Identification of 24-O-β-d-Glycosides and 7-Deoxy-Analogues of Okadaic Acid and Dinophysistoxin-1 and -2 in Extracts from Dinophysis Blooms, Dinophysis and Prorocentrum Cultures, and Shellfish in Europe, North America and Australasia

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
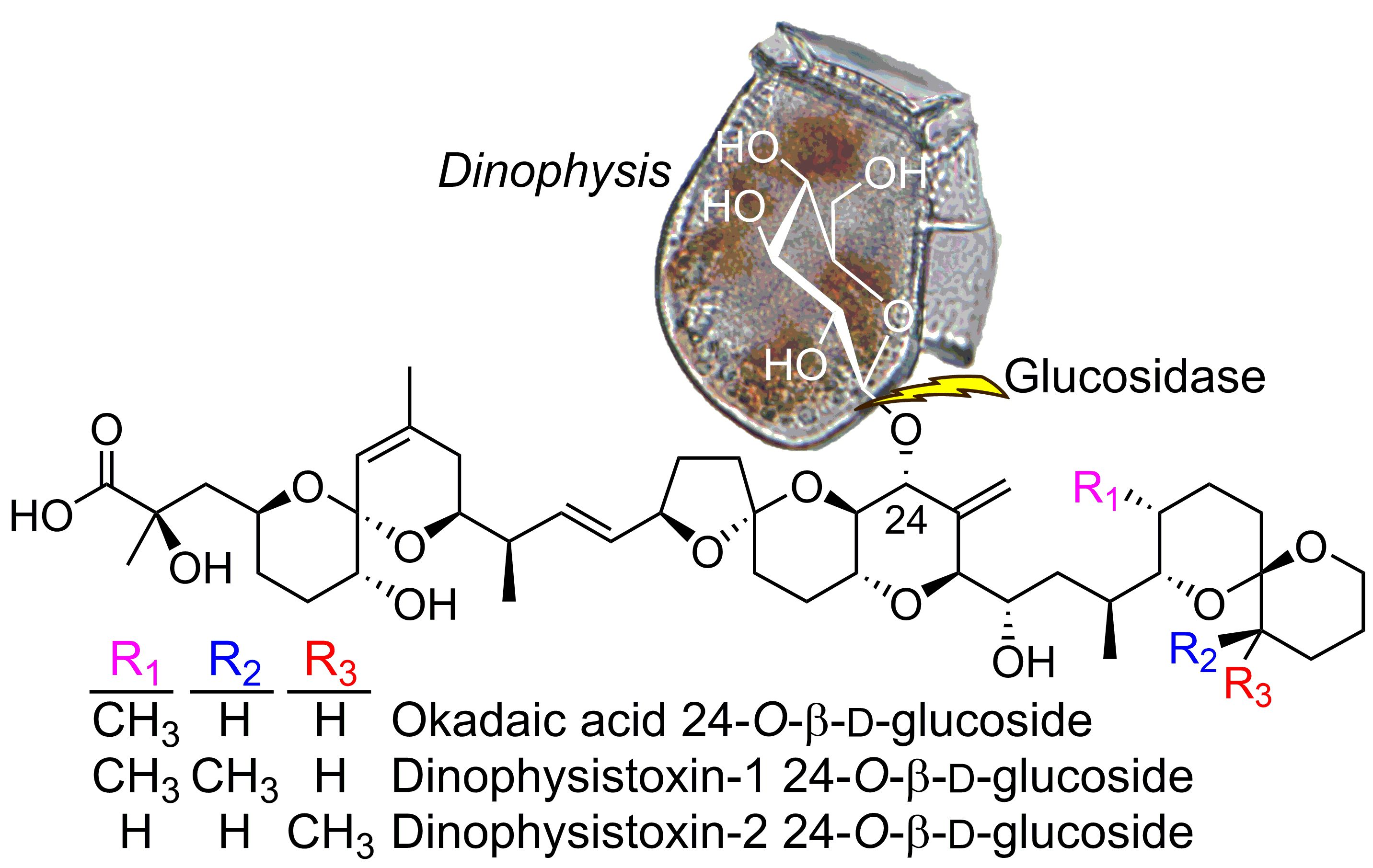
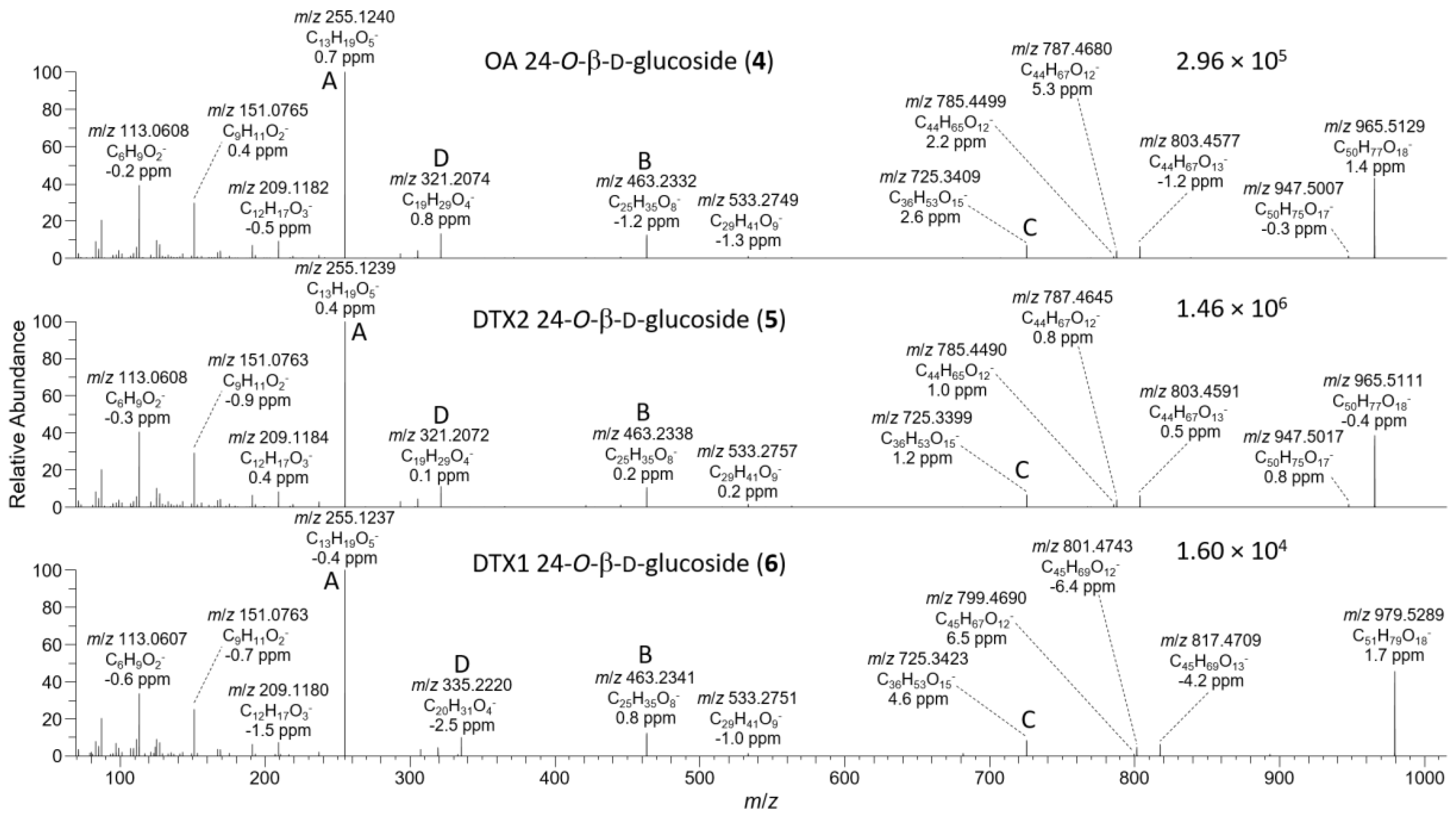
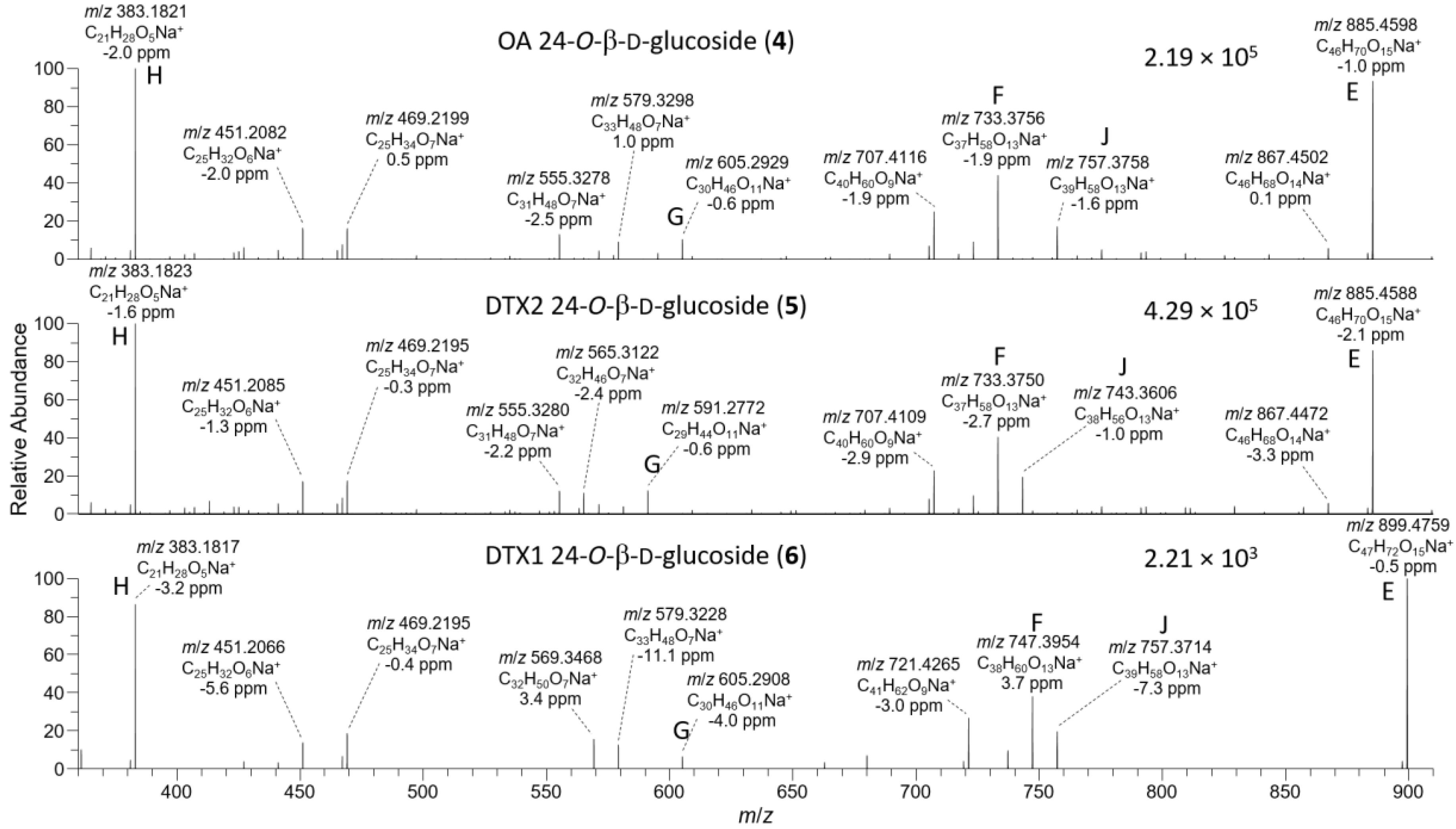
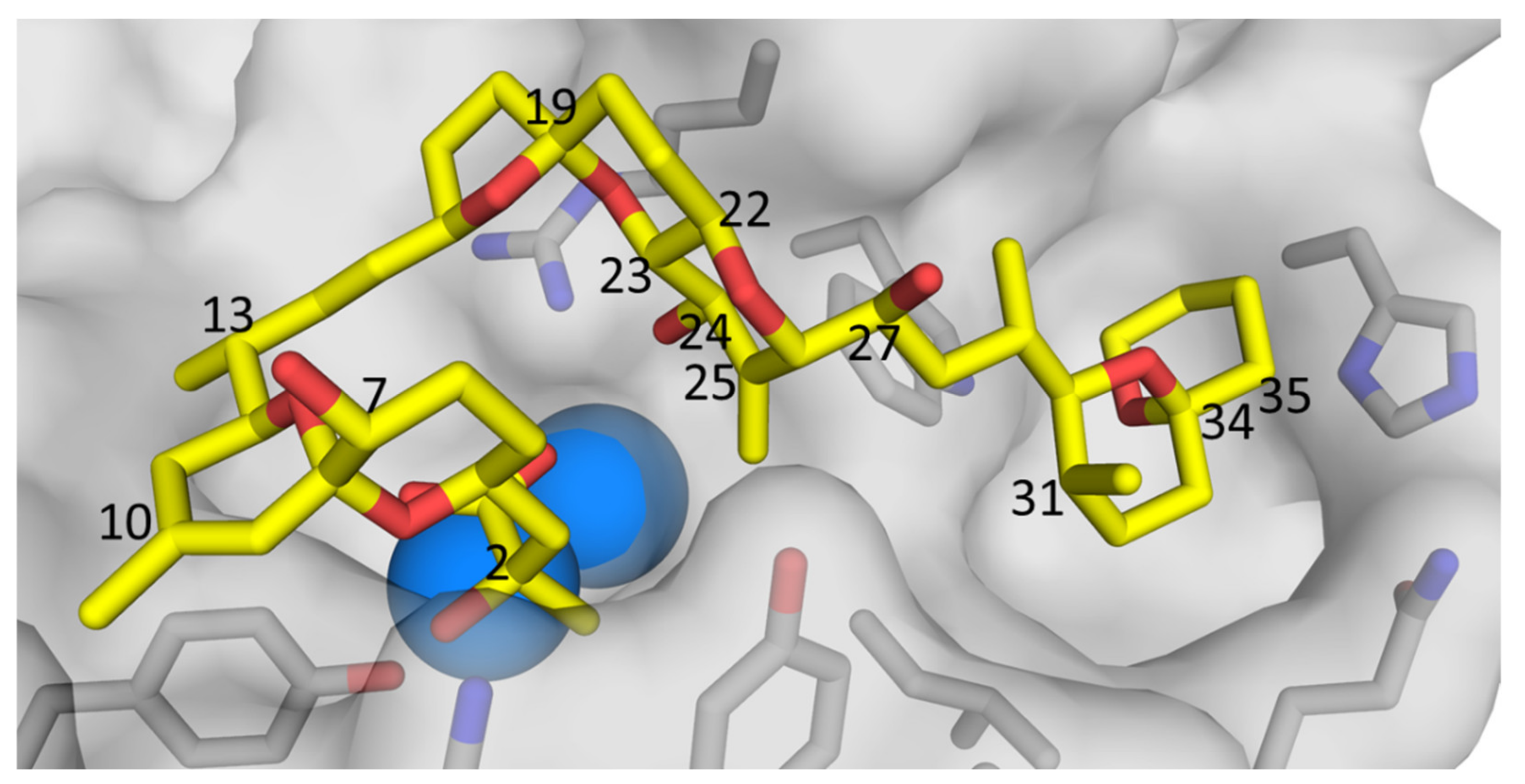
2.1. Mass Spectrometric Analysis of Glucosides 4 and 5


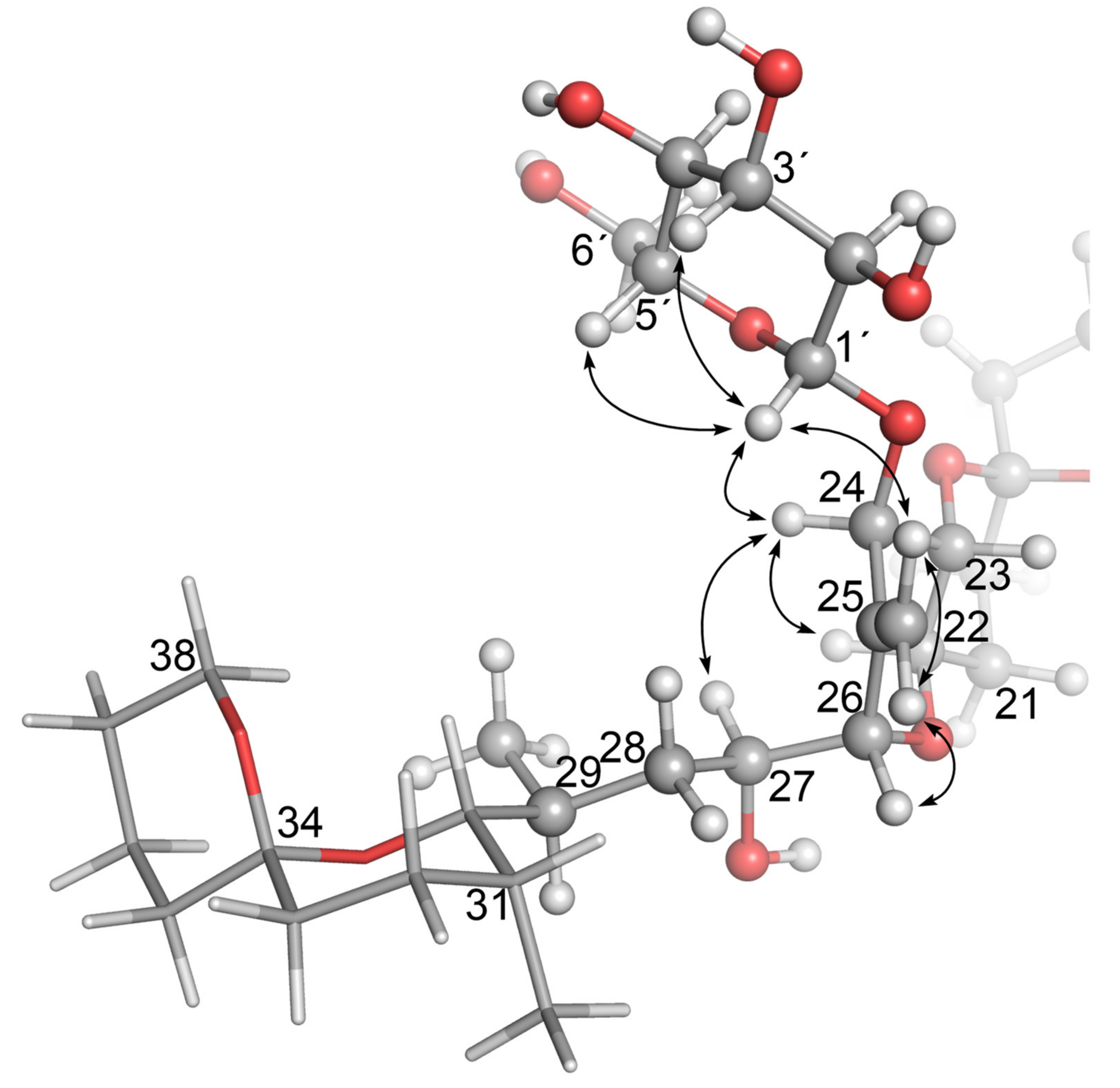
2.2. NMR Structure Elucidation of Glucosides 4 and 5
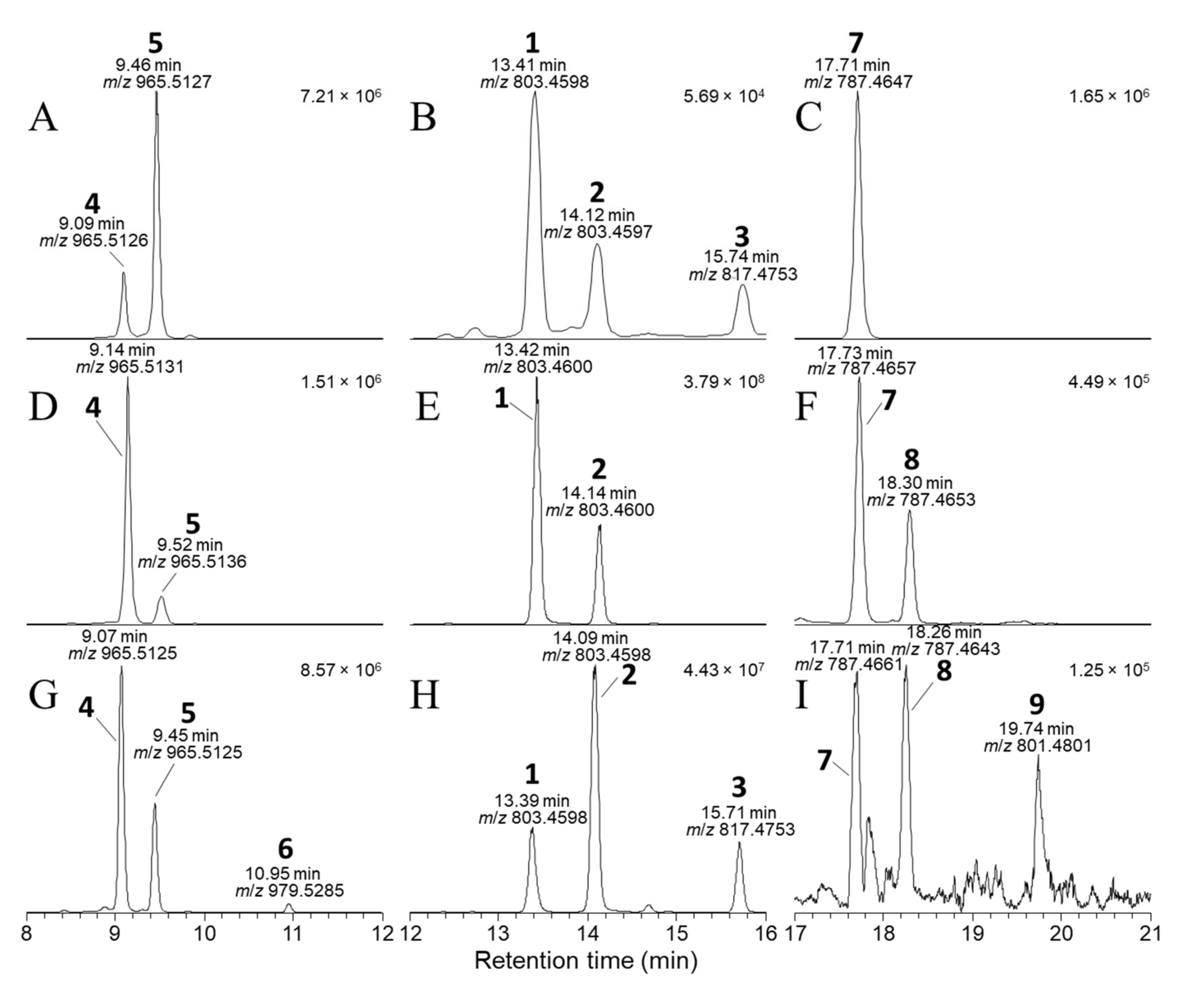
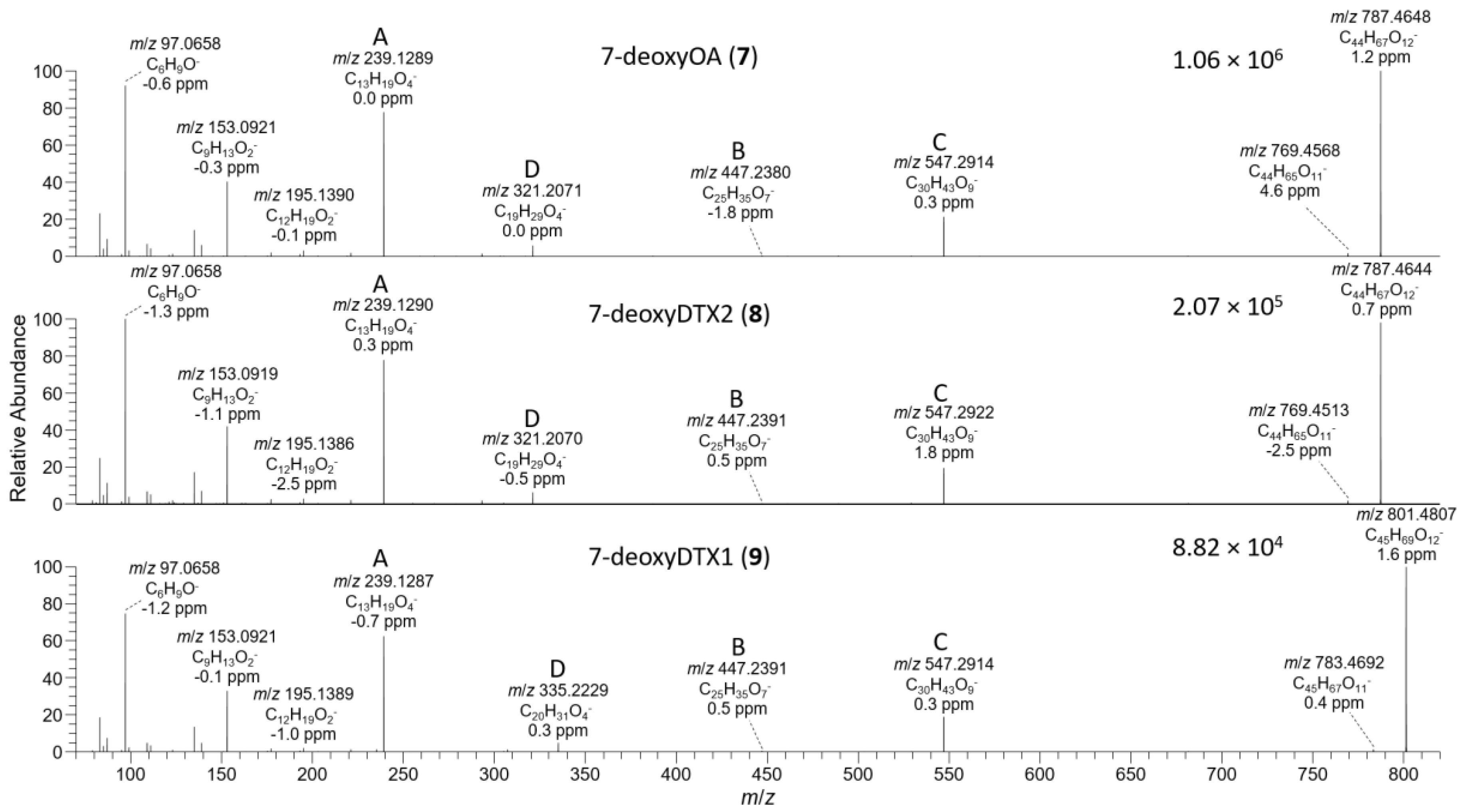
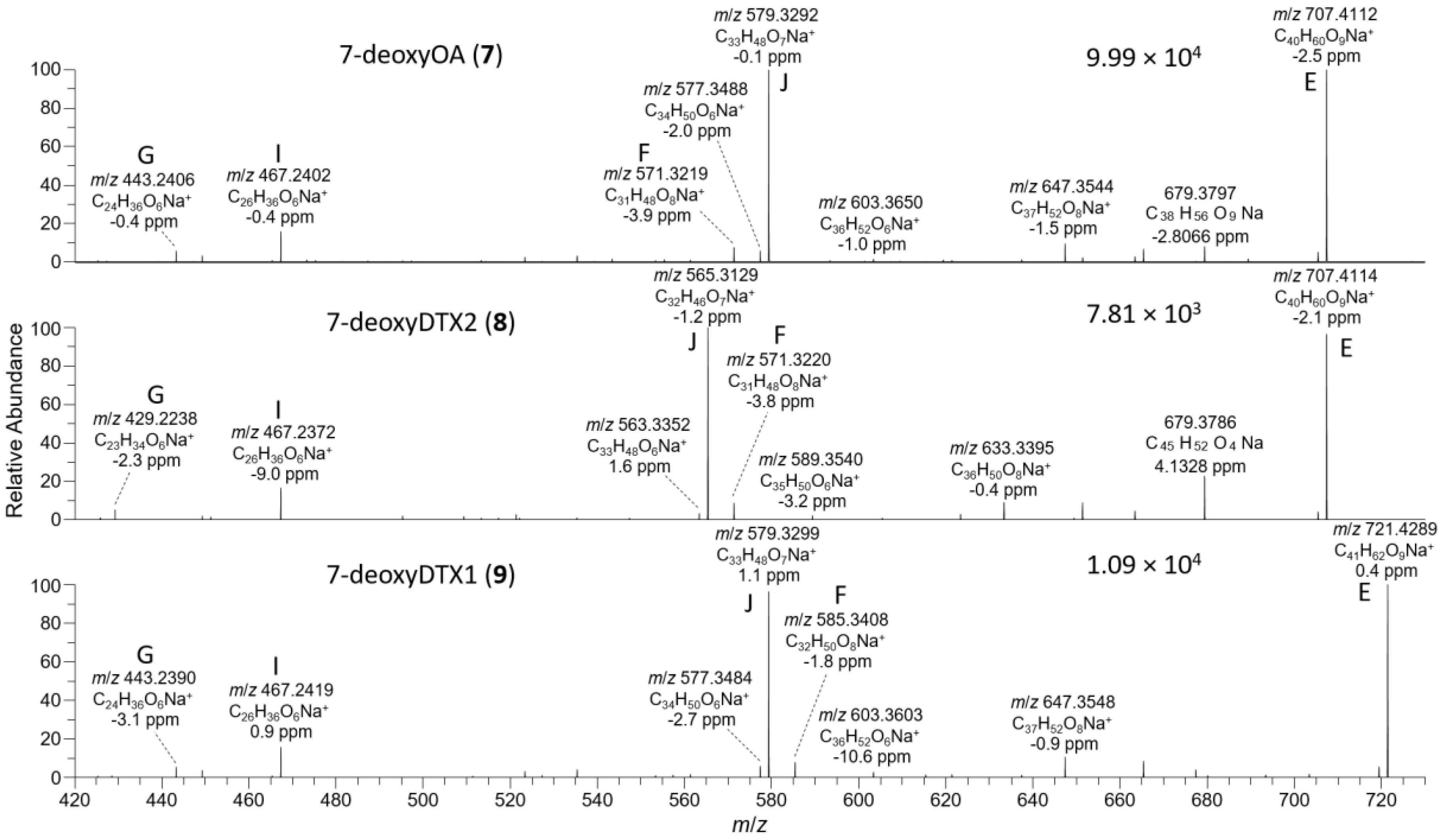
2.3. LC–MSn and LC–HRMS/MS Detection of DTX-1 24-O-β-d-Glucoside (6)
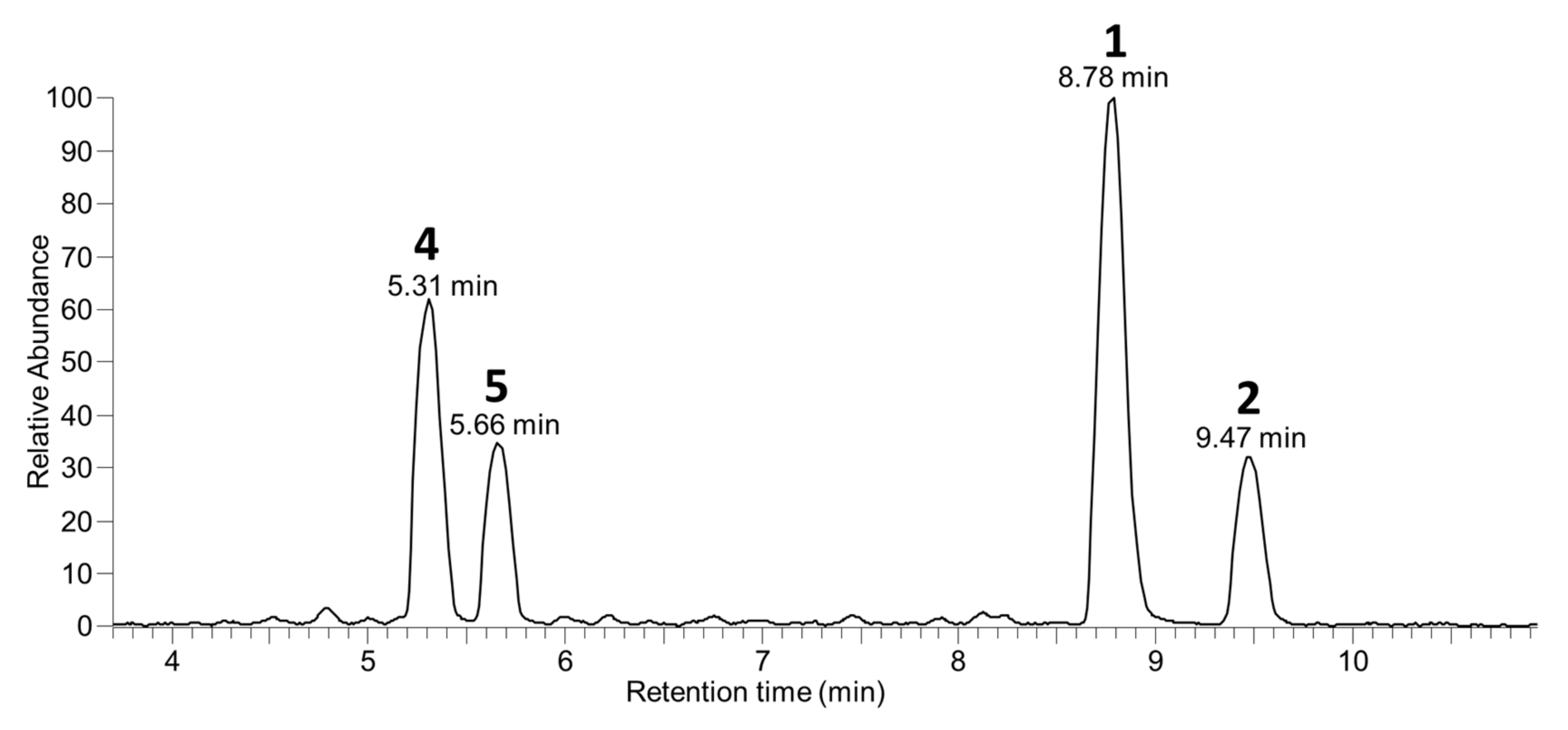
2.4. Detection of OA/DTX Analogues in Dinophysis and P. lima Cultures
2.5. Detection of 1–9 in Blue Mussels from Canada, Ireland, Norway, and New Zealand
3. Conclusions
4. Materials and Methods
4.1. Reagents
4.2. Shellfish Samples
4.3. Passive Sampler Extracts
4.4. Isolation of Glucosides 4 and 5
4.5. Dinophysis Cultures
4.6. Cultured P. lima Extracts
4.7. LC–MSn (Method A)
4.8. LC–HRMS (Method B)
4.9. LC–HRMS (Method C)
4.10. LC–HRMS (Method D)
4.11. LC–MS/MS (Method E)
4.12. NMR Spectroscopy
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reguera, B.; Velo-Suárez, L.; Raine, R.; Park, M.G. Harmful Dinophysis species: A review. Harmful Algae 2012, 14, 87–106. [Google Scholar] [CrossRef]
- Hu, W.; Xu, J.; Sinkkonen, J.; Wu, J. Polyketides from marine dinoflagellates of the genus Prorocentrum, biosynthetic origin and bioactivity of their okadaic acid analogues. Mini Rev. Med. Chem. 2010, 10, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J. Accumulation of Dinophysis toxins in bivalve molluscs. Toxins 2018, 10, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Union. Commission decision of 15 March 2002 laying down detailed rules for the implementation of Council Directive 91/492/EEC as regards the maximum permitted levels and the methods for analysis of certain marine biotoxins in bivalve molluscs, echinoderms, tunicates and marine gastropods (2002/225/EC). Off. J. Eur. Communities 2002, L75, 62–64. [Google Scholar]
- Bates, S.S.; Beach, D.G.; Comeau, L.A.; Haigh, N.; Lewis, N.I.; Locke, A.; Martin, J.L.; McCarron, P.; McKenzie, C.H.; Michel, C.; et al. Marine harmful algal blooms and phycotoxins of concern to Canada. Available online: http://waves-vagues.dfo-mpo.gc.ca/Library/4088319x.pdf (accessed on 22 April 2021).
