In Vitro and In Vivo Antifibrotic Effects of Fraxetin on Renal Interstitial Fibrosis via the ERK Signaling Pathway

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
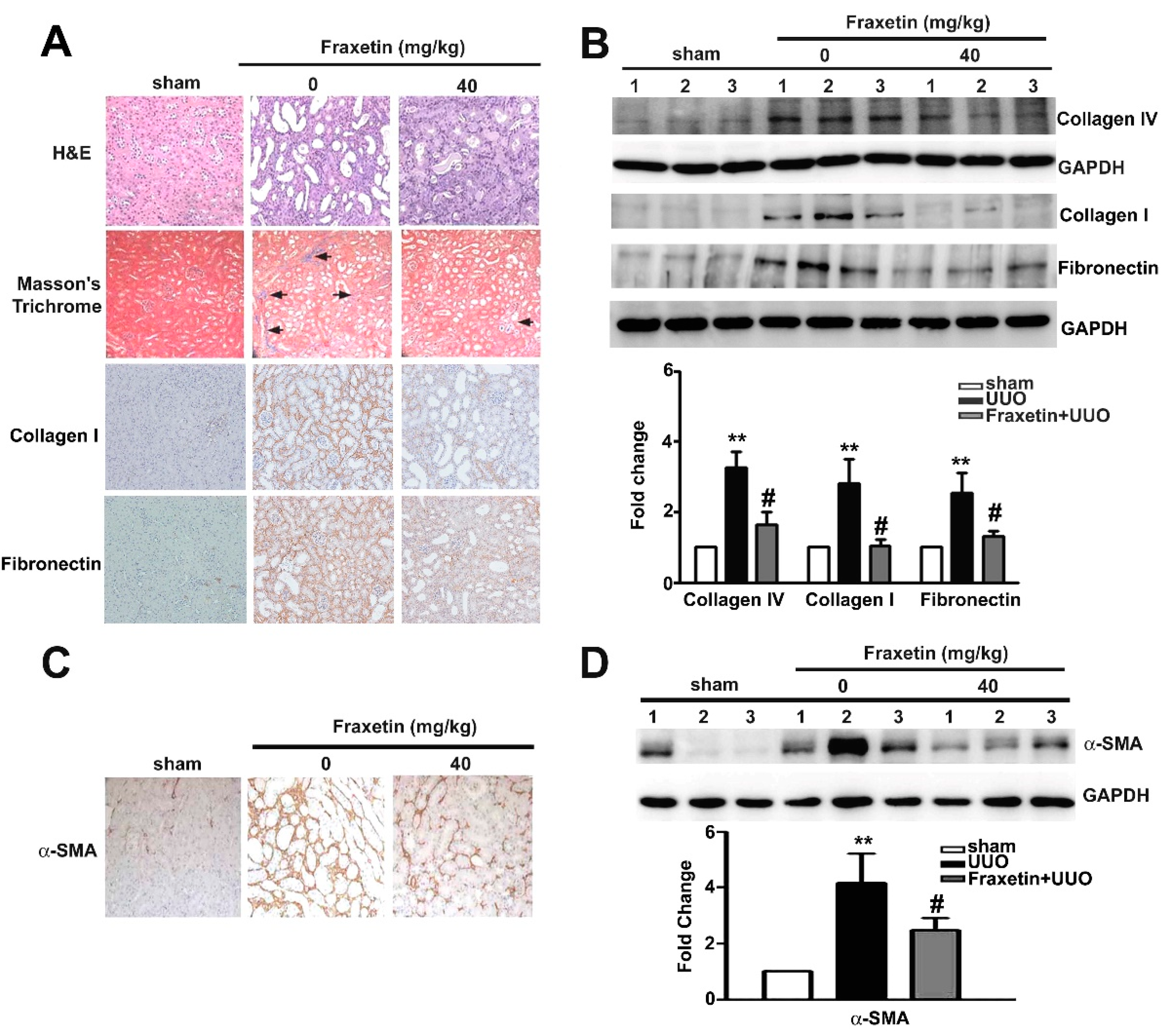
2.1. Fraxetin Inhibits Renal Interstitial Fibrosis in UUO Mice
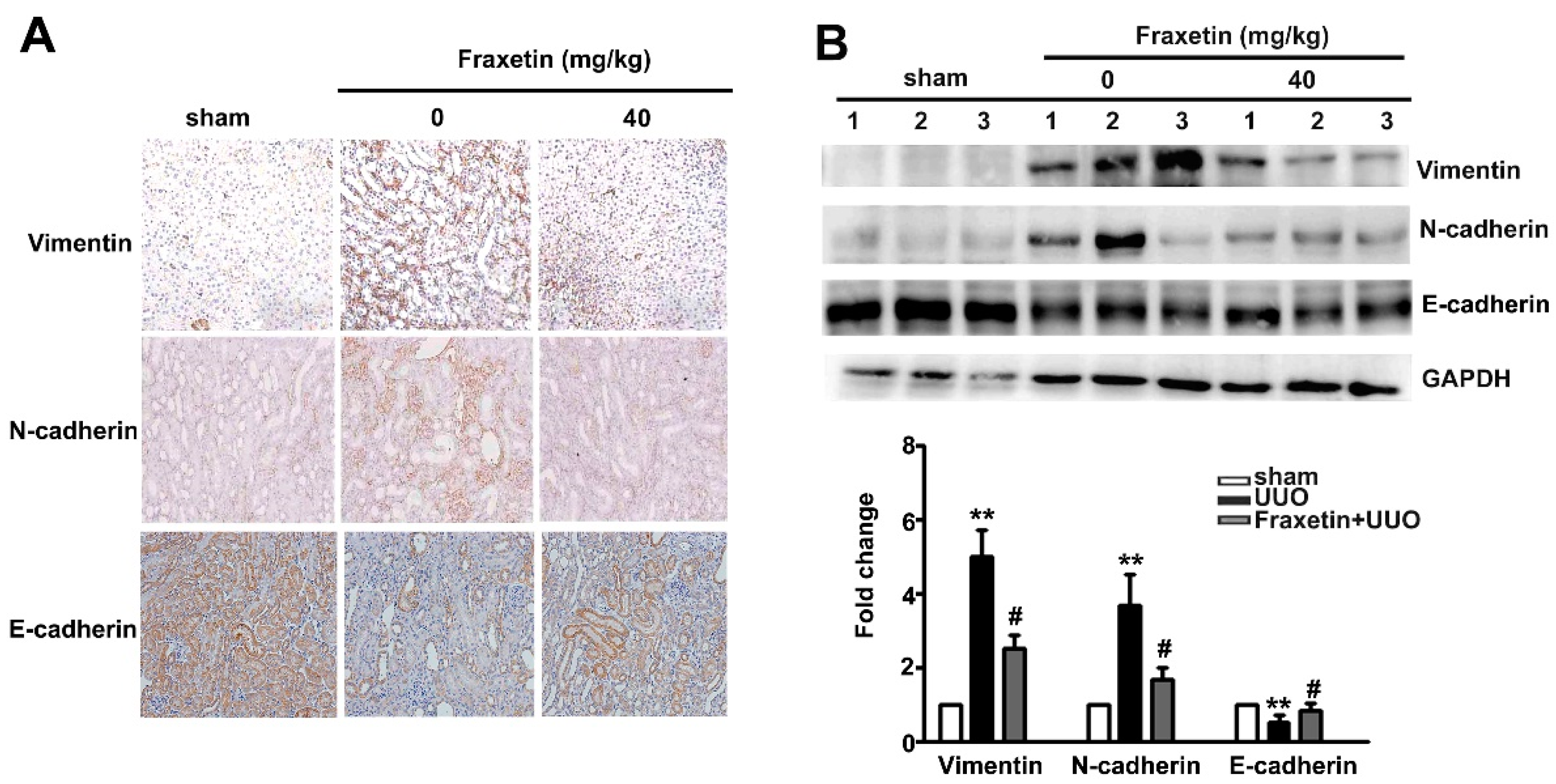
2.2. Fraxetin Decreased the Expression of EMT-Related Proteins in UUO Mice
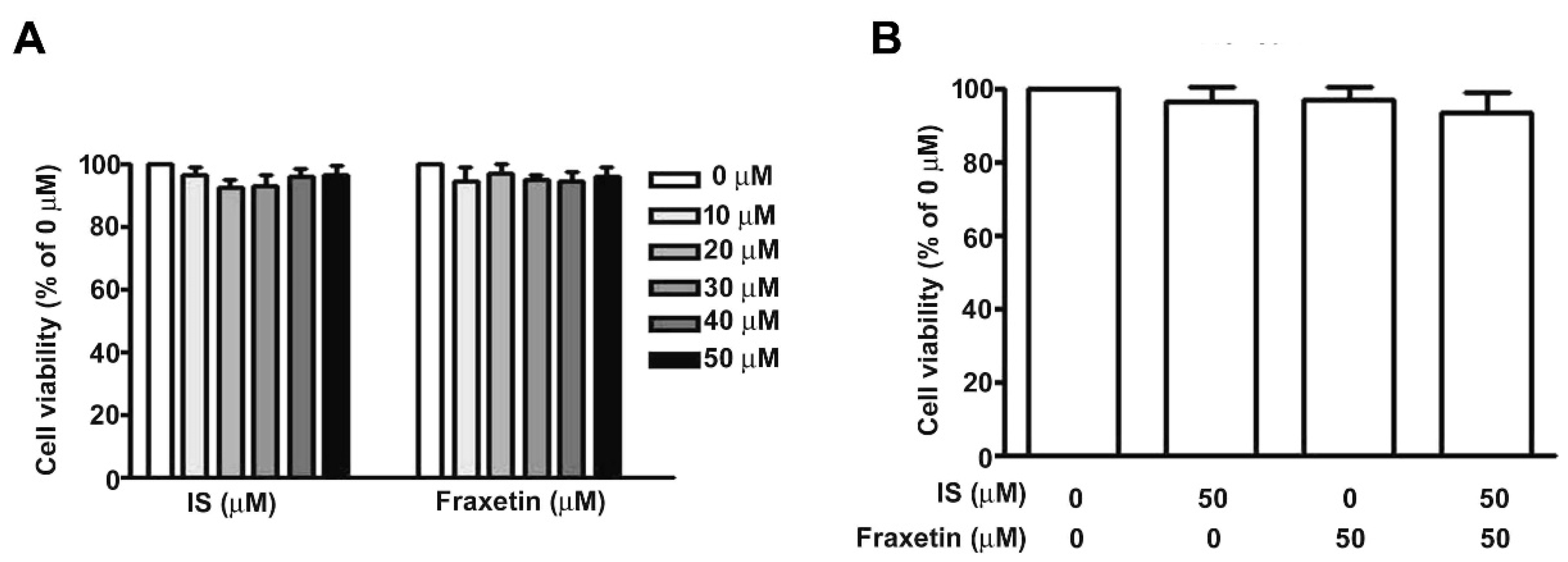
2.3. Effect of IS Combined with Fraxetin on Viability of MES13 Cells
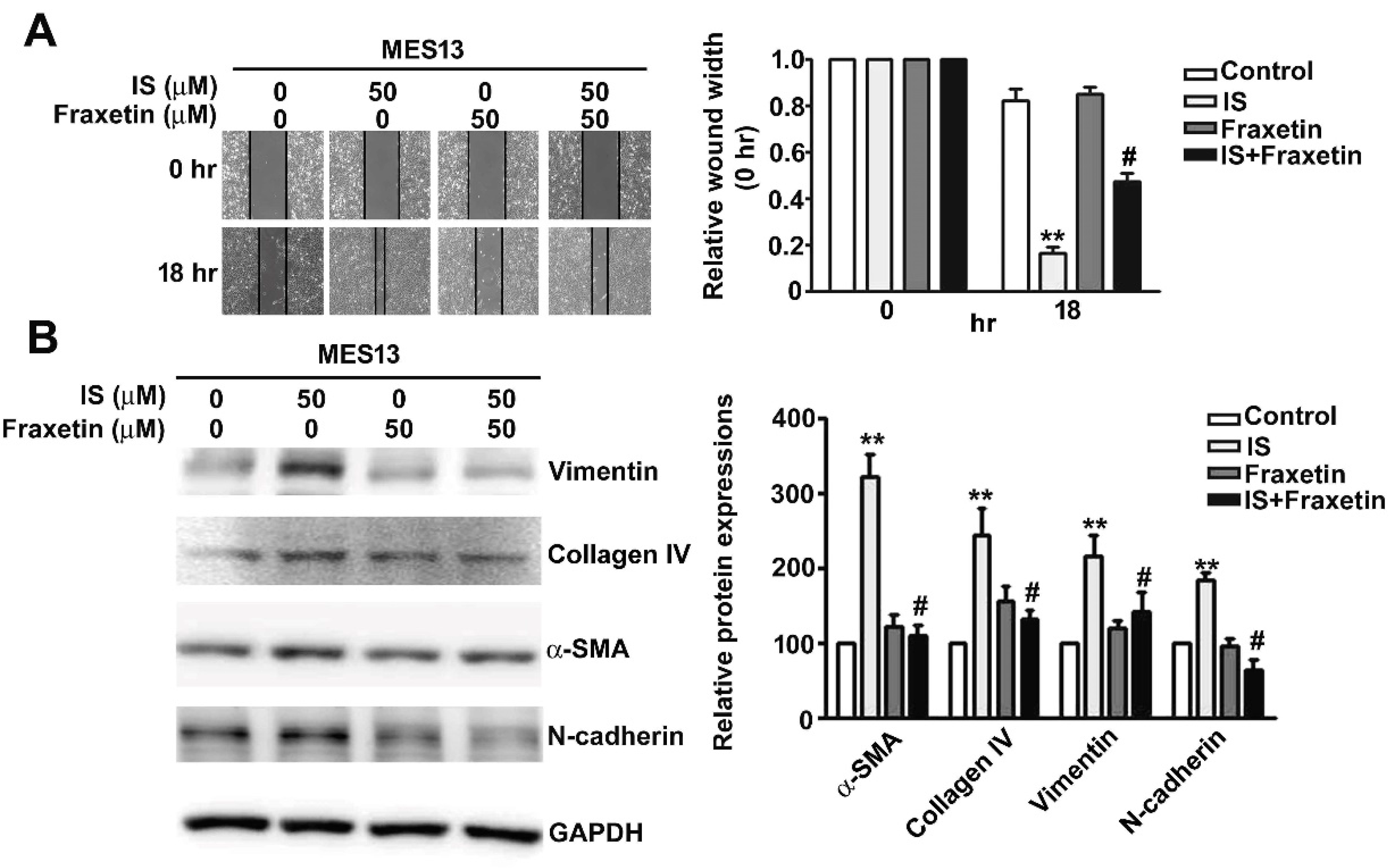
2.4. Effect of Fraxetin on IS-Induced Motility of MES13 Cells
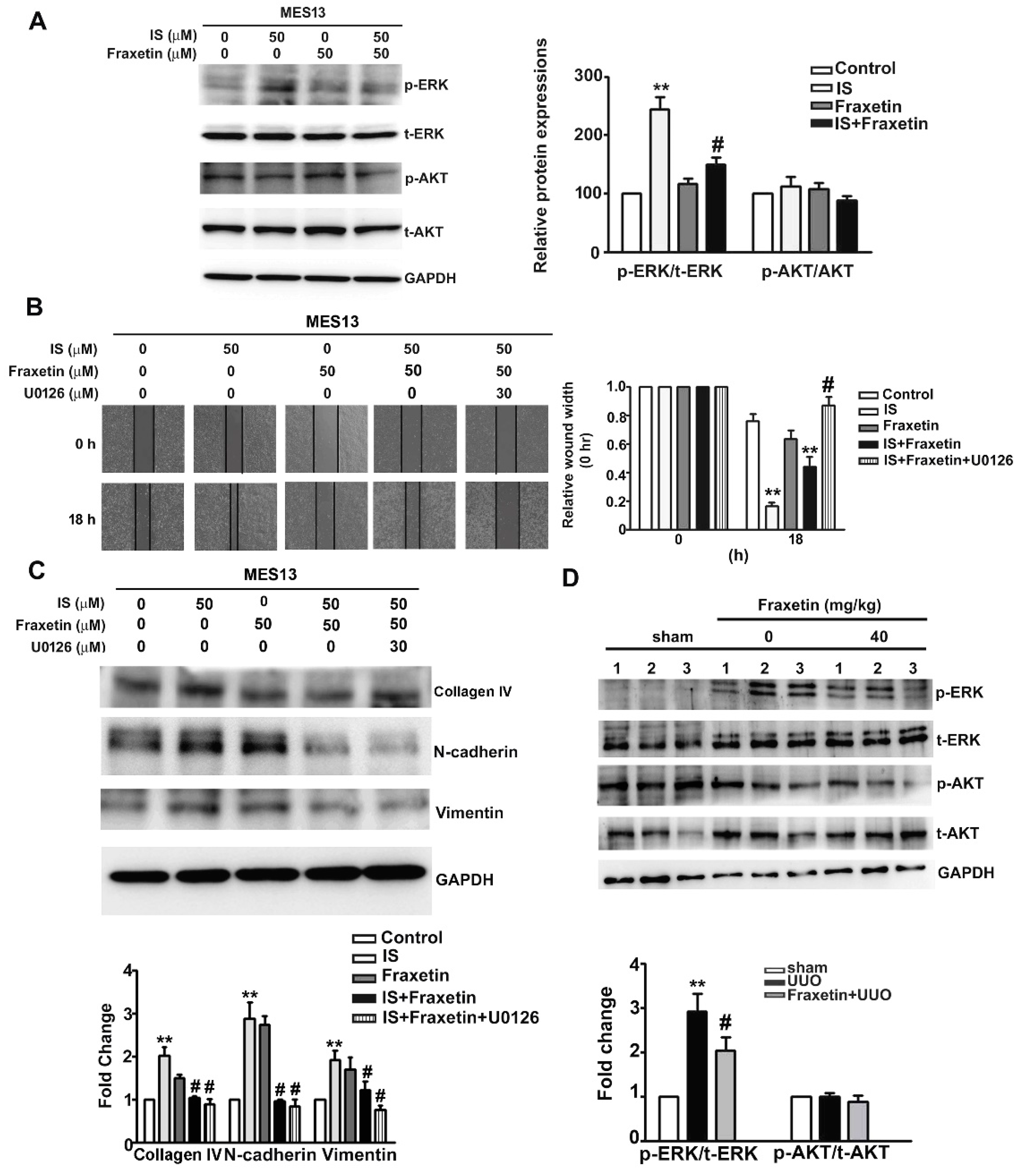
2.5. Effect of Fraxetin on Renal Fibrosis Involves ERK Phosphoryation Pathways in Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagent and Chemical Drug
4.2. Cell Viability Assay
4.3. Cell Motility Assay
4.4. Western Blotting
4.5. Animal Model and Experimental Procedure
4.6. Immunohistochemistry Assay and Masson’s Trichrome Staining
4.7. Statistical Analysis
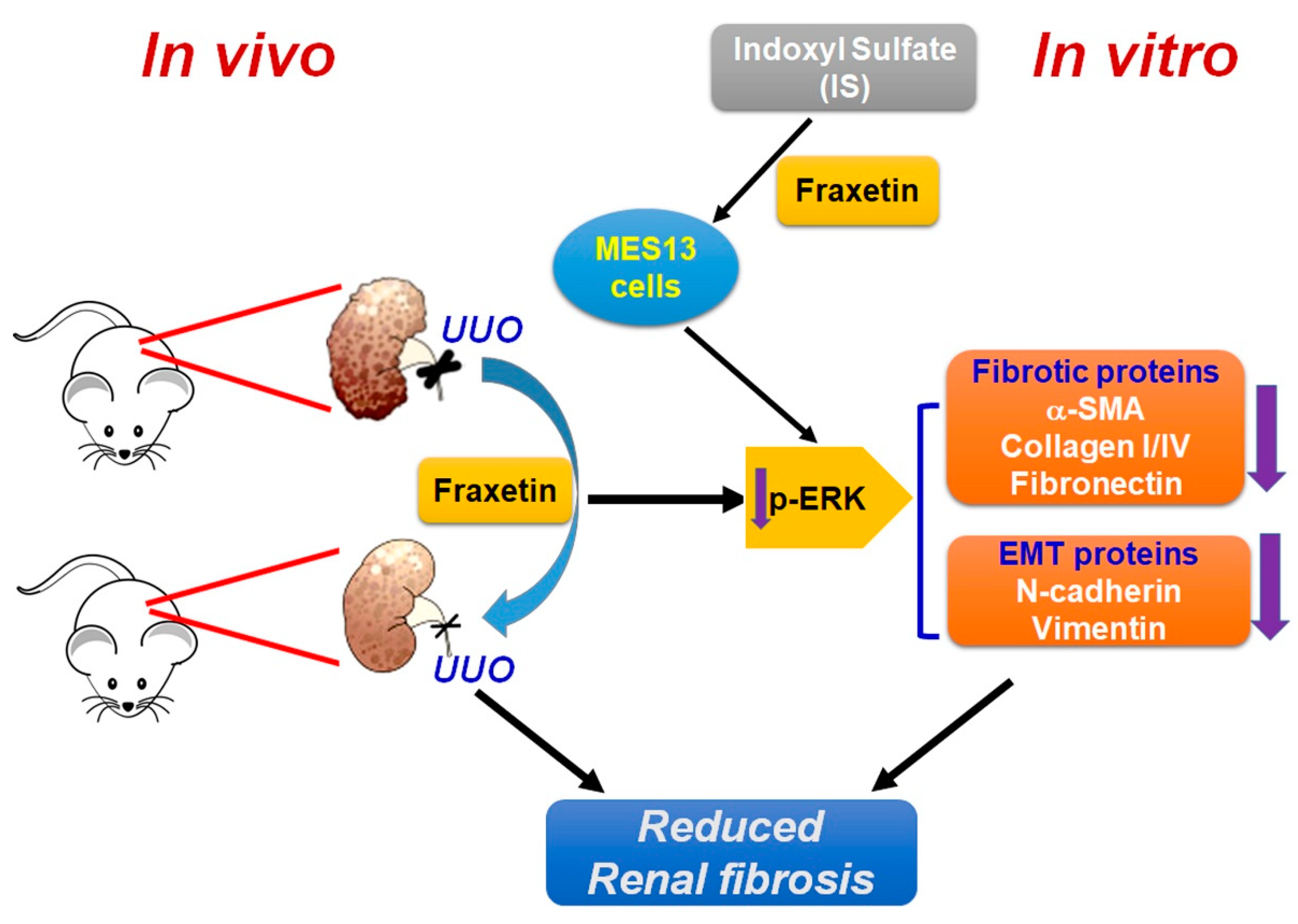
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hsu, C.C.; Hwang, S.J.; Wen, C.P.; Chang, H.Y.; Chen, T.; Shiu, R.S.; Horng, S.S.; Chang, Y.K.; Yang, W.C. High prevalence and low awareness of CKD in Taiwan: A study on the relationship between serum creatinine and awareness from a nationally representative survey. Am. J. Kidney Dis. 2006, 48, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, C.P.; Cheng, T.Y.; Tsai, M.K.; Chang, Y.C.; Chan, H.T.; Tsai, S.P.; Chiang, P.H.; Hsu, C.C.; Sung, P.K.; Hsu, Y.H.; et al. All-cause mortality attributable to chronic kidney disease: A prospective cohort study based on 462 293 adults in Taiwan. Lancet 2008, 371, 2173–2182. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Cravedi, P.; Remuzzi, G. Mechanisms and treatment of CKD. J. Am. Soc. Nephrol. 2012, 23, 1917–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.R.; Yang, Y.; Wang, S.C.; Chiu, P.F.; Chou, W.Y.; Lin, C.Y.; Chang, J.M.; Chen, T.W.; Ferng, S.H.; Lin, C.L. Effectiveness of multidisciplinary care for chronic kidney disease in Taiwan: A 3-year prospective cohort study. Nephrol. Dialysis Transplant. 2013, 28, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, S.J.; Tsai, J.C.; Chen, H.C. Epidemiology, impact and preventive care of chronic kidney disease in Taiwan. Nephrology 2010, 15, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Essawy, M.; Soylemezoglu, O.; Muchaneta-Kubara, E.C.; Shortland, J.; Brown, C.B.; el Nahas, A.M. Myofibroblasts and the progression of diabetic nephropathy. Nephrol. Dialysis Transplant. 1997, 12, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, I.S.; Burrows, C.; Shanks, J.H.; Venning, M.; McWilliam, L.J. Interstitial myofibroblasts: Predictors of progression in membranous nephropathy. J. Clin. Pathol. 