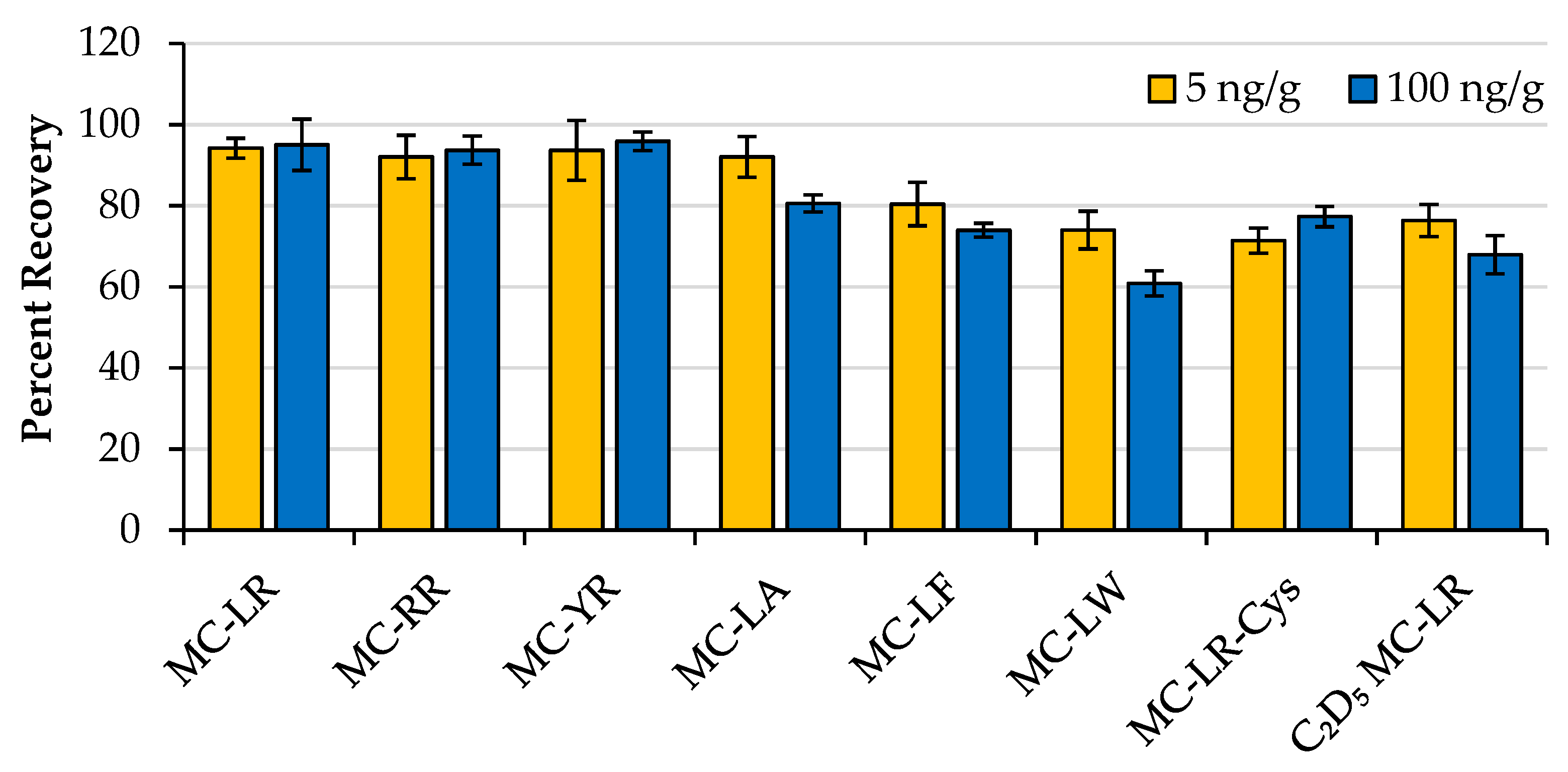{kind=link}
| MC Congener | LOD* (ng/g) | LOQ (ng/g) | Wild-type Calibration Curves | Leprdb/J Calibration Curves | ||
|---|---|---|---|---|---|---|
| Equation | R² | Equation | R² | |||
| MC-LR | 0.08 | 0.25 | y = 0.7133x + 0.0013 | 0.997 | y = 1.2856x + 0.1187 | 0.995 |
| MC-RR | 0.15 | 0.50 | y = 1.7273x − 0.0625 | 0.997 | y = 2.0529x − 0.0126 | 0.995 |
| MC-YR | 0.15 | 0.50 | y = 0.5487x + 0.0247 | 0.999 | y = 0.5702x + 0.0057 | 0.998 |
| MC-LA | 0.23 | 0.75 | y = 1.2411x − 0.0357 | 0.995 | y = 0.9152x + 0.0077 | 0.997 |
| MC-LF | 0.75 | 2.50 | y = 0.4632x − 0.1415 | 0.993 | y = 0.4404x − 0.0193 | 0.994 |
| MC-LW | 0.75 | 2.50 | y = 0.5882x − 0.0726 | 0.999 | y = 0.4380x − 0.0005 | 0.999 |
| MC-LR-Cys | 0.23 | 0.75 | y = 1.4500x − 0.1208 | 0.997 | y = 2.4115x − 0.0030 | 0.999 |
| Mouse | Time after Final Gavage | MC-LR Concentration (ng/g) | MC-LR-Cys Concentration (ng/g) | ||||
|---|---|---|---|---|---|---|---|
| Individual | Average | Standard Deviation | Individual | Average | Standard Deviation | ||
| 1 | 2 h | 0.73 | 0.81 | 0.48 | 40.85 | 38.43 | 14.64 |
| 2 | * | 31.49 | |||||
| 3 | 0.44 | 23.18 | |||||
| 4 | 1.51 | 62.01 | |||||
| 5 | 0.56 | 34.63 | |||||
| 6 | 4 h | 0.55 | 0.68 | 0.15 | 48.60 | 90.18 | 26.45 |
| 7 | 0.87 | 97.24 | |||||
| 8 | 0.75 | 81.32 | |||||
| 9 | * | 111.23 | |||||
| 10 | 0.56 | 112.49 | |||||
| 11 | 48 h | 0.40 | 0.33 | 0.05 | 20.25 | 52.04 | 25.40 |
| 12 | 0.30 | 80.62 | |||||
| 13 | 0.35 | 33.94 | |||||
| 14 | 0.32 | 52.77 | |||||
| 15 | 0.26 | 72.63 | |||||
| Mouse | MC-LR Dosage | Time after Final Gavage | MC-LR Concentration (ng/g) | MC-LR-Cys Concentration (ng/g) | ||||
|---|---|---|---|---|---|---|---|---|
| Individual | Average | Standard Deviation | Individual | Average | Standard Deviation | |||