- European Union. Regulation (EC) No 853/2004 of the European Parliament and of the Council of 29 April 2004 laying down specific hygiene rules for food of animal origin. Off. J. Eur. Union 2004, L139, 55–205. [Google Scholar]
- Hu, T.; Marr, J.C.; de Freitas, A.S.W.; Quilliam, M.A.; Walter, J.A.; Wright, J.L.C.; Pleasance, S. New diol esters isolated from cultures of the dinoflagellates Prorocentrum lima and Prorocentrum concavum. J. Nat. Prod. 1992, 55, 1631–1637. [Google Scholar] [CrossRef]
- Cruz, P.G.; Daranas, A.H.; Fernández, J.J.; Souto, M.L.; Norte, M. DTX5c, a new OA sulphate ester derivative from cultures of Prorocentrum belizeanum. Toxicon 2006, 47, 920–924. [Google Scholar] [CrossRef]
- Miles, C.O.; Wilkins, A.L.; Hawkes, A.D.; Jensen, D.J.; Cooney, J.M.; Larsen, K.; Petersen, D.; Rise, F.; Beuzenberg, V.; MacKenzie, A.L. Isolation and identification of a cis-C8-diol-ester of okadaic acid from Dinophysis acuta in New Zealand. Toxicon 2006, 48, 195–203. [Google Scholar] [CrossRef]
- Takai, A.; Murata, M.; Isobe, M.; Mieskes, G.; Yasumoto, T. Inhibitory effect of okadaic acid derivatives on protein phosphatases. A study on structure–affinity relationship. Biochem. J. 1992, 284, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Holmes, M.J.; Lee, F.C.; Khoo, H.W.; Teo, S.L.M. Production of 7-deoxy-okadaic acid by a New Caledonian strain of Prorocentrum lima (Dinophyceae). J. Phycol. 2001, 37, 280–288. [Google Scholar] [CrossRef]
- Beach, D.G.; Crain, S.; Lewis, N.; LeBlanc, P.; Hardstaff, W.R.; Perez, R.A.; Giddings, S.D.; Martinez-Farina, C.F.; Stefanova, R.; Burton, I.W.; et al. Development of certified reference materials for diarrhetic shellfish poisoning toxins, part 1: Calibration solutions. J. AOAC Int. 2016, 99, 1151–1162. [Google Scholar] [CrossRef] [PubMed]
- Cruz, P.G.; Daranas, A.H.; Fernandez, J.J.; Norte, M. 19-epi-Okadaic acid, a novel protein phosphatase inhibitor with enhanced selectivity. Org. Lett. 2007, 9, 3045–3048. [Google Scholar] [CrossRef] [PubMed]
- Kilcoyne, J.; Burrell, S.; Nulty, C.; Salas, R.; Wright, E.J.; Rajotte, I.; Miles, C.O. Improved isolation procedures for okadaic acid group toxins from shellfish (Mytilus edulis) and microalgae (Prorocentrum lima). Mar. Drugs 2020, 18, 647. [Google Scholar] [CrossRef]
- EFSA (The European Food Safety Authority). Opinion of the Scientific Panel on Contaminants in the Food Chain on a request from the European Commission on marine biotoxins in shellfish—Okadaic acid and analogues. EFSA J. 2008, 589, 1–62. [Google Scholar]
