Shining a Light on Colibactin Biology


1
Department of Medicine, University of Florida, Gainesville, FL 32610, USA
2
Department of Infectious Diseases and Inflammation, University of Florida, Gainesville, FL 32610, USA
*
Author to whom correspondence should be addressed.
Toxins 2021, 13(5), 346; https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13050346
Submission received: 6 April 2021
/
Revised: 6 May 2021
/
Accepted: 10 May 2021
/
Published: 12 May 2021
(This article belongs to the Special Issue Escherichia coli Toxins and Intestinal Diseases)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Colibactin is a secondary metabolite encoded by the pks gene island identified in several Enterobacteriaceae, including some pathogenic Escherichia coli (E. coli) commonly enriched in mucosal tissue collected from patients with inflammatory bowel disease and colorectal cancer. E. coli harboring this biosynthetic gene cluster cause DNA damage and tumorigenesis in cell lines and pre-clinical models, yet fundamental knowledge regarding colibactin function is lacking. To accurately assess the role of pks+ E. coli in cancer etiology, the biological mechanisms governing production and delivery of colibactin by these bacteria must be elucidated. In this review, we will focus on recent advances in our understanding of colibactin’s structural mode-of-action and mutagenic potential with consideration for how this activity may be regulated by physiologic conditions within the intestine.
Keywords:
colibactin; genotoxin; pks; Escherichia coli; colorectal cancer; DNA damage; mutation; microbiome; inflammation; APCKey Contribution: Colibactin is a potential carcinogenic metabolite produced by pks+ E. coli and few other Enterobacteriaceae. Complex biological and cellular events influence pks+ E. coli-derived genotoxicity and, consequently, carcinogenesis.
1. Introduction
Escherichia coli (E. coli) are a group of Gram-negative facultative anaerobic bacteria isolated from multiple body sites of almost every individual, paradoxically implicated in both intestinal homeostasis and various pathologies depending on species and subspecies classification [1]. Certain E. coli strains, primarily from the B2-phylogroup, harbor a pathogenic island termed pks. This biosynthetic gene cluster is widely distributed throughout various Enterobacteriaceae and encodes for a secondary metabolite named colibactin, putatively acquired through horizontal gene transfer as part of a mobile genetic element [2]. The widespread distribution, prevalence, and evolutionary persistence of pks genes in this bacterial family suggest that this biosynthetic gene cluster may promote host fitness. In contrast, these compounds have deleterious effects in eukaryotic cells, inhibiting cell-cycle progression and inducing DNA damage [3,4]. Recently, the structure of colibactin has been identified [5]. Colibactin contains dual electrophilic warheads each capable of binding DNA, suggesting that colibactin causes DNA damage by inducing interstrand DNA-crosslinks [5,6,7].
The production of a genotoxic cyclomodulin by a common commensal microbe (E. coli) led to the hypothesis that these microbes may play a causative role in a subset of colorectal cancer (CRC) cases, as exposure to environmental genotoxins may promote specific somatic mutations implicated in a variety of cancers [8]. In murine models of colitis-associated cancer, administration of pks+ E. coli promotes tumor formation [4]. Furthermore, pks+ E. coli are identified in tumor biopsies from CRC (66.7%) and IBD cases (40%) at a higher rate than healthy individuals (20.8%) [4]. Subsequent studies utilizing in vitro models ascertained a unique mutational signature occurring after exposure to pks+ E. coli consistent with the hypothesized nucleotide binding activity of the recently proposed colibactin structure, and identified these mutational signatures in approximately 5% of colorectal cancer patients [9]. Collectively, these studies have generated a high amount of research aimed at elucidating colibactin’s mutagenic potential in the context of CRC etiology. However, colibactin itself is a highly unstable molecule and cancer development is dependent on specific biological context, leading to some debate regarding its role as an initiator of tumorigenesis [10]. Thus, a number of recent studies have focused on identifying how colibactin regulation and pks+ E. coli biology may influence the oncogenic capacity of the compound [11,12,13,14,15,16]. In this review, we will summarize how advances in our understanding of colibactin’s structural mode-of-action have begun to clarify the role pks+ E. coli may play in CRC, and how new findings describing pks regulation shed a light on how physiologic contexts may influence its carcinogenic potential.
2. Colibactin Genotoxicity
Bacterial genotoxins are secondary metabolites which directly damage host DNA. Such compounds have the potential to trigger genomic instability, as cells with deficiencies in DNA repair pathways or are utilizing error-prone repair pathways have a high risk of mutational acquisition following exposure to genotoxic stressors in their environment [17]. Because genetic mutations are a hallmark of almost all cancers [18] and pks+ E. coli promote tumor formation in pre-clinical models [4], it is reasonable to theorize that colonization by microbes producing colibactin may be linked to higher cancer risk. However, until recently colibactin’s mode-of-action remained unclear. In this section, we will review how recent advances in structural characterization of this compound have helped elucidate the mutagenic potential of pks+ E. coli, providing a putative mechanism for carcinogenic phenotypes observed nearly a decade earlier.
2.1. Structure and Alkylating Activity
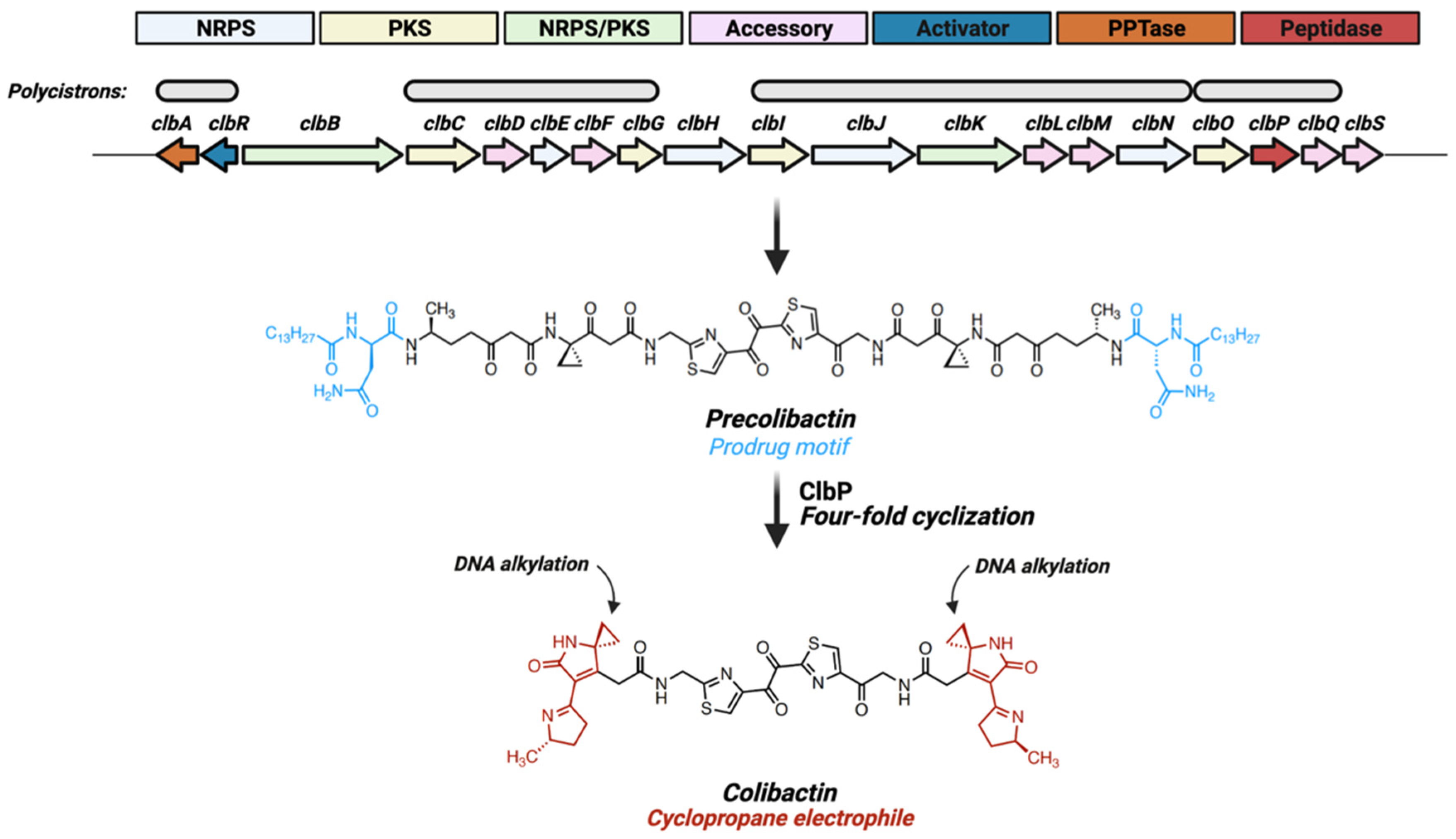
Colibactin is a secondary metabolite produced by the 54-kb pks gene island encoding for a hybrid non-ribosomal peptide synthetase (NRPS)—polyketide synthetase (PKS) assembly line and accessory proteins (clbA—S; Figure 1) [3]. This NRPS-PKS protein assembly produces linear biosynthetic intermediates termed precolibactin which are characterized by a N-myristoyl-D-Asn prodrug motif (Figure 1). Precolibactins are translocated into the periplasm via the multi-drug and toxin extrusion transporter ClbM [19] and then converted to genotoxic colibactin through the removal of prodrug motifs via the membrane-bound peptidase ClbP [20]. Removal of prodrug motifs causes spontaneous four-fold cyclization of linear precolibactins to yield the bioactive colibactin structure (Figure 1) [21]. Colibactin is composed of two nearly symmetrical subunits, each containing an electrophilic cyclopropane warhead which binds adenine residues on DNA to generate interstrand cross-links (Figure 1) [5,21,22]. The incorporation of prodrug motifs in colibactin biosynthesis may be part of a self-resistance mechanism common to toxin biosynthesis pathways in multiple bacterial species [20]. However, studies utilizing clbP mutants to characterize various shunt precolibactins demonstrated that a subset of these molecules, in the presence of copper, generate interstrand cross-links (ICLs) in linearized plasmid DNA [7], a phenomena still not fully understood. For several years, colibactin’s structure and mode-of-action remained elusive due to its resilience to traditional isolation techniques and high instability. Recent studies have utilized DNA-adducts formed during exposure of pks+ bacteria to linearized plasmids [5] or within gnotobiotic mice [22] to characterize the structure of DNA-bound colibactin, providing the first insight into colibactin’s molecular structure and identifying DNA-reactive electrophilic sites capable of alkylating DNA by ring-opening addition [5]. In silico modeling utilizing structures derived from adductomics suggest colibactin has a high binding affinity for adenine rich motifs (AAAATT) within the DNA minor groove [23]. These findings support colibactin’s proposed activity as a DNA cross-linking agent and have helped clarify the molecular mechanisms of colibactin-associated genotoxicity. The next section will address mammalian host response mechanisms associated with pks+ E. coli exposure.