1997, 50, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Beshay, O.N.; Ewees, M.G.; Abdel-Bakky, M.S.; Hafez, S.; Abdelrehim, A.B.; Bayoumi, A.M.A. Resveratrol reduces gentamicin-induced EMT in the kidney via inhibition of reactive oxygen species and involving TGF-beta/Smad pathway. Life Sci. 2020, 258, 118178. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Wang, W.; Yin, P.; Gao, J.; Na, L.; Sun, Y.; Wang, Z.; Zhang, Z.; Zhao, C. Ruxolitinib Alleviates Renal Interstitial Fibrosis in UUO Mice. Int. J. Biol. Sci. 2020, 16, 194–203. [Google Scholar] [CrossRef]
- Annaldas, S.; Saifi, M.A.; Khurana, A.; Godugu, C. Nimbolide ameliorates unilateral ureteral obstruction-induced renal fibrosis by inhibition of TGF-beta and EMT/Slug signalling. Mol. Immunol. 2019, 112, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, S.; Garibaldi, S.; Saio, M.; Ghigliotti, G.; Picciotto, D.; Ameri, P.; Garibotto, G.; Barisione, C.; Verzola, D. Indoxyl Sulfate Induces Renal Fibroblast Activation through a Targetable Heat Shock Protein 90-Dependent Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 2050183. [Google Scholar] [CrossRef]
- Sun, C.Y.; Chang, S.C.; Wu, M.S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Yu, M.A.; Ryu, E.S.; Jang, Y.H.; Kang, D.H. Indoxyl sulfate-induced epithelial-to-mesenchymal transition and apoptosis of renal tubular cells as novel mechanisms of progression of renal disease. Lab. Investig. 