- Yanagi, T.; Murata, M.; Torigoe, K.; Yasumoto, T. Biological activities of semisynthetic analogs of dinophysistoxin-3, the major diarrhetic shellfish toxin. Agric. Biol. Chem. 1989, 53, 525–529. [Google Scholar] [CrossRef]
- Twiner, M.J.; Doucette, G.J.; Pang, Y.; Fang, C.; Forsyth, C.J.; Miles, C.O. Structure–activity relationship studies using natural and synthetic okadaic acid/dinophysistoxin toxins. Mar. Drugs 2016, 14, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munday, R. Is protein phosphatase inhibition responsible for the toxic effects of okadaic acid in animals? Toxins 2013, 5, 267–285. [Google Scholar] [CrossRef] [Green Version]
- Larsen, K.; Petersen, D.; Wilkins, A.L.; Samdal, I.A.; Sandvik, M.; Rundberget, T.; Goldstone, D.; Arcus, V.; Hovgaard, P.; Rise, F.; et al. Clarification of the C-35 stereochemistries of dinophysistoxin-1 and dinophysistoxin-2 and its consequences for binding to protein phosphatase. Chem. Res. Toxicol. 2007, 20, 868–875. [Google Scholar] [CrossRef]
- Abal, P.; Louzao, M.C.; Suzuki, T.; Watanabe, R.; Vilariño, N.; Carrera, C.; Botana, A.M.; Vieytes, M.R.; Botana, L.M. Toxic action reevaluation of okadaic acid, dinophysistoxin-1 and dinophysistoxin-2: Toxicity equivalency factors based on the oral toxicity study. Cell. Physiol. Biochem. 2018, 49, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Garibo, D.; de la Iglesia, P.; Diogène, J.; Campàs, M. Inhibition equivalency factors for dinophysistoxin-1 and dinophysistoxin-2 in protein phosphatase assays: Applicability to the analysis of shellfish samples and comparison with LC−MS/MS. J. Agric. Food Chem. 2013, 61, 2572–2579. [Google Scholar] [CrossRef]
- Dounay, A.B.; Urbanek, R.A.; Sabes, S.F.; Forsyth, C.J. Total synthesis of the marine natural product 7-deoxy-okadaic acid: A potent inhibitor of serine/threonine-specific protein phosphatases. Angew. Chem. Int. Ed. 1999, 38, 2258–2262. [Google Scholar] [CrossRef]
- Sklarz, B. Organic chemistry of periodates. Q. Rev. Chem. Soc. 1967, 21, 3–28. [Google Scholar] [CrossRef]
- Rundberget, T.; Sandvik, M.; Larsen, K.; Pizarro, G.M.; Reguera, B.; Castberg, T.; Gustad, E.; Loader, J.I.; Rise, F.; Wilkins, A.L.; et al. Extraction of microalgal toxins by large-scale pumping of sea water in Spain and Norway, and isolation of okadaic acid and dinophysistoxin-2. Toxicon 2007, 50, 960–970. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T.; Murata, M.; Oshima, Y.; Sano, M.; Matsumoto, G.; Clardy, J. Diarrhetic shellfish toxins. Tetrahedron 1985, 41, 1019–1025. [Google Scholar] [CrossRef]