2.2. Genotoxic Activity in Mammalian Cells
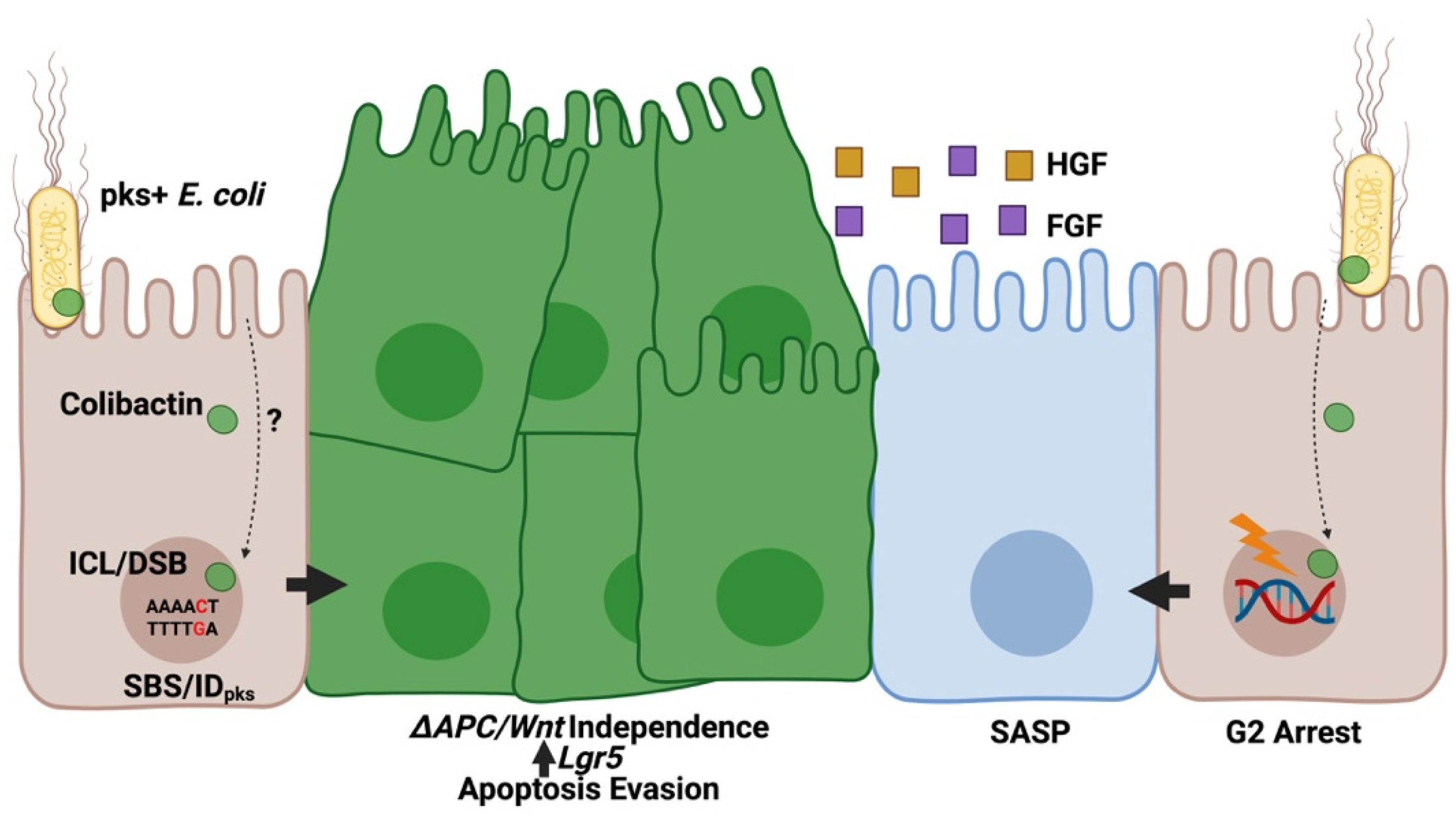
Several studies have demonstrated the genotoxic property of pks+ E. coli and have identified signaling pathways associated with this cellular response. For example, E. coli harboring a bacterial artificial chromosome encoding the complete pks gene island (BACpks) activate classical ataxia telangiectasia (ATM) and Rad53 related (ATR) dependent DNA damage responses (DDR) in infected HeLa cells, characterized by phosphorylation of checkpoint kinase 2 (CHK2), replication protein A (RPA), γH2AX, and the accumulation of phosphorylated cyclin dependent kinase 1 (CDC2) resulting in G2-cell cycle arrest (Figure 2) [3]. Pks-induced DDR activation in HeLa cells promotes recruitment of the interstrand cross-link (ICL) repair protein FANCD2 to phosphorylated γH2AX sites [6], suggesting involvement of the Fanconi-anemia (FA) pathway in repairing colibactin-generated ICLs. While these results are consistent with colibactin’s proposed alkylating activity, several other studies suggest infection with pks+ E. coli also induces double-strand breaks (DSBs), which can be directly visualized by neutral comet assay after infection [3]. In CHO cells, exposure to pks+ E. coli causes increased recruitment of the non-homologous end joining repair protein Ku80 [24], a pathway primarily involved in the resolution of DSBs rather than ICLs. Structural elucidation of colibactin implies that the compound is likely not involved in the direct formation of DSBs, suggesting that DNA breaks may instead be the product of endogenous changes occurring after the formation of alkylated lesions generated by colibactin-mediated cross-linking. Activation of FA pathways may indirectly cause DSBs, as cells excise cross-linked DNA using homologous cellular pathways (e.g., γH2AX) before undergoing repair [25]. Alternatively, the internalization of certain pks+ adherent invasive E. coli (AIEC) strains may promote the production of reactive oxygen species (ROS) which cause DSBs [26] expected to trigger the apurinic/apyrimidinic endonuclease system (APE1). It is unclear if ROS production is dependent on pks or simply a response to bacterial internalization which may exacerbate DNA damage caused by colibactin.
Perhaps the most cryptic aspect of colibactin’s genotoxic mechanisms is the method by which these molecules translocate from the bacterial periplasm to the host nucleus (Figure 2). It is known that cell-to-cell contact is necessary for genotoxicity after pks+ infection in vitro, as separation of mammalian cells and bacteria by cell-impermeable membranes attenuates colibactin’s cytotoxic effects [3]. It seems likely that the compounds instability may prevent remote delivery by pathogenic bacteria but remains unclear if specific secretion systems are necessary to deliver colibactin to host cells. So far, no essential transporters for pks+ genotoxicity have been identified. Interestingly, early experiments inhibiting bacterial internalization did not attenuate BACpks genotoxicity [3]. In contrast, a clinical pks+ AIEC isolate from a CRC patient was shown to invade and replicate within epithelial cells, with a concomitant increase in tumor formation [27]. In murine models administered this strain, oncogenic activity could be attenuated by either effective host autophagy or mutation of the pks editing thioesterase clbQ required for colibactin production [27], suggesting a potential association between intracellular invasion and pks+ AIEC’s tumorigenic efficacy. However, it remains unclear if this internalization enhances colibactin’s effects or is simply a byproduct of lowered host defense (in this case, genetically modified autophagy-deficient mice) while autophagy promotes DDR through a different mechanism. Since release of bacterial outer member vesicles have been linked to DNA damage [28], it would be interesting to investigate this mode of bacterial-host communication in pks+ E. coli mediated genotoxicity.
2.3. Pks-Induced Mutagenesis
In mammalian cells, exposure to pks+ E. coli leads to the accumulation of chromosomal aberrations and aneuploidy, suggesting colibactin exposure may be linked to oncogenic mutation [24]. Acute exposure of human colonic organoids to pks+ E. coli induces functional mutations related to the p53 and Wnt signaling pathways [29]. Importantly, colonic organoid cultures continue to proliferate in the absence of the traditionally essential growth factors Wnt3a and CHIR99021, both regulators of the adenomatous polyposis coli (APC) signaling pathways mutated in 80% of CRC cases [29]. These derived Wnt-independent cultures expressed higher stem-cell transcriptional signatures (e.g., Lgr5, Fzd7, Sox9) and avoided p53-mediated apoptosis following administration of the p53 pathway activator Nutlin-3a, providing direct evidence of oncogenic mutation following pks+ E. coli infection (Figure 2) [29]. Two independent groups identified mutational signatures arising from pks+ E. coli infection in vitro [9,23]. Pleguezuelos-Manazno, Puschhof, and Huber et al. [9] performed whole-genome sequencing of clonal organoids after chronic exposure to pks+ E. coli. These investigators identified a mutational signature characterized by a high number of single-base substitutions (SBS-pks, T > N) and less common short insertion-deletions (ID-pks) within AT-rich DNA regions. Similarly, Dziubanska-Kusibab et al. [23] demonstrated specific enrichment of SBSs within AT-rich pentanucleotide sequences located in the DNA minor groove following short-term exposure of Caco-2 cells to pks+ E. coli. During chronic infection, a majority of SBS-pks motifs occurred within the coding DNA strand and mutations matching either SBS-pks and ID-pks motifs were identified in 5% and 4.4%, respectively, of CRC tumors from an independent database of 2208 predominately primary sites [9]. In these cases the gene with the highest number of mutations matching the pks-target motif was the APC gene (5.3%), which is frequently mutated in CRC cases [9]. Interestingly, the incidence of SBS/ID-pks motifs in CRC cases is much lower than the incidence of pks+ E. coli identified from colorectal tumors (67% [4]), further implying that the carcinogenic capacity of these microbes is derived from the combinatory production of colibactin and other aspects of intestinal ecology (see commentary [30]). Collectively, these findings provide direct evidence of the mutagenic potential of colibactin in the context of CRC (Figure 2). Importantly, presence of pks+ bacteria alone is likely insufficient for cancer development, and the colibactin mutational signature is found in a large number of healthy individuals as well as CRC patients [31], suggesting typically commensal pks+ bacteria may only exert a carcinogenic influence under specific conditions.