2012, 92, 488–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Wang, R.; Li, S.; Wang, Y.; Song, F.; Gu, Y.; Yuan, Y. Antifibrotic effects of Fraxetin on carbon tetrachloride-induced liver fibrosis by targeting NF-kappaB/IkappaBalpha, MAPKs and Bcl-2/Bax pathways. Pharmacol. Rep. 2019, 71, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Molina-Jimenez, M.F.; Sanchez-Reus, M.I.; Cascales, M.; Andres, D.; Benedi, J. Effect of fraxetin on antioxidant defense and stress proteins in human neuroblastoma cell model of rotenone neurotoxicity. Comparative study with myricetin and N-acetylcysteine. Toxicol. Appl. Pharmacol. 2005, 209, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Molina-Jimenez, M.F.; Sanchez-Reus, M.I.; Andres, D.; Cascales, M.; Benedi, J. Neuroprotective effect of fraxetin and myricetin against rotenone-induced apoptosis in neuroblastoma cells. Brain Res. 2004, 1009, 9–16. [Google Scholar] [CrossRef]
- Thuong, P.T.; Pokharel, Y.R.; Lee, M.Y.; Kim, S.K.; Bae, K.; Su, N.D.; Oh, W.K.; Kang, K.W. Dual anti-oxidative effects of fraxetin isolated from Fraxinus rhinchophylla. Biol. Pharm. Bull. 2009, 32, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Murali, R.; Srinivasan, S.; Ashokkumar, N. Antihyperglycemic effect of fraxetin on hepatic key enzymes of carbohydrate metabolism in streptozotocin-induced diabetic rats. Biochimie 2013, 95, 1848–1854. [Google Scholar] [CrossRef]
- Chen, X.; Ying, X.; Zhang, W.; Chen, Y.; Shi, C.; Hou, Y.; Zhang, Y. The hepatoprotective effect of fraxetin on carbon tetrachloride induced hepatic fibrosis by antioxidative activities in rats. Int. Immunopharmacol. 2013, 17, 543–547. [Google Scholar] [CrossRef]