- Torgersen, T.; Miles, C.O.; Rundberget, T.; Wilkins, A.L. New esters of okadaic acid in seawater and blue mussels (Mytilus edulis). J. Agric. Food Chem. 2008, 56, 9628–9635. [Google Scholar] [CrossRef]
- Schmitz, F.J.; Prasad, R.S.; Gopichand, Y.; Hossain, M.B.; van der Helm, D.; Schmidt, P. Acanthifolicin, a new episulfide-containing polyether carboxylic acid from extracts of the marine sponge Pandaros acanthifolium. J. Am. Chem. Soc. 1981, 103, 2467–2469. [Google Scholar] [CrossRef]
- Tachibana, K.; Scheuer, P.J.; Tsukitani, Y.; Kikuchi, H.; Van Engen, D.; Clardy, J.; Gopichand, Y.; Schmitz, F.J. Okadaic acid, a cytotoxic polyether from two marine sponges of the genus Halichondria. J. Am. Chem. Soc. 1981, 103, 2469–2471. [Google Scholar] [CrossRef]
- Wright, E.; Reeves, K.L.; Giddings, S.D.; Quilliam, M.A.; McCarron, P. Certificate of Analysis. CRM-FDMT1, A Freeze-Dried Mussel Tissue Certified Reference Material for Multiple Marine Toxins; Biotoxin Metrology Technical Report CRM-FDMT1-20070717; National Research Council: Halifax, NS, Canada, 2016. [Google Scholar]
- McCarron, P.; Emteborg, H.; Nulty, C.; Rundberget, T.; Loader, J.I.; Teipel, K.; Miles, C.O.; Quilliam, M.A.; Hess, P. A mussel tissue certified reference material for multiple phycotoxins. Part 1: Design and preparation. Anal. Bioanal. Chem. 2011, 400, 821–833. [Google Scholar] [CrossRef] [Green Version]
- Wright, E.J.; McCarron, P. A mussel tissue certified reference material for multiple phycotoxins. Part 5: Profiling by liquid chromatography–high-resolution mass spectrometry. Anal. Bioanal. Chem. 2021, 413, 2055–2069. [Google Scholar] [CrossRef] [PubMed]
- McCarron, P.; Wright, E.; Emteborg, H.; Quilliam, M.A. A mussel tissue certified reference material for multiple phycotoxins. Part 4: Certification. Anal. Bioanal. Chem. 2017, 409, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, B.K. Mass Spectral Characterisation of Three New Groups of Algal Toxins. Master’s Thesis, University of Waikato, Hamilton, New Zealand, 2009. [Google Scholar]
- MacKenzie, L.; Beuzenberg, V.; Holland, P.; McNabb, P.; Suzuki, T.; Selwood, A. Pectenotoxin and okadaic acid-based toxin profiles in Dinophysis acuta and Dinophysis acuminata from New Zealand. Harmful Algae 2005, 4, 75–85. [Google Scholar] [CrossRef]
- Nagai, H.; Satake, M.; Yasumoto, T. Antimicrobial activities of polyether compounds of dinoflagellate origins. J. Appl. Phycol. 1990, 2, 305–308. [Google Scholar] [CrossRef]
- Valdiglesias, V.; Prego-Faraldo, M.V.; Pásaro, E.; Méndez, J.; Laffon, B. Okadaic acid: More than a diarrheic toxin. Mar. Drugs 2013, 11, 4328–4349. [Google Scholar] [CrossRef] [Green Version]