3. Colibactin Activity in Physiologic Context
In order to exert a mutagenic influence on the host epithelium in vivo, experimental evidence suggests pks+ E. coli likely requires two physiologic factors, namely: (1) transcriptional activation of all clb genes (besides clbS), and (2) cell-to-cell contact, i.e., a close association between colonizing pks+ strains and host epithelial cells [3]. Bacterial metabolism and biosynthesis of specific compounds is regulated through transcriptional mechanisms influenced by metabolic conditions in the environment [32,33,34]. Thus, an understanding of how pks+ E. coli regulate colibactin with respect to environmental condition, particularly during opportunistic expansion near the host epithelium, is critical to assessing the carcinogenic potential of these bacteria. In this section, we will summarize the current framework of colibactin regulation and how its genotoxic potential may be functionally influenced by environmental condition. Finally, we will summarize a newly emerging research area focused on characterizing colibactin’s effects extending beyond direct DNA damage.
3.1. Metabolic Regulation of pks Genes
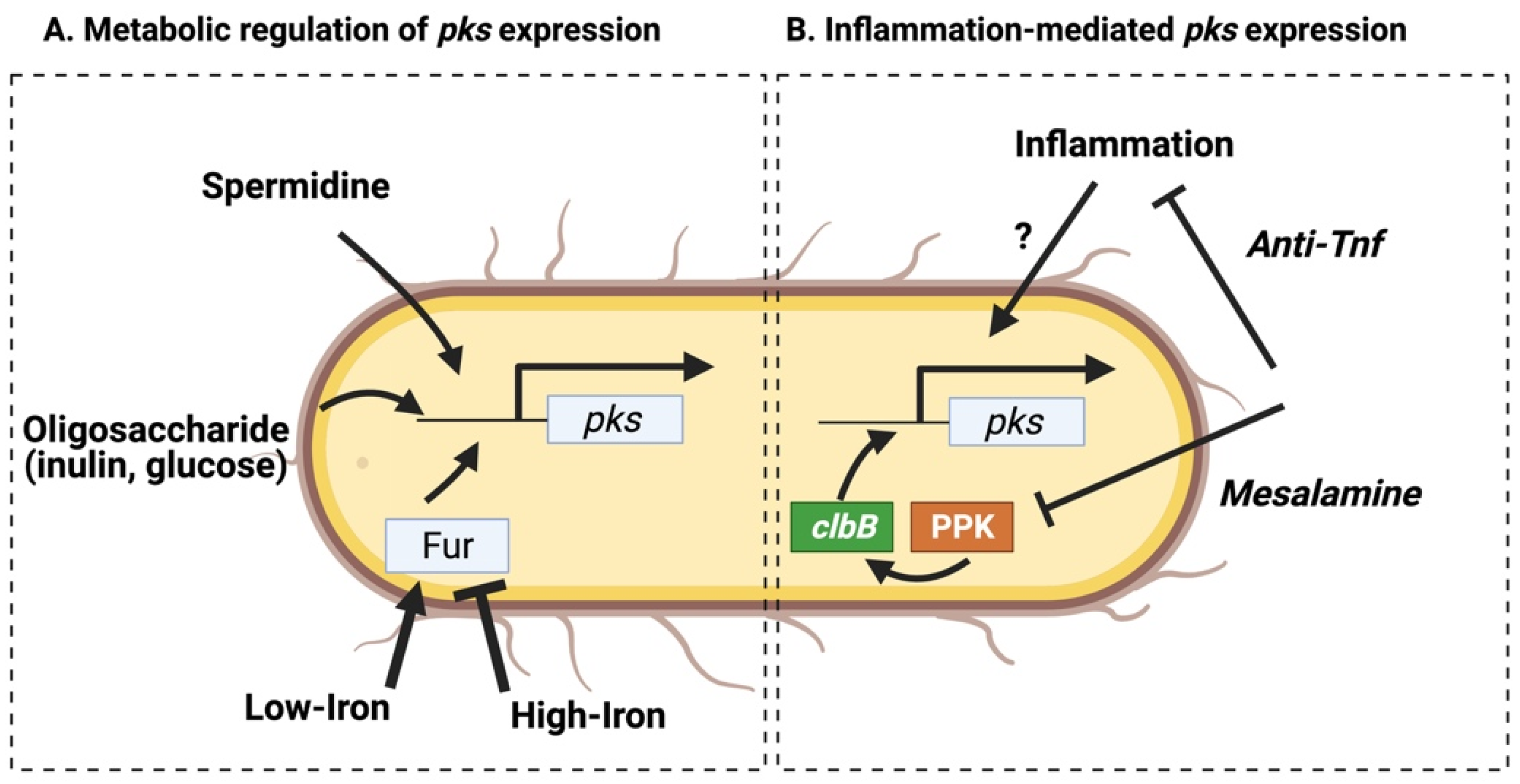
The pks operon consists of four polycistronic transcripts (of seven total) oriented in a single direction, with the exception being a single polycistron encoding the pantetheinyltransferase (PPTase) (clbA) and the LuxR-type transcriptional activator (clbR) (Figure 1) [35] that promote the expression of downstream pks genes [11]. Transcription of these regulatory elements may govern colibactin production, and their expression levels are directly influenced by environmental metabolites (Figure 3a). With respect to the dominant intestinal taxa (e.g., Bacteroidetes, Firmicutes), E. coli have relatively limited glycolytic capacity, instead relying on an abundant siderophoric repertoire to opportunistically thrive in iron-limited conditions [36]. These pathways are controlled by the ferric iron uptake regulator (Fur) that is activated in low-iron conditions and binds directly to the promoter region of the pks gene clbA [15], which in turn acts as both an additional siderophore and an initiator of colibactin biosynthesis [16]. The regulatory pks gene clbR, which acts as a transcription factor for clbB, is similarly upregulated in low iron conditions [11]. As expected given these findings, when pks+ E. coli are cultured in high-iron conditions production of colibactin is inhibited [12]. Additionally, the colibactin transmembrane peptidase ClbP is required for production of the siderophore microcin in pks+ E. coli Nissle 1917 (EcN), and is necessary for colonization in the presence of other opportunistic pathogens such as Salmonella Typhimurium [13]. Thus, pks genes may provide an evolutionary advantage to E. coli strains by enhancing iron-scavenging capabilities by acting as or contributing to the maturation of critical siderophores.
Other metabolites derived endogenously from pks+ E. coli or the host diet similarly alter transcription of clb genes. Spermidine, a polyamine produced during bacterial metabolism or scavenged from the environment is necessary for the production of colibactin [14]. Administration of oligosaccharide prebiotics, such as inulin or glucose, similarly enhance clbA transcription and pks+ genotoxicity [37]. In contrast, metabolites promoting pks transcription can be attenuated by inhibitory factors (such as ferrous sulfate [37]), suggesting that regulation of colibactin production is thus intrinsically linked to metabolic conditions within the intestinal lumen or the mucosal lining. These findings are particularly relevant when considering how the carcinogenic activity of pks+ E. coli may be enhanced in colitis or CRC patients, who often present with anemia [38] or other disruptions to metabolic homeostasis which may alter pks transcription. However, since direct measurement of colibactin level is currently unavailable, the consequences of transcriptional changes in individual pks genes on cellular genotoxicity in vivo is unclear.
3.2. Inflammation
Inflammatory bowel disease (IBD) creates a pro-neoplastic environment within the colonic epithelium [39] that promotes dysplasia and the development of colitis-associated cancer (CAC) [40]. Inflammation heavily modulates microbial balance in the gut, typically associated with increased Proteobacteria/Enterobacteriaceae/E. coli prevalence. For example, chemically-induced inflammation following dextran sodium sulfate (DSS) treatment reduces populations of anaerobic Bacteroidetes by approximately 70% while increasing levels of various aerobic species by approximately 25% [41]. In this acute model, the abundance of a nonpathogenic E. coli strain doubled in DSS treated mice, and colonization in E. coli-naïve mice corresponded with a decrease in mucosal Bacteroidales colonization [41]. Several epidemiological studies have established a link between IBD or CRC and increased mucosal colonization by pks+ E. coli, accounting for as much as 13% (IBD) or 26% (CRC) of all E. coli strains isolated from these patients [4,42,43]. These findings suggest tumorigenesis may be driven, at least partially, by selective enrichment of pks+ strains within the colonic mucosa. This hypothesis is borne out by studies in murine models of CAC utilizing azoxymethane treated Il10−/− mice, showing that the presence of pks+ E. coli promotes DNA damage and neoplastic transformation under inflammatory conditions [4] abrogated in Rag2−/− mice with no inflammatory response [44]. In similar models of CAC, limiting pks+ E. coli colonization during inflammation by inhibiting nitrate reductase activity abrogates pks-associated tumorigenesis [45]. However, the current clinical impracticality of eradicating E. coli populations in colitis patients renders this an unlikely preventative treatment option to limit tumor progression in IBD patient.