- Chen, X.; Ying, X.; Sun, W.; Zhu, H.; Jiang, X.; Chen, B. The therapeutic effect of fraxetin on ethanol-induced hepatic fibrosis by enhancing ethanol metabolism, inhibiting oxidative stress and modulating inflammatory mediators in rats. Int. Immunopharmacol. 2018, 56, 98–104. [Google Scholar] [CrossRef]
- Bascands, J.L.; Schanstra, J.P. Obstructive nephropathy: Insights from genetically engineered animals. Kidney Int. 2005, 68, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Loeffler, I.; Wolf, G. Epithelial-to-Mesenchymal Transition in Diabetic Nephropathy: Fact or Fiction? Cells 2015, 4, 631–652. [Google Scholar] [CrossRef]
- Liu, W.C.; Tomino, Y.; Lu, K.C. Impacts of Indoxyl Sulfate and p-Cresol Sulfate on Chronic Kidney Disease and Mitigating Effects of AST-120. Toxins 2018, 10, 367. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.F.; Ewart, L.M.; Masterson, E.; Tennant, S.; Grebnev, G.; Prunotto, M.; Pomposiello, S.; Conde-Knape, K.; Martin, F.M.; Godson, C. BMP7-induced-Pten inhibits Akt and prevents renal fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3095–3104. [Google Scholar] [CrossRef]