- Maynes, J.T.; Bateman, K.S.; Cherney, M.M.; Das, A.K.; Luu, H.A.; Holmes, C.F.B.; James, M.N.C. Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J. Biol. Chem. 2001, 276, 44078–44082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, Y.; Xu, Y.; Chen, Y.; Jeffrey, P.D.; Chao, Y.; Lin, Z.; Li, Z.; Strack, S.; Stock, J.B.; Shi, Y. Structure of protein phosphatase 2A core enzyme bound to tumor-inducing toxins. Cell 2006, 127, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Huhn, J.; Jeffrey, P.D.; Larsen, K.; Rundberget, T.; Rise, F.; Cox, N.R.; Arcus, V.; Shi, Y.; Miles, C.O. A structural basis for the reduced toxicity of dinophysistoxin-2. Chem. Res. Toxicol. 2009, 22, 1782–1786. [Google Scholar] [CrossRef]
- Kilcoyne, J.; McCarron, P.; Twiner, M.J.; Rise, F.; Hess, P.; Wilkins, A.L.; Miles, C.O. Identification of 21,22-dehydroazaspiracids in mussels (Mytilus edulis) and in vitro toxicity of azaspiracid-26. J. Nat. Prod. 2018, 81, 885–893. [Google Scholar] [CrossRef] [Green Version]
- McNabb, P.; Selwood, A.I.; Holland, P.T. Multiresidue method for determination of algal toxins in shellfish: Single-laboratory validation and interlaboratory study. J. AOAC 2005, 88, 761–772. [Google Scholar] [CrossRef] [Green Version]
- McKenzie, C.H.; Paranjape, M.; Gilgan, M.W.; Powell, C.; Quilliam, M.A. A Dinophysis norvegica bloom and its implications in the occurrence of a diarrhetic shellfish poisoning episode in mussels from Newfoundland in the late autumn of 1993. In Proceedings of the Fourth Canadian Workshop on Harmful Marine Algae, St. Andrews, NB, Canada, 3–5 May 1994; Forbes, J.R., Ed.; Institute of Ocean Sciences: Sidney, BC, Canada, 1994; p. 26. [Google Scholar]
- Rourke, W.A.; Justason, A.; Martin, J.L.; Murphy, C.J. Shellfish toxin uptake and depuration in multiple Atlantic Canadian molluscan species: Application to selection of sentinel species in monitoring programs. Toxins 2021, 13, 168. [Google Scholar] [CrossRef] [PubMed]
- McCarron, P.; Giddings, S.D.; Reeves, K.L.; Hess, P.; Quilliam, M.A. A mussel (Mytilus edulis) tissue certified reference material for the marine biotoxins azaspiracids. Anal. Bioanal. Chem. 2015, 407, 2985–2996. [Google Scholar] [CrossRef]
- Fux, E.; Marcaillou, C.; Mondeguer, F.; Bire, R.; Hess, P. Field and mesocosm trials on passive sampling for the study of adsorption and desorption behaviour of lipophilic toxins with a focus on OA and DTX1. Harmful Algae 2008, 7, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Fux, E.; Bire, R.; Hess, P. Comparative accumulation and composition of lipophilic marine biotoxins in passive samplers and in mussels (M. edulis) on the West Coast of Ireland. Harmful Algae 2009, 8, 523–537. [Google Scholar] [CrossRef]