Alternatively, elevated pks expression in inflamed tissue may be attenuated as a byproduct of IBD treatments which dampen inflammatory cascades or directly inhibit clb gene transcription. In murine CAC models gavaged with pks+ E. coli, inflammation promotes transcription of multiple clb genes involved in colibactin biosynthesis during tumor initiation [44]. A recent study by Yang et al. [46] demonstrates that anti-TNF treatment prevents colonic inflammation and subsequent tumor development in both chemically-induced (DSS/ApcMin/+) and spontaneous (Il10−/− ApcMin/+) CAC models by modulating microbiota composition and transcription, while co-housing anti-TNF treated with control mice prevented these microbial change and inhibited the anti-tumor effects of TNF neutralization [38]. Interestingly, targeting inflammation with anti-TNF antibody did not alter pks+ E. coli colonization level, pointing to a change in microbial activity [46]. Related to this, the anti-inflammatory drug mesalamine inhibits the microbial enzyme polyphosphate kinase (PPK), sensitizing bacteria to oxidative stress and inhibiting proliferation in vitro [47]. In a small cohort of human test subjects, mesalamine reduced microbial polyP accumulation, suggesting PPK inhibition reduces microbial metabolism in a clinical setting [47]. One would expect that these effects would extend to virulence associated with pks+ E. coli, and the evidence suggests mesalamine directly inhibits colibactin production and ICL formation in vitro [48]. These studies suggests that anti-inflammatory treatment may limit the risk of CRC by acting on the host to limit inflammation-induced dysplasia while simultaneously limiting production of carcinogenic toxins released by opportunistically pathogenic pks+ E. coli (summarized in Figure 3b). Thus, a more nuanced understanding of how modulating the inflammatory environment alters colibactin concentrations may help direct treatment options in IBD patients with high levels of colibactin-producing bacteria, who may be at a higher risk for cancer progression.
3.3. Mucosal Adherence and Biofilm Formation
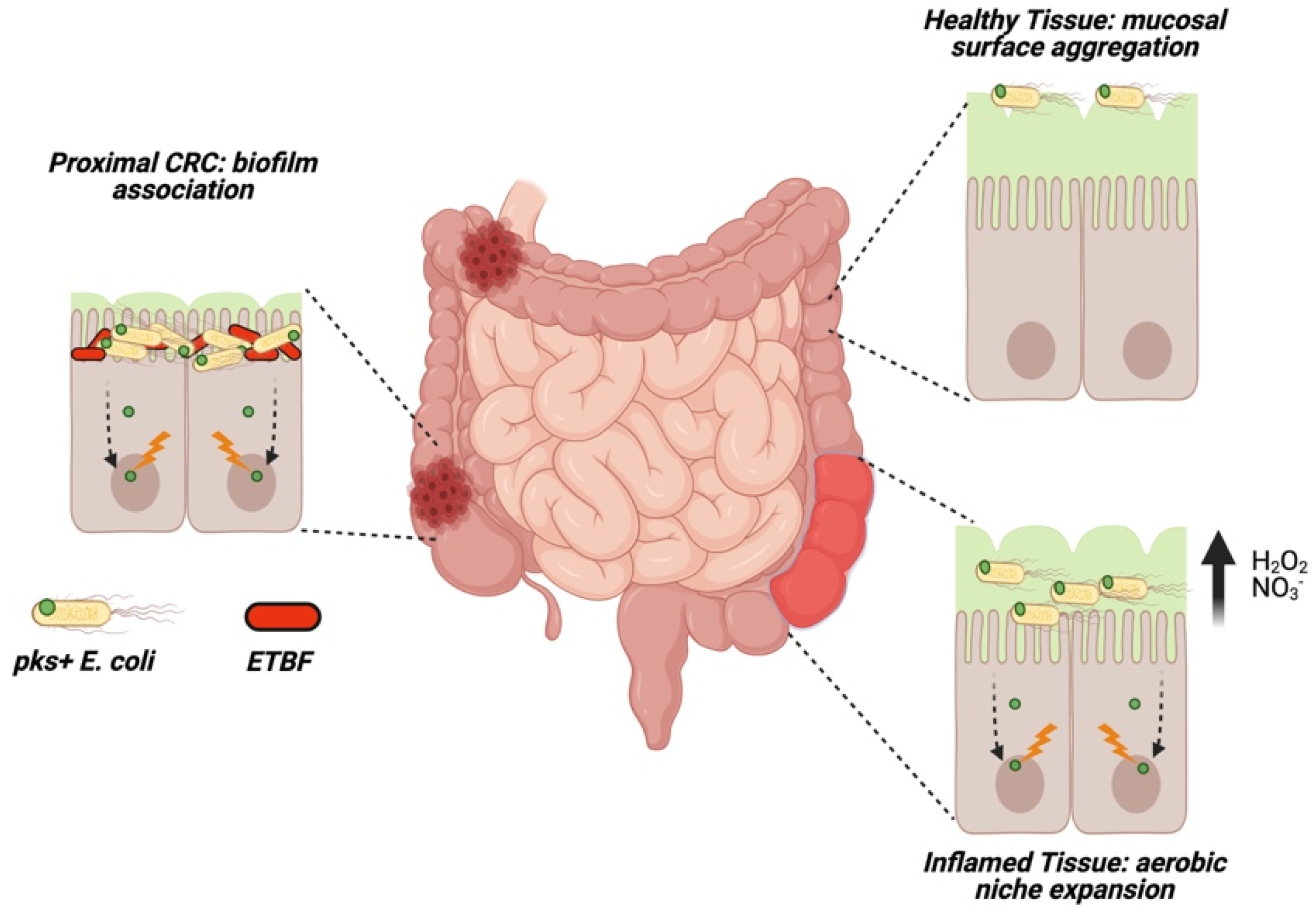
In vitro evidence that cell-to-cell contact is necessary for colibactin’s genotoxic activity suggests that delivery of the molecule in vivo may necessitate pks+ bacteria somehow bypassing the protective mucosal barrier to interact directly with the host epithelium. E. coli possess various fimbral adhesins which promote binding and internalization at the colonic mucosa [49], suggesting pks+ E. coli may colonize and invade the mucosa increasing potential direct contact with epithelial cells. In tumor tissue, this is often the case: biopsies from CRC patients show a higher level of mucosally-invasive E. coli relative to normal adjacent tissue where E. coli aggregate at the mucosal surface (Figure 4) [50,51], and tumors with high microsatellite instability exhibit higher rates of mucosally-invasive E. coli [26]. Clinical E. coli isolates exhibiting high mucosal invasion show high binding affinity to mucus secreting cells in vitro, with pks+ strains inducing high levels of DNA damage exacerbated by mucosal disruption [52]. During inflammation, host production of nitric oxides and reactive oxygen species increases mucosal oxygenation, creating a microaerobic niche in which E. coli preferentially colonize (Figure 4) [53,54]. These changes may promote proliferation of mucosal pks+ E. coli species during inflammation, providing a mechanism for colibactin induced oncogenesis in CAC.
At the mucosal interface, aggregating bacteria can encapsulate themselves in a self-secreted extracellular matrix (i.e., biofilm) to avoid perturbations from host defenses or the intestinal environment [55]. Biofilm structures are often found in patients with intestinal disease, and promote intramucosal invasion [56]. In E. coli strains producing genotoxins, biofilm association increases mucosal invasion depth which may facilitate the delivery of virulent small molecules to host epithelial cells (Figure 4). In two geographically distinct cohorts from the USA and Malaysia, right-sided (proximal) CRC cases almost universally (89%) harbored biofilms both at the tumor site and throughout the normal mucosa [57]. While bacterial diversity in normal and tumor tissue did not differ, biofilm-covered CRCs exhibited higher bacterial invasion into the underlying tumor tissue [57].
Several studies have used a combination of in-situ hybridization and 16S rRNA gene sequencing to investigate the composition of mucosal biofilms in CRC patients, frequently identifying Proteobacteria in mucosal biofilms from both tumor and normal adjacent tissue, with a high prevalence of pks+ E. coli (68%) in proximal CRC biofilms [58,59]. Biofilms harvested from both normal and tumor tissue from CRC increase mucosal invasion by gut microbes and induce a higher tumor burden in murine models [60], suggesting some element of core biofilm structure, a commonly distributed biofilm-associated species, or a combination of these factors, promotes carcinogenesis. A causative role for pks+ E. coli seems likely given their ubiquitous distribution throughout biofilms isolated from CRC patients. Co-infection with the biofilm initiating strain enterotoxigenic Bacteroides fragilis (ETBF) and pks+ E. coli in specific pathogen free (SPF) mice demonstrate that biofilm association promotes mucosal invasion, expansion of pks+ E. coli populations, and a concomitant increase in γH2AX histological staining and tumor burden relative to monoassociation [58]. ETBF may further promote interaction between pks+ E. coli and host epithelial cells by degrading the protective mucus layer through an undetermined mechanism (see commentary [61]), minimizing a crucial barrier for E. coli adherence. Importantly, deletion of B. fragilis toxin (ETBF) or colibactin (E. coli) diminished tumor burdens, suggesting the existence of a cooperative network between these toxins [58]. However, the use of SPF mice in these experiments makes it difficult to distinguish if additional bacteria are involved in biofilm initiation and tumor development.
Altogether, these findings suggest that pks+ E. coli association with microbial biofilms enhances colibactin’s carcinogenic activity, possibly by increasing proximity to host epithelial cells and enhancing total abundance of these bacteria within the mucosa. In these scenarios, normally innocuous genotoxins secreted by the bacteria may occur in elevated doses near replicating host cells resulting in carcinogenic activity and CRC initiation or progression.