- Hsieh, Y.H.; Syu, R.J.; Lee, C.C.; Lin, S.H.; Lee, C.H.; Cheng, C.W.; Tsai, J.P. Arecoline induces epithelial mesenchymal transition in HK2 cells by upregulating the ERK-mediated signaling pathway. Environ. Toxicol. 2020, 35, 1007–1014. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.B.; Qu, X.; Caruana, G.; Li, J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 2016, 92, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Humphreys, B.D. Kidney pericytes: Roles in regeneration and fibrosis. Semin. Nephrol. 2014, 34, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Bolati, D.; Shimizu, H.; Higashiyama, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate induces epithelial-to-mesenchymal transition in rat kidneys and human proximal tubular cells. Am. J. Nephrol. 2011, 34, 318–323. [Google Scholar] [CrossRef]
- Shimizu, H.; Bolati, D.; Higashiyama, Y.; Nishijima, F.; Shimizu, K.; Niwa, T. Indoxyl sulfate upregulates renal expression of MCP-1 via production of ROS and activation of NF-kappaB, p53, ERK, and JNK in proximal tubular cells. Life Sci. 2012, 90, 525–530. [Google Scholar] [CrossRef]
- Bolati, D.; Shimizu, H.; Niwa, T. AST-120 ameliorates epithelial-to-mesenchymal transition and interstitial fibrosis in the kidneys of chronic kidney disease rats. J. Renal Nutr. 2012, 22, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.C.; Sun, H.L.; Tsai, C.H.; Kuo, C.W.; Liu, K.L.; Lii, C.K.; Huang, C.S.; Li, C.C. 1,25(OH)2 D3 attenuates indoxyl sulfate-induced epithelial-to-mesenchymal cell transition via inactivation of PI3K/Akt/beta-catenin signaling in renal tubular epithelial cells. Nutrition 2020, 69, 110554. [Google Scholar] [CrossRef]