- Hernández-Urcera, J.; Rial, P.; García-Portela, M.; Lourés, P.; Kilcoyne, J.; Rodríguez, F.; Fernández-Villamarín, A.; Reguera, B. Notes on the cultivation of two mixotrophic Dinophysis species and their ciliate prey Mesodinium rubrum. Toxins 2018, 10, 505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, M.D.; Selvin, R.C.; Claus, W.; Guillard, R.R.L. Media for the culture of oceanic ultraphytoplankton. J. Phycol. 2007, 23, 633–638. [Google Scholar] [CrossRef]
- Guillard, R.R.L.; Hargraves, E.P. Stichochrysis immobilis is a diatom, not a chrysophyte. Phycologia 1993, 32, 234–236. [Google Scholar] [CrossRef]
- Bravo, I.; Fernández, M.L.; Ramilo, I.; Martıínez, A. Toxin composition of the toxic dinoflagellate Prorocentrum lima isolated from different locations along the Galician coast (NW Spain). Toxicon 2001, 39, 1537–1545. [Google Scholar] [CrossRef]
- Guillard, R.R.L.; Morton, S.L. Culture methods. In Manual on Harmful Marine Microalgae, 2nd ed.; Hallegraeff, G.M., Anderson, D.M., Cembella, A.D., Enevoldsen, H.O., Eds.; UNESCO Publishing: Paris, France, 2003; Volume 11, pp. 77–97. [Google Scholar]
- Jackson, A.E.; Marr, J.C.; McLachlan, J.L. The production of diarrhetic shellfish toxins by an isolate of Prorocentrum lima from Nova Scotia, Canada. In Toxic Phytoplankton Blooms in the Sea; Smayda, T.J., Shimizu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 1993; pp. 513–518. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tRb | Ion A | Ion B | Ion C | Ion D | Ion E | Ion F | Ion G | Ion H | Ion I | Ion J | Ion K | Ion L | |
| 1 | 13.42 | 255.1238 | 463.2337 | 563.2862 | 321.2071 | 723.4079 | 571.3241 | 443.2404 | 383.1829 | 483.2353 | 595.3241 | 263.1618 | 151.0730 |
| 2 | 14.15 | 255.1238 | 463.2337 | 563.2862 | 321.2071 | 723.4079 | 571.3241 | 429.2248 | 383.1829 | 483.2353 | 581.3085 | 263.1618 | 165.0886 |
| 3 | 15.75 | 255.1238 | 463.2337 | 563.2862 | 335.2228 | 737.4235 | 585.3398 | 443.2404 | 383.1829 | 483.2353 | 595.3241 | 277.1774 | 165.0886 |
| 4 | 9.12 | 255.1238 | 463.2337 | 725.3390 | 321.2071 | 885.4607 | 733.3770 | 605.2932 | 383.1829 | 645.2881 | 757.3770 | 263.1618 | 151.0730 |
| 5 | 9.48 | 255.1238 | 463.2337 | 725.3390 | 321.2071 | 885.4607 | 733.3770 | 591.2776 | 383.1829 | 645.2881 | 743.3613 | 263.1618 | 165.0886 |