3.4. Modulation of Tumor Microenvironment
While colibactin can directly promote oncogenic transformation by DNA alkylation and subsequent mutagenesis in epithelial cells [6,9,23,29], its role in CRC progression extends beyond these activities by helping to establish a pro-carcinogenic environment supporting tumor growth [62,63,64]. After pks-induced DNA damage, cells may undergo apoptosis, transformation, or senescence. Adoption of a senescent state induces a senescence-associated secretory phenotype (SASP) characterized by increased secretion of various growth factors that promote proliferation of nearby cells (Figure 2) [62,63]. These changes may occur near cells that have acquired pks-generated mutations in the APC or p53 pathways after colibactin exposure [29], further promoting unrestricted growth and tumor proliferation. A recent study demonstrated that the pks+ E. coli strain 11G5 translocates to mesenteric lymph nodes in ApcMin/+ mice, with a concomitant reduction of some cytotoxic T cell lineages and an increase in regulatory T cells within infected lymph nodes [64]. Furthermore, levels of cytotoxic T cells are reduced within the colonic mucosa of 11G5-colonized mice and invasive margins from tumor biopsies from CRC patients colonized by pks+ E. coli [64], suggesting that colibactin-producing microbes may create a pro-carcinogenic environment within the gut by modulating immune cell activity. Collectively, these observations suggest that colibactin-producing E. coli may create a “perfect storm” of tumorigenic potential, simultaneously promoting oncogenic transformation in epithelial cells and generating populations of bystander tumor-promoting cells while restricting immune activation.
Colibactin may also influence interactions between pks+ E. coli and other intestinal bacteria, altering the complex ecological networks observed within gut microbiota communities [65]. Shunt precolibactins have been shown to have mild inhibitory effects on the growth of a common probiotic, Bacillus subtilis, in vitro [66]. The effects of colibactin on microbial communities in vivo remain poorly characterized, but at least one study has directly demonstrated that colonization by pks+ E. coli in murine models reduces the relative abundance of Firmicutes and Clostridia [67], taxa which represent a significant proportion of microbes in healthy individuals [68]. Disruption of the ratio of Firmicutes to Bacteroidetes has been used as a marker of various metabolic disorders [69] and a general measure of dysbiosis. The exact nature of how colibactin production may alter transcriptional regulation or abundance of gut microbes is not well understood. However, the relevance of microbial dysbiosis in the etiology of various cancers is well-defined [70,71,72]. Future studies focusing on how colibactin influences multi-kingdom interactions in the gut may elucidate novel pathways by which pks+ bacterial colonization deleteriously affects their host.
4. Conclusions
Overrepresentation of pks+ E. coli in CRC cases has led to the hypothesis that these bacteria may directly promote tumorigenesis. Several biological characteristics of colibactin supports a role for this secondary molecule in carcinogenesis. First, colibactin contains two electrophilic cyclopropane warheads that bind adenine-rich motifs in host DNA, creating inter-strand cross-links that stall cell cycle progression and activate DNA damage response pathways. Infection with pks+ E. coli causes somatic mutation that induces oncogenic transformation in colonoids and promotes tumor development in vivo. However, the prevalence of pks+ E. coli colonization far exceeds the observed proportion of individuals carrying mutational signatures attributed to the genotoxin, suggesting physiological context dictates the compounds genotoxic activity. This raises an important question: is the presence of pks genes enough to produce colibactin with functional genotoxic effect on host epithelium? An interesting case-study can be made by investigating the genotoxic potential of the commercially available probiotic strain E. coli Nissle 1917 (EcN), that has a long history of use as a beneficial probiotic [73] despite carrying the pks biosynthetic gene cluster [3]. Counterintuitively, the pks peptidase ClbP is actually essential for the antibacterial activity of EcN, contributing to the maturation of siderophores that enable this probiotic to outcompete pathogenic bacteria [13]. Whether or not EcN infection results in DNA damage is debated. While one recent study reported no genotoxic effects of EcN were observable in cell lines or murine models [74], others have demonstrated genotoxicity in mammalian cell lines attenuated by clbA mutation [13]. Thus, more work is necessary to determine if these strains produce physiologically active colibactin, and if long-term intake of this probiotic may increase cancer risk.
Pks+ bacteria may have relevance to cancer outside of the intestine. These bacteria frequently colonize extraintestinal sites and colibactin mutational signatures have been reported in a subset of patients with head and neck, urinary tract, neuroendocrine tumors, and ovarian cancer [9]. A higher concentration of colibactin biosynthetic byproducts in the urine of patients with urinary tract infection (UTI) relative to healthy individuals, and an archetypal UTI strain of pks+ E. coli causes DNA damage within the regenerative compartment of the bladder after transurethral infection in mice, suggesting a potential role for these microbes in bladder cancer [75]. Two separate studies currently published as pre-prints have identified a high proportion of pks+ E. coli in association with DNA damage in biopsies from prostate cancer patients [76] and pks+ Klebsiella pneumoniae in a subset of hospital patients with liver abscess [77]. These findings suggest that colibactin may play a role in a variety of cancers beyond CRC. Although solid evidence supports the case for colibactin as a carcinogen, the oncogenic potential of pks+ E. coli may be influenced by many factors altering colibactin production and localization within the intestinal tract. Colibactin’s instability suggests it may only exhibit oncogenic activity while in close association with host epithelial cells, and limits mechanistic studies aimed at studying the effects of the toxin without influence from other aspects of bacterial infection or unrelated virulence factors. Because all colibactin-related studies utilize pks or clb mutants (e.g., [3,5,6,13]) which may influence other biosynthetic pathways in ways we do not completely understand [78], it is difficult to attribute phenotypes directly to colibactin. Recent structural characterization of colibactin may facilitate the production of synthetic compounds which can be used to answer questions specifically regarding structure-activity relationship. These synthetic compounds would allow for more comprehensive and accurate screening methods capable of identifying pathways involved in DNA repair following exposure to pks+ E. coli, or for the derivation of targeted therapeutics which inhibit genotoxic activity by directly inhibiting colibactin’s mechanism-of-action. Furthermore, it remains unclear how this large transitory molecule migrates from the bacterial periplasm to the host nucleus or which DNA repair pathways eukaryotic cells use to repair cross-links formed after colibactin exposure. Future studies should focus on clarifying fundamental aspects of colibactin’s biology such as this, which remain mysterious and may inform future therapeutic approaches aimed at reducing pks-associated cancer risk. Pertinent research questions and potential clinical applications involving pks+ E. coli are summarized in Figure 5.
The most evident question is related to colibactin’s function within the microbiota. What biological advantage is gained by E. coli strains carrying the pks island? One would assume that the biosynthetic gene cluster confers a fitness advantage, but data supporting this function are limited. The extent of colibactin’s activity on other microbes (bacteria, fungi, archaea, and viruses) inhabiting the intestine is unclear at best. It is intriguing to note that certain secondary metabolites generated from biosynthetic gene clusters are implicated in bacteria anti-phage defense response [79], thereby favoring fitness to bacteria carrying this weaponry. Whether the secondary metabolite colibactin enhances bacterial fitness through anti-phage responses is currently unknown. On the flip side, colibactin analogs may find applications as chemotherapeutic agents. For example, the NPRS/PKS molecule bleomycin, which induces DNA damage is currently used as anti-cancer drug for various form of cancers [80]. There is a long road ahead before colibactin finds application in chemotherapy and for now, all efforts are focused on its cancer promoting ability.
Evidence suggests that pks-derived mutational signatures are acquired during childhood [9,31] and thus may contribute towards oncogenic transformation which does not appear for decades, until additional factors promote tumorigenesis. The ubiquity of B2 phylogroup E. coli in the human microbiome, especially at early age [81], raise the concern of long-term consequences on DNA integrity in asymptomatic hosts. However, until further clinical evidence assigns a clear carcinogenic label to pks+ E. coli, the need for childhood screening is nonexistent. If this moment ever arises, the challenge would be to design precision microbiome interventions since antibiotic approach would likely do more harm than good in this young population. Engineered probiotics [82] may be designed to specifically target pks+ microbes or produce metabolites which directly interrupt colibactin activity. Such small molecule inhibitors may come from a self-resistance component of the pks island itself, the cyclopropane hydrolase ClbS, which disrupts active cyclopropane sites in colibactin [83,84]. Phage screening may identify viruses capable of targeted depletion of specific microbial species [85], and screening pks+ isolates from individuals against phage libraries may allow for the creation of personalized phage cocktails with high activity for patient-specific strains. Such an approach has been used in pre-clinical model of CRC driven by pks+ E. coli [86]. In addition, predatory bacteria against AIEC such as Bdellovibrio bacteriovorus could represent another means to selectively deplete pks+ E.coli from a complex community [87]. In conclusion, it has been a remarkable journey from pks/colibactin’s discovery to molecular structure characterization and potential in vivo function. Yet, much remains to be addressed regarding this fascinating molecule and the years to come promise to be exciting for this field of research.