- Martin-Aragon, S.; Benedi, J.M.; Villar, A.M. Modifications on antioxidant capacity and lipid peroxidation in mice under fraxetin treatment. J. Pharm. Pharmacol. 1997, 49, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Molina-Jimenez, M.F.; Sanchez-Reus, M.I.; Benedi, J. Effect of fraxetin and myricetin on rotenone-induced cytotoxicity in SH-SY5Y cells: Comparison with N-acetylcysteine. Eur. J. Pharmacol. 2003, 472, 81–87. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, S.; Guo, L.; Wang, R.; Chen, J.; Shen, J. NMDA receptor-mediated CaMKII/ERK activation contributes to renal fibrosis. BMC Nephrol. 2020, 21, 392. [Google Scholar] [CrossRef]
- Liu, X.; Chen, J.; Sun, N.; Li, N.; Zhang, Z.; Zheng, T.; Li, Z. Ginsenoside Rb1 ameliorates autophagy via the AMPK/mTOR pathway in renal tubular epithelial cells in vitro and in vivo. Int. J. Biol. Macromol. 2020, 163, 996–1009. [Google Scholar] [CrossRef]
- Li, Z.; Liu, X.; Wang, B.; Nie, Y.; Wen, J.; Wang, Q.; Gu, C. Pirfenidone suppresses MAPK signalling pathway to reverse epithelial-mesenchymal transition and renal fibrosis. Nephrology 2017, 22, 589–597. [Google Scholar] [CrossRef]
- Lin, S.H.; Chiou, S.J.; Ho, W.T.; Chuang, C.T.; Chuang, L.Y.; Guh, J.Y. Arecoline-induced pro-fibrotic proteins in LLC-PK1 cells are dependent on c-Jun N-terminal kinase. Toxicology 2016, 344–346, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Chien, H.J.; Ying, T.H.; Hsieh, S.C.; Lin, C.L.; Yu, Y.L.; Kao, S.H.; Hsieh, Y.H. Alpha-Mangostin