| 6 | 11.01 | 255.1238 | 463.2337 | 725.3390 | 335.2228 | 899.4763 | 747.3926 | 605.2932 | 383.1829 | 645.2881 | 757.3770 | 277.1774 | 165.0886 |
| 7 | 17.68 | 239.1289 | 447.2388 | 547.2913 | 321.2071 | 707.4130 | 571.3241 | 443.2404 | 367.1880 | 467.2404 | 579.3292 | 263.1618 | 151.0730 |
| 8 | 18.25 | 239.1289 | 447.2388 | 547.2913 | 321.2071 | 707.4130 | 571.3241 | 429.2248 | 367.1880 | 467.2404 | 565.3136 | 263.1618 | 165.0886 |
| 9 | 19.76 | 239.1289 | 447.2388 | 547.2913 | 335.2228 | 721.4286 | 585.3398 | 443.2404 | 367.1880 | 467.2404 | 579.3292 | 277.1774 | 165.0886 |
| OA (1) | 24-GlcOA (4) | 24-GlcDTX2 (5) | DTX2 (2) | |||||
|---|---|---|---|---|---|---|---|---|
| Atom | 13C | 1H | 13C | 1HG | 13C | 1H | 13C | 1H |
| 1 | 182.8 | 182.1 | 183.3 | 180.6 | ||||
| 2 | 76.3 | 76.5 | 76.2 | 76.1 | ||||
| 3 | 46.7 | 1.65, 1.91 | 46.8 | 1.69, 1.88 | 46.7 | 1.68, 1.89 | 45.7 | 1.65, 2.01 |
| 4 | 68.9 | 4.08 | 68.7 | 4.09 | 68.7 | 4.09 | 69.3 | 4.08 |
| 5 | 33.4 | 1.30, 1.89 | 33.6 | 1.30, 1.79 | 33.6 | 1.31, 1.81 | 33.4 | 1.34, 1.79 |
| 6 | 28.3 | 1.66, 1.94 | 28.4 | 1.65, 1.96 | 28.3 | 1.65, 1.97 | 28.0 | 1.65, 1.98 |
| 7 | 73.4 | 3.35 | 73.4 | 3.36 | 73.4 | 3.36 | 73.1 | 3.37 |
| 8 | 97.5 | 97.5 | 97.6 | 97.6 | ||||
| 9 | 123.8 | 5.26 | 123.9 | 5.28 | 123.8 | 5.28 | 123.2 | 5.29 |
| 10 | 139.3 | 139.2 | 139.2 | 139.9 | ||||
| 11 | 34.0 | 1.82, 1.98 | 34.1 | 1.83, 1.97 | 34.1 | 1.84, 1.97 | 34.1 | 1.88, 1.96 |
| 12 | 71.8 | 3.87 | 71.8 | 3.91 | 71.8 | 3.91 | 71.9 | 3.75 |
| 13 | 43.2 | 2.35 | 43.5 | 2.36 | 43.4 | 2.36 | 43.3 | 2.34 |
| 14 | 137.2 | 5.95 | 137.3 | 5.95 | 137.2 | 5.93 | 137.4 | 5.83 |
| 15 | 132.0 | 5.50 | 131.8 | 5.50 | 131.8 | 5.49 | 132.3 | 5.52 |
| 16 | 80.6 | 4.66 | 80.7 | 4.60 | 80.7 | 4.60 | 80.5 | 4.62 |
| 17 | 31.5 | 1.60, 2.21 | 31.6 | 1.61, 2.18 | 31.6 | 1.61, 2.18 | 31.5 | 1.61, 2.20 |
| 18 | 38.1 | 1.85, 2.00 | 38.1 | 1.89, 2.00 | 38.1 | 1.89, 2.02 | 38.0 | 1.85, 2.00 |
| 19 | 107.1 | 107.4 | 107.4 | 107.1 | ||||
| 20 | 34.2 | 1.84–1.87 | 34.3 | 1.85–1.92 | 34.3 | 1.85–1.92 | 34.1 | 1.82–1.89 |
| 21 | 27.7 | 1.80, 1.90 | 28.0 | 1.80, 1.91 | 28.0 | 1.81, 1.90 | 27.7 | 1.79, 1.90 |
| 22 | 71.4 | 3.63 | 71.6 | 3.72 | 71.6 | 3.72 | 71.4 | 3.64 |
| 23 | 78.3 | 3.42 | 76.9 | 3.59 | 76.8 | 3.59 | 78.1 | 3.40 |
| 24 | 72.1 | 4.07 | 75.9 | 4.44 | 75.5 | 4.44 | 72.2 | 4.10 |
| 25 | 147.1 | 143.9 | 144.1 | 147.4 | ||||
| 26 | 86.4 | 3.94 | 86.8 | 3.93 | 87.0 | 3.94 | 86.4 | 3.97 |
| 27 | 66.3 | 4.08 | 65.9 | 4.14 | 66.1 | 4.13 | 67.0 | 4.07 |
| 28 | 36.8 | 0.96, 1.37 | 37.0 | 0.92, 1.41 | 38.9 | 1.37, 1.42 | 38.6 | 1.37, 1.44 |