Author Contributions
M.W.D., writing—original draft preparation; C.J., writing—review and editing; supervision; funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health, grant number R01CA215553-O1A1, University of Florida Health Cancer Center Funds and University of Florida Department of Medicine Gatorade Funds.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The authors gratefully acknowledge Kevin M. Wernke for critical revision of sections related to colibactin structure and diagrammatic representations of the molecule. Figures were made using BioRender.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Tenaillon, O.; Skurnik, D.; Picard, B.; Denamur, E. The population genetics of commensal Escherichia coli. Nat. Rev. Microbiol. 2010, 8, 207–217. [Google Scholar] [CrossRef]
- Messerer, M.; Fischer, W.; Schubert, S. Investigation of horizontal gene transfer of pathogenicity islands in Escherichia coli using next-generation sequencing. PLoS ONE 2017, 12, e0179880. [Google Scholar] [CrossRef] [Green Version]
- Nougayrède, J.-P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, M.; Kim, C.S.; Healy, A.R.; Wernke, K.M.; Wang, Z.; Frischling, M.C.; Shine, E.E.; Wang, W.; Herzon, S.B.; Crawford, J.M. Structure elucidation of colibactin and its DNA cross-links. Science 2019, 365, eaax2685. [Google Scholar] [CrossRef] [PubMed]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrède, J.-P. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. mBio 2018, 9, e02393-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vizcaino, M.I.; Crawford, J.M. The colibactin warhead crosslinks DNA. Nat. Chem. 2015, 7, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Rosendahl Huber, A.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Wassenaar, T.M. E. coli and colorectal cancer: A complex relationship that deserves a critical mindset. Crit. Rev. Microbiol. 2018, 44, 619–632. [Google Scholar] [CrossRef] [Green Version]
- Wallenstein, A.; Rehm, N.; Brinkmann, M.; Selle, M.; Bossuet-Greif, N.; Sauer, D.; Bunk, B.; Spröer, C.; Wami, H.T.; Homburg, S.; et al. ClbR Is the Key Transcriptional Activator of Colibactin Gene Expression in Escherichia coli. mSphere 2020, 5, e00591-20. [Google Scholar] [CrossRef]
- Tronnet, S.; Garcie, C.; Brachmann, A.O.; Piel, J.; Oswald, E.; Martin, P. High iron supply inhibits the synthesis of the genotoxin colibactin by pathogenic Escherichia coli through a non-canonical Fur/RyhB-mediated pathway. Pathog. Dis. 2017, 75, ftx066. [Google Scholar] [CrossRef]
- Massip, C.; Branchu, P.; Bossuet-Greif, N.; Chagneau, C.V.; Gaillard, D.; Martin, P.; Boury, M.; Sécher, T.; Dubois, D.; Nougayrède, J.-P.; et al. Deciphering the interplay between the genotoxic and probiotic activities of Escherichia coli Nissle 1917. PLoS Pathog. 2019, 15, e1008029. [Google Scholar] [CrossRef]
- Chagneau, C.V.; Garcie, C.; Bossuet-Greif, N.; Tronnet, S.; Brachmann, A.O.; Piel, J.; Nougayrède, J.-P.; Martin, P.; Oswald, E. The polyamine spermidine modulates the production of the bacterial genotoxin colibactin. mSphere 2019, 4, e00414-19. [Google Scholar] [CrossRef] [Green Version]
- Tronnet, S.; Garcie, C.; Rehm, N.; Dobrindt, U.; Oswald, E.; Martin, P. Iron Homeostasis Regulates the Genotoxicity of Escherichia coli That Produces Colibactin. Infect. Immun. 2016, 84, 3358–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, P.; Marcq, I.; Magistro, G.; Penary, M.; Garcie, C.; Payros, D.; Boury, M.; Olier, M.; Nougayrède, J.-P.; Audebert, M.; et al. Interplay between siderophores and colibactin genotoxin biosynthetic pathways in Escherichia coli. PLoS Pathog. 2013, 9, e1003437. [Google Scholar] [CrossRef]
- Volkova, N.V.; Meier, B.; González-Huici, V.; Bertolini, S.; Gonzalez, S.; Vöhringer, H.; Abascal, F.; Martincorena, I.; Campbell, P.J.; Gartner, A.; et al. Mutational signatures are jointly shaped by DNA damage and repair. Nat. Commun. 2020, 11, 2169. [Google Scholar] [CrossRef]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Mousa, J.J.; Yang, Y.; Tomkovich, S.; Shima, A.; Newsome, R.C.; Tripathi, P.; Oswald, E.; Bruner, S.D.; Jobin, C. MATE transport of the E. coli-derived genotoxin colibactin. Nat. Microbiol. 2016, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, C.A.; Balskus, E.P. A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J. Am. Chem. Soc. 2013, 135, 3359–3362. [Google Scholar] [CrossRef]
- Wernke, K.M.; Xue, M.; Tirla, A.; Kim, C.S.; Crawford, J.M.; Herzon, S.B. Structure and bioactivity of colibactin. Bioorg. Med. Chem. Lett. 2020, 30, 127280. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef] [PubMed]
- Dziubańska-Kusibab, P.J.; Berger, H.; Battistini, F.; Bouwman, B.A.M.; Iftekhar, A.; Katainen, R.; Cajuso, T.; Crosetto, N.; Orozco, M.; Aaltonen, L.A.; et al. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat. Med. 2020, 26, 1063–1069. [Google Scholar] [CrossRef]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, N.; Takahashi, A.; Ono, K.; Ohnishi, T. DNA damage induced by alkylating agents and repair pathways. J. Nucleic Acids 2010, 2010, 543531. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, O.D.K.; Scanlon, K.M.; Donnenberg, M.S. An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. mBio 2013, 4, e00152-13. [Google Scholar] [CrossRef] [Green Version]
- Lucas, C.; Salesse, L.; Hoang, M.H.T.; Bonnet, M.; Sauvanet, P.; Larabi, A.; Godfraind, C.; Gagnière, J.; Pezet, D.; Rosenstiel, P.; et al. Autophagy of Intestinal Epithelial Cells Inhibits Colorectal Carcinogenesis Induced by Colibactin-Producing Escherichia coli in ApcMin/+ Mice. Gastroenterology 2020, 158, 1373–1388. [Google Scholar] [CrossRef] [Green Version]
- Murase, K.; Martin, P.; Porcheron, G.; Houle, S.; Helloin, E.; Pénary, M.; Nougayrède, J.-P.; Dozois, C.M.; Hayashi, T.; Oswald, E. HlyF Produced by Extraintestinal Pathogenic Escherichia coli Is a Virulence Factor That Regulates Outer Membrane Vesicle Biogenesis. J. Infect. Dis. 2016, 213, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat. Commun. 2021, 12, 1003. [Google Scholar] [CrossRef]
- Yang, Y.; Jobin, C. A mutational signature that can be made by a bacterium arises in human colon cancer. Nature 2020, 580, 194–195. [Google Scholar] [CrossRef] [Green Version]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Turnbough, C.L. Regulation of bacterial gene expression by transcription attenuation. Microbiol. Mol. Biol. Rev. 2019, 83, e00019-19. [Google Scholar] [CrossRef]
- Raibaud, O.; Schwartz, M. Positive control of transcription initiation in bacteria. Annu. Rev. Genet. 1984, 18, 173–206. [Google Scholar] [CrossRef]
- Trastoy, R.; Manso, T.; Fernández-García, L.; Blasco, L.; Ambroa, A.; Pérez Del Molino, M.L.; Bou, G.; García-Contreras, R.; Wood, T.K.; Tomás, M. Mechanisms of bacterial tolerance and persistence in the gastrointestinal and respiratory environments. Clin. Microbiol. Rev. 2018, 31, e00023-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homburg, S.; Oswald, E.; Hacker, J.; Dobrindt, U. Expression analysis of the colibactin gene cluster coding for a novel polyketide in Escherichia coli. FEMS Microbiol. Lett. 2007, 275, 255–262. [Google Scholar] [CrossRef]
- Li, H.; Limenitakis, J.P.; Fuhrer, T.; Geuking, M.B.; Lawson, M.A.; Wyss, M.; Brugiroux, S.; Keller, I.; Macpherson, J.A.; Rupp, S.; et al. The outer mucus layer hosts a distinct intestinal microbial niche. Nat. Commun. 2015, 6, 8292. [Google Scholar] [CrossRef]
- Oliero, M.; Calvé, A.; Fragoso, G.; Cuisiniere, T.; Hajjar, R.; Dobrindt, U.; Santos, M.M. Oligosaccharides increase the genotoxic effect of colibactin produced by pks+ Escherichia coli strains. BMC Cancer 2021, 21, 172. [Google Scholar] [CrossRef]
- Kaitha, S.; Bashir, M.; Ali, T. Iron deficiency anemia in inflammatory bowel disease. World J. Gastrointest. Pathophysiol. 2015, 6, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Mark-Christensen, A.; Laurberg, S.; Haboubi, N. Dysplasia in inflammatory bowel disease: Historical review, critical histopathological analysis, and clinical implications. Inflamm. Bowel Dis. 2018, 24, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Terzić, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- Lupp, C.; Robertson, M.L.; Wickham, M.E.; Sekirov, I.; Champion, O.L.; Gaynor, E.C.; Finlay, B.B. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007, 2, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Raisch, J.; Buc, E.; Bonnet, M.; Sauvanet, P.; Vazeille, E.; de Vallée, A.; Déchelotte, P.; Darcha, C.; Pezet, D.; Bonnet, R.; et al. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J. Gastroenterol. 2014, 20, 6560–6572. [Google Scholar] [CrossRef]
- Buc, E.; Dubois, D.; Sauvanet, P.; Raisch, J.; Delmas, J.; Darfeuille-Michaud, A.; Pezet, D.; Bonnet, R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE 2013, 8, e56964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, J.C.; Gharaibeh, R.Z.; Mühlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; McCafferty, J.; Fodor, A.A.; Jobin, C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 2014, 5, 4724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Miyata, N.; Winter, M.G.; Arenales, A.; Hughes, E.R.; Spiga, L.; Kim, J.; Sifuentes-Dominguez, L.; Starokadomskyy, P.; Gopal, P.; et al. Editing of the gut microbiota reduces carcinogenesis in mouse models of colitis-associated colorectal cancer. J. Exp. Med. 2019, 216, 2378–2393. [Google Scholar] [CrossRef]
- Yang, Y.; Gharaibeh, R.Z.; Newsome, R.C.; Jobin, C. Amending microbiota by targeting intestinal inflammation with TNF blockade attenuates development of colorectal cancer. Nat. Cancer 2020, 1, 723–734. [Google Scholar] [CrossRef]