attenuates stemness and enhances cisplatin-induced cell death in cervical cancer stem-like cells through induction of mitochondrial-mediated apoptosis. J. Cell. Physiol. 2020, 235, 5590–5601. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.W.; Chu, C.Y.; Yu, C.L.; Lee, C.C.; Hsu, L.S.; Chen, Y.S.; Hsieh, Y.H.; Tsai, J.P. Endothelial Cell-Specific Molecule 1 Promotes Endothelial to Mesenchymal Transition in Renal Fibrosis. Toxins 2020, 12, 506. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsieh, Y.-H.; Hung, T.-W.; Chen, Y.-S.; Huang, Y.-N.; Chiou, H.-L.; Lee, C.-C.; Tsai, J.-P. In Vitro and In Vivo Antifibrotic Effects of Fraxetin on Renal Interstitial Fibrosis via the ERK Signaling Pathway. Toxins 2021, 13, 474. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070474
Hsieh Y-H, Hung T-W, Chen Y-S, Huang Y-N, Chiou H-L, Lee C-C, Tsai J-P. In Vitro and In Vivo Antifibrotic Effects of Fraxetin on Renal Interstitial Fibrosis via the ERK Signaling Pathway. Toxins. 2021; 13(7):474. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070474
Chicago/Turabian StyleHsieh, Yi-Hsien, Tung-Wei Hung, Yong-Syuan Chen, Yi-Ning Huang, Hui-Ling Chiou, Chu-Che Lee, and Jen-Pi Tsai. 2021. "In Vitro and In Vivo Antifibrotic Effects of Fraxetin on Renal Interstitial Fibrosis via the ERK Signaling Pathway" Toxins 13, no. 7: 474. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13070474