| 29 | 32.3 | 1.89 | 32.5 | 1.87 | 35.3 | 1.82 | 35.3 | 1.83 |
| 30 | 76.8 | 3.27 | 76.8 | 3.27 | 75.0 | 3.42 | 75.0 | 3.42 |
| 31 | 28.8 | 1.81 | 28.9 | 1.82 | 28.8 | 1.27, 1.52 | 28.7 | 1.27, 1.52 |
| 32 | 27.5 | 1.38, 2.00 | 27.6 | 1.38, 2.00 | 20.1 | 1.61, 1.74 | 20.1 | 1.74, 1.60 |
| 33 | 31.2 | 1.36, 1.59 | 31.3 | 1.32, 1.59 | 33.4 | 1.17, 1.67 | 33.4 | 1.16, 1.68 |
| 34 | 97.0 | 97.0 | 99.2 | 99.3 | ||||
| 35 | 37.0 | 1.43, 1.61 | 37.1 | 1.41, 1.61 | 37.4 | 1.62 | 37.4 | 1.61 |
| 36 | 19.8 | 1.53, 1.88 | 19.9 | 1.53, 1.88 | 26.9 | 1.32, 2.17 | 26.7 | 1.29, 2.17 |
| 37 | 26.5 | 1.49–1.55 | 26.6 | 1.49–1.56 | 20.9 | 1.28, 1.78 | 21.0 | 1.78, 1.26 |
| 38 | 61.3 | 3.51, 3.71 | 61.4 | 3.52, 3.71 | 61.4 | 3.51, 3.70 | 61.4 | 3.51, 3.70 |
| 25 =CH2 | 112.6 | 5.04, 5.38 | 114.6 | 5.11, 5.67 | 114.3 | 5.10, 5.69 | 112.1 | 5.04, 5.36 |
| 10-Me | 23.1 | 1.72 | 23.2 | 1.72 | 23.2 | 1.73 | 23.1 | 1.75 |
| 13-Me | 17.1 | 1.11 | 17.1 | 1.11 | 17.2 | 1.11 | 16.8 | 1.07 |
| 29-Me | 16.6 | 1.05 | 17.0 | 1.07 | 14.5 | 0.96 | 14.4 | 0.95 |
| 2-Me | 27.9 | 1.31 | 27.6 | 1.32 | 27.5 | 1.32 | 27.7 | 1.34 |
| 31-Me | 11.1 | 0.93 | 11.2 | 0.94 | ||||
| 35-Me | 14.6 | 1.00 | 14.7 | 0.99 | ||||
| OA 24-O-β-d-Glucoside (4) | DTX2 24-O-β-d-Glucoside (5) | |||||
|---|---|---|---|---|---|---|
| atom | 13C | 1H | Mult, J (Hz) | 13C | 1H | Mult, J (Hz) |
| 1′ | 102.5 | 4.39 | d, 7.8 a | 102.5 | 4.39 | d, 7.8 a |
| 2′ | 75.0 | 3.39 | dd, 7.8, 9.0 b | 75.0 | 3.39 | dd, 7.8, 9.0 b |
| 3′ | 78.1 | 3.35 | ~t, J = 9.0 b | 78.2 | 3.32 | ~t, 9.0 b |
| 4′ | 71.9 | 3.28 | ~t, 9.0 b | 71.9 | 3.29 | ~t, 9.0 b |
| 5′ | 78.5 | 3.20 | m | 78.4 | 3.19 | m |
| 6′ | 63.1 | 3.64 | dd, 12.0, 5.5 b | 63.1 | 3.64 | dd, 12.0, 5.5 b |
| 3.83 | dd, 12.0, 2.2 b | 3.83 | dd, 12.0, 2.2 b | |||
| Percentage of Sum of 1–9 | Sum 1–9 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample | Description | Origin b | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ng/mL | µg/g |
| Mixed standard | Std of 4, 5 and 7 | N/A | 0.4 | 0.2 | 0.07 | 21 | 72 | ND | 6.1 | ND | ND | 2100 | |
| RM-Multi-toxin | Multitoxin RM | N/A | 30 | 31 | 40 | ND | ND | ND | ND | ND | ND | 750 | |
| D. acuta | VGO1065/1 | ES | 71 | 28 | 0.2 | 0.3 | 0.05 | ND | 0.09 | 0.04 | ND | 38,000 | |
| D. caudata (conc.) | VGO1396/2 | ES | 77 | 12 | 10 | 0.5 | ND | ND | ND | ND | ND | 5.2 | |
| D. caudata (conc.) | VGO1396/4 | ES | 68 | 14 | 17 | 0.3 | ND | ND | ND | ND | ND | 2.0 | |
| D. acuminata | VGO1349/3 | ES | 99 | ND | 0.01 | 0.6 | 0.01 | ND | 0.1 | ND | ND | 2000 | |
| D. acuminata | VGO1349/5 | ES | 100 | ND | 0.03 | 0.07 | ND | ND | 0.