- Dahl, J.-U.; Gray, M.J.; Bazopoulou, D.; Beaufay, F.; Lempart, J.; Koenigsknecht, M.J.; Wang, Y.; Baker, J.R.; Hasler, W.L.; Young, V.B.; et al. The anti-inflammatory drug mesalamine targets bacterial polyphosphate accumulation. Nat. Microbiol. 2017, 2, 16267. [Google Scholar] [CrossRef]
- Tang-Fichaux, M.; Chagneau, C.V.; Bossuet-Greif, N.; Nougayrède, J.-P.; Oswald, É.; Branchu, P. The Polyphosphate Kinase of Escherichia coli Is Required for Full Production of the Genotoxin Colibactin. mSphere 2020, 5, e01195-20. [Google Scholar] [CrossRef] [PubMed]
- Dumych, T.; Yamakawa, N.; Sivignon, A.; Garenaux, E.; Robakiewicz, S.; Coddeville, B.; Bongiovanni, A.; Bray, F.; Barnich, N.; Szunerits, S.; et al. Oligomannose-Rich Membranes of Dying Intestinal Epithelial Cells Promote Host Colonization by Adherent-Invasive E. coli. Front. Microbiol. 2018, 9, 742. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, O.D.K.; Short, A.J.; Donnenberg, M.S.; Bader, S.; Harrison, D.J. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS ONE 2009, 4, e5517. [Google Scholar] [CrossRef] [Green Version]
- Martin, H.M.; Campbell, B.J.; Hart, C.A.; Mpofu, C.; Nayar, M.; Singh, R.; Englyst, H.; Williams, H.F.; Rhodes, J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 2004, 127, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Reuter, C.; Alzheimer, M.; Walles, H.; Oelschlaeger, T.A. An adherent mucus layer attenuates the genotoxic effect of colibactin. Cell Microbiol. 2018, 20, 12812. [Google Scholar] [CrossRef] [Green Version]
- Chanin, R.B.; Winter, M.G.; Spiga, L.; Hughes, E.R.; Zhu, W.; Taylor, S.J.; Arenales, A.; Gillis, C.C.; Büttner, L.; Jimenez, A.G.; et al. Epithelial-Derived Reactive Oxygen Species Enable AppBCX-Mediated Aerobic Respiration of Escherichia coli during Intestinal Inflammation. Cell Host Microbe 2020, 28, 780–788.e5. [Google Scholar] [CrossRef]
- Winter, S.E.; Winter, M.G.; Xavier, M.N.; Thiennimitr, P.; Poon, V.; Keestra, A.M.; Laughlin, R.C.; Gomez, G.; Wu, J.; Lawhon, S.D.; et al. Host-derived nitrate boosts growth of E. coli in the inflamed gut. Science 2013, 339, 708–711. [Google Scholar] [CrossRef] [Green Version]
- Tytgat, H.L.P.; Nobrega, F.L.; van der Oost, J.; de Vos, W.M. Bowel Biofilms: Tipping Points between a Healthy and Compromised Gut? Trends Microbiol. 2019, 27, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Motta, J.-P.; Allain, T.; Green-Harrison, L.E.; Groves, R.A.; Feener, T.; Ramay, H.; Beck, P.L.; Lewis, I.A.; Wallace, J.L.; Buret, A.G. Iron sequestration in microbiota biofilms as A novel strategy for treating inflammatory bowel disease. Inflamm. Bowel Dis. 2018, 24, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Mark Welch, J.L.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326. [Google Scholar] [CrossRef] [Green Version]
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; DeStefano Shields, C.E.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microb. 2017, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Tomkovich, S.; Dejea, C.M.; Winglee, K.; Drewes, J.L.; Chung, L.; Housseau, F.; Pope, J.L.; Gauthier, J.; Sun, X.; Mühlbauer, M.; et al. Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J. Clin. Investig. 2019, 129, 1699–1712. [Google Scholar] [CrossRef] [PubMed]
- Tomkovich, S.; Jobin, C. Microbial networking in cancer: When two toxins collide. Br. J. Cancer 2018, 118, 1407–1409. [Google Scholar] [CrossRef] [Green Version]
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Déchelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef]
- Lopès, A.; Billard, E.; Casse, A.H.; Villéger, R.; Veziant, J.; Roche, G.; Carrier, G.; Sauvanet, P.; Briat, A.; Pagès, F.; et al. Colibactin-positive Escherichia coli induce a procarcinogenic immune environment leading to immunotherapy resistance in colorectal cancer. Int. J. Cancer 2020, 146, 3147–3159. [Google Scholar] [CrossRef] [Green Version]
- Rao, C.; Coyte, K.Z.; Bainter, W.; Geha, R.S.; Martin, C.R.; Rakoff-Nahoum, S. Multi-kingdom ecological drivers of microbiota assembly in preterm infants. Nature 2021, 591, 633–638. [Google Scholar] [CrossRef]
- Vizcaino, M.I.; Engel, P.; Trautman, E.; Crawford, J.M. Comparative metabolomics and structural characterizations illuminate colibactin pathway-dependent small molecules. J. Am. Chem. Soc. 2014, 136, 9244–9247. [Google Scholar] [CrossRef]
- Tronnet, S.; Floch, P.; Lucarelli, L.; Gaillard, D.; Martin, P.; Serino, M.; Oswald, E. The genotoxin colibactin shapes gut microbiota in mice. mSphere 2020, 5, e00589-20. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The firmicutes/bacteroidetes ratio: A relevant marker of gut dysbiosis in obese patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, X.; Xu, H.; Li, S.; Cheuk-Hay Lau, H.; Chen, Q.; Zhang, B.; Zhao, L.; Chen, H.; Jao-Yiu Sung, J.; et al. Microbial community heterogeneity within colorectal neoplasia and its correlation with colorectal carcinogenesis. Gastroenterology 2021. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, R.M.; Jobin, C. Microbiota in pancreatic health and disease: The next frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M. Clinical use of E. coli Nissle 1917 in inflammatory bowel disease. Inflamm. Bowel Dis. 2008, 14, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Dubbert, S.; Klinkert, B.; Schimiczek, M.; Wassenaar, T.M.; von Bünau, R. No Genotoxicity Is Detectable for Escherichia coli Strain Nissle 1917 by Standard In Vitro and In Vivo Tests. Eur. J. Microbiol. Immunol. 2020, 10, 11–19. [Google Scholar] [CrossRef]
- Chagneau, C.V.; Massip, C.; Bossuet-Greif, N.; Fremez, C.; Motta, J.-P.; Shima, A.; Besson, C.; Le Faouder, P.; Cénac, N.; Roth, M.-P.; et al. coli induces DNA damage in the bladder. PLoS Pathog. 2021, 17, e1009310. [Google Scholar] [CrossRef]
- Shrestha, E.; Coulter, J.B.; Guzman, W.; Ozbek, B.; Mummert, L.; Ernst, S.E.; Maynard, J.P.; Meeker, A.K.; Heaphy, C.M.; Haffner, M.C.; et al. Oncogenic Gene Fusions in Non-Neoplastic Precursors as Evidence that Bacterial Infection Initiates Prostate Cancer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bohrhunter, J.; Taffner, S.; Wang, J.; Hardy, D.; Pecora, N. Genomic Characterization of Tissue Invasive Klebsiella pneumoniae Complex in a Hospital System with a Focus on Species Distribution and Hypervirulence. bioRxiv 2020. [Google Scholar] [CrossRef]
- Balskus, E.P. Colibactin: Understanding an elusive gut bacterial genotoxin. Nat. Prod. Rep. 2015, 32, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Kronheim, S.; Daniel-Ivad, M.; Duan, Z.; Hwang, S.; Wong, A.I.; Mantel, I.; Nodwell, J.R.; Maxwell, K.L. A chemical defence against phage infection. Nature 2018, 564, 283–286. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Du, L.; Sanchez, C.; Edwards, D.J.; Chen, M.; Murrell, J.M. The biosynthetic gene cluster for the anticancer drug bleomycin from Streptomyces verticillus ATCC15003 as a model for hybrid peptide-polyketide natural product biosynthesis. J. Ind. Microbiol. Biotechnol. 2001, 27, 378–385. [Google Scholar] [CrossRef]
- Secher, T.; Brehin, C.; Oswald, E. Early settlers: Which E. coli strains do you not want at birth? Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G123–G129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, G.; Bikard, D. Editing the microbiome the CRISPR way. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2019, 374, 20180103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, P.; Shine, E.E.; Healy, A.R.; Kim, C.S.; Herzon, S.B.; Bruner, S.D.; Crawford, J.M. Clbs is a cyclopropane hydrolase that confers colibactin resistance. J. Am. Chem. Soc. 2017, 139, 17719–17722. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Dubois, D.; Petit, C.; Tronnet, S.; Martin, P.; Bonnet, R.; Oswald, E.; Nougayrède, J.-P. Escherichia coli ClbS is a colibactin resistance protein. Mol. Microbiol. 2016, 99, 897–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divya Ganeshan, S.; Hosseinidoust, Z. Phage Therapy with a Focus on the Human Microbiota. Antibiotics 2019, 8, 131. [Google Scholar] [CrossRef] [Green Version]
- Gogokhia, L.; Buhrke, K.; Bell, R.; Hoffman, B.; Brown, D.G.; Hanke-Gogokhia, C.; Ajami, N.J.; Wong, M.C.; Ghazaryan, A.; Valentine, J.F.; et al. Expansion of bacteriophages is linked to aggravated intestinal inflammation and colitis. Cell. Host Microbe 2019, 25, 285–299.e8. [Google Scholar] [CrossRef] [Green Version]
- Bonfiglio, G.; Neroni, B.; Radocchia, G.; Pompilio, A.; Mura, F.; Trancassini, M.; Di Bonaventura, G.; Pantanella, F.; Schippa, S. Growth Control of Adherent-Invasive Escherichia coli (AIEC) by the Predator Bacteria Bdellovibrio bacteriovorus: A New Therapeutic Approach for Crohn’s Disease Patients. Microorganisms 2019, 8, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Colibactin is a product of the 54-kb pks gene island. The pks gene island consists of 19 clb genes transcribed in four polycistronic and three cistronic elements, encoding various non-ribosomal peptide synthetase (NRPS), polyketide synthetase (PKS), hybrid NRPS-PKS or accessory proteins. Colibactin production is regulated by the LuxR-type transcriptional activator ClbR and the pantetheinyltransferase (PPTase) ClbA. Following transcriptional activation, a biosynthetic scaffold coordinates production of a linear intermediate (precolibactin) harboring a N-myristoyl-D-Asn motif. The transmembrane peptidase ClbP removes these prodrug motifs, inducing spontaneous dual two-fold cyclizing events resulting in production of the bioactive colibactin molecule, characterized by two electrophilic cyclopropane warheads with high binding-affinity for adenine residues within AAWWTT nucleotide motifs.