| 1 | 50 µg/kg | 2 h | 14.21 | 12.64 | 5.37 | 1.17 | 2.77 | 1.38 |
| 2 | 8.37 | 4.38 | ||||||
| 3 | 8.41 | 2.21 | ||||||
| 4 | 19.57 | 3.31 | ||||||
| 5 | 4 h | 11.45 | 12.17 | 5.01 | 2.16 | 2.90 | 1.00 | |
| 6 | 15.55 | 3.16 | ||||||
| 7 | 14.51 | 4.15 | ||||||
| 8 | 3.63 | 1.65 | ||||||
| 9 | 7.18 | 3.41 | ||||||
| 10 | 100 µg/kg | 2 h | 12.95 | 16.38 | 3.96 | 5.10 | 7.23 | 2.03 |
| 11 | 14.78 | 7.44 | ||||||
| 12 | 22.05 | * | ||||||
| 13 | 15.72 | 9.14 | ||||||
| 14 | 4 h | 18.54 | 13.50 | 4.42 | 13.83 | 11.24 | 2.37 | |
| 15 | 10.29 | 9.16 | ||||||
| 16 | 11.66 | 10.75 | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baliu-Rodriguez, D.; Kucheriavaia, D.; Palagama, D.S.W.; Lad, A.; O’Neill, G.M.; Birbeck, J.A.; Kennedy, D.J.; Haller, S.T.; Westrick, J.A.; Isailovic, D. Development and Application of Extraction Methods for LC-MS Quantification of Microcystins in Liver Tissue. Toxins 2020, 12, 263. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040263
Baliu-Rodriguez D, Kucheriavaia D, Palagama DSW, Lad A, O’Neill GM, Birbeck JA, Kennedy DJ, Haller ST, Westrick JA, Isailovic D. Development and Application of Extraction Methods for LC-MS Quantification of Microcystins in Liver Tissue. Toxins. 2020; 12(4):263. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040263
Chicago/Turabian StyleBaliu-Rodriguez, David, Daria Kucheriavaia, Dilrukshika S. W. Palagama, Apurva Lad, Grace M. O’Neill, Johnna A. Birbeck, David J. Kennedy, Steven T. Haller, Judy A. Westrick, and Dragan Isailovic. 2020. "Development and Application of Extraction Methods for LC-MS Quantification of Microcystins in Liver Tissue" Toxins 12, no. 4: 263. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins12040263