Figure 1.
Colibactin is a product of the 54-kb pks gene island. The pks gene island consists of 19 clb genes transcribed in four polycistronic and three cistronic elements, encoding various non-ribosomal peptide synthetase (NRPS), polyketide synthetase (PKS), hybrid NRPS-PKS or accessory proteins. Colibactin production is regulated by the LuxR-type transcriptional activator ClbR and the pantetheinyltransferase (PPTase) ClbA. Following transcriptional activation, a biosynthetic scaffold coordinates production of a linear intermediate (precolibactin) harboring a N-myristoyl-D-Asn motif. The transmembrane peptidase ClbP removes these prodrug motifs, inducing spontaneous dual two-fold cyclizing events resulting in production of the bioactive colibactin molecule, characterized by two electrophilic cyclopropane warheads with high binding-affinity for adenine residues within AAWWTT nucleotide motifs.

Figure 2.
Pks+ E. coli promote tumor formation in the colonic epithelium. During infection with pks+ E. coli, colibactin molecules translocate to the host nucleus via an undetermined mechanism, where the compound generates inter-strand crosslinks (ICLs) in adenine-rich nucleotide motifs and double-strand DNA breaks (DSBs). Errors during DNA repair following pks-induced damage result in the accumulation of a specific mutational signature characterized by T > N single base substitutions (SBSpks) or insertion/deletions of varying length (IDpks). Exposure to pks+ E. coli induces oncogenic phenotypes characterized by enhanced proliferation and Wnt independence. Unrepaired lesions cause cell-cycle arrest. Arrested cells adopt a senescence-associated secretory phenotype (SASP) resulting in enhanced growth factor production (hepatocyte growth factor [HGF], fibroblast growth factor [FGF]) which promote proliferation of nearby cells.
Figure 2.
Pks+ E. coli promote tumor formation in the colonic epithelium. During infection with pks+ E. coli, colibactin molecules translocate to the host nucleus via an undetermined mechanism, where the compound generates inter-strand crosslinks (ICLs) in adenine-rich nucleotide motifs and double-strand DNA breaks (DSBs). Errors during DNA repair following pks-induced damage result in the accumulation of a specific mutational signature characterized by T > N single base substitutions (SBSpks) or insertion/deletions of varying length (IDpks). Exposure to pks+ E. coli induces oncogenic phenotypes characterized by enhanced proliferation and Wnt independence. Unrepaired lesions cause cell-cycle arrest. Arrested cells adopt a senescence-associated secretory phenotype (SASP) resulting in enhanced growth factor production (hepatocyte growth factor [HGF], fibroblast growth factor [FGF]) which promote proliferation of nearby cells.

Figure 3.
Transcriptional regulation of clb genes. (A) Transcription of the regulatory clb genes (clbA and clbR) is increased in low-iron or oligosaccharide rich environments. The endo- or exogenously derived polyamine spermidine is necessary for pks transcription. (B) Inflammation promotes the transcription of several individual clb components. Administration of anti-inflammatory drugs such as anti-TNF attenuate these effects and limit pks-associated tumorigenicity in vivo. Anti-polyphosphate kinase (PPK) inhibitors downregulate clbB expression and the genotoxicity of pks+ E. coli.
Figure 3.
Transcriptional regulation of clb genes. (A) Transcription of the regulatory clb genes (clbA and clbR) is increased in low-iron or oligosaccharide rich environments. The endo- or exogenously derived polyamine spermidine is necessary for pks transcription. (B) Inflammation promotes the transcription of several individual clb components. Administration of anti-inflammatory drugs such as anti-TNF attenuate these effects and limit pks-associated tumorigenicity in vivo. Anti-polyphosphate kinase (PPK) inhibitors downregulate clbB expression and the genotoxicity of pks+ E. coli.

Figure 4.
The oncogenic capacity of pks+ Escherichia coli is enhanced by increased mucosal invasion and biofilm association. In healthy tissue, E. coli typically aggregate at the mucosal surface. In biofilm-covered CRCs, pks+ E. coli form biofilms in cooperation with enterotoxigenic Bacteroides fragilis (ETBF), increasing depth of mucosal invasion. During inflammation, a microaerobic niche formed within the mucosa (derived from host nitric oxides or peroxides) facilitates the expansion of mucosal E. coli populations.
Figure 4.
The oncogenic capacity of pks+ Escherichia coli is enhanced by increased mucosal invasion and biofilm association. In healthy tissue, E. coli typically aggregate at the mucosal surface. In biofilm-covered CRCs, pks+ E. coli form biofilms in cooperation with enterotoxigenic Bacteroides fragilis (ETBF), increasing depth of mucosal invasion. During inflammation, a microaerobic niche formed within the mucosa (derived from host nitric oxides or peroxides) facilitates the expansion of mucosal E. coli populations.

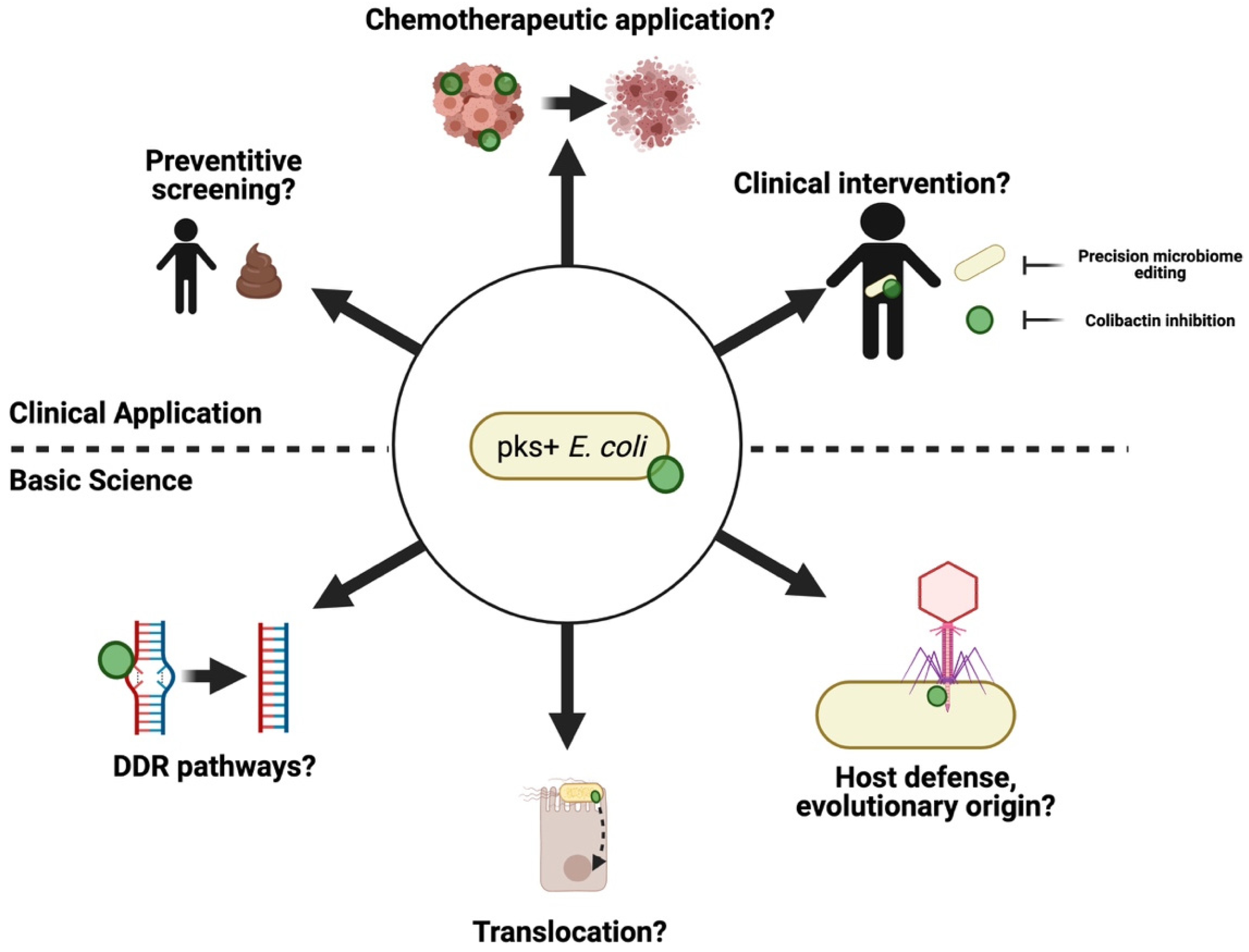
Figure 5.
Future areas of research involving pks+ E. coli.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dougherty, M.W.; Jobin, C. Shining a Light on Colibactin Biology. Toxins 2021, 13, 346. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13050346
AMA Style
Dougherty MW, Jobin C. Shining a Light on Colibactin Biology. Toxins. 2021; 13(5):346. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13050346
Chicago/Turabian StyleDougherty, Michael W., and Christian Jobin. 2021. "Shining a Light on Colibactin Biology" Toxins 13, no. 5: 346. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13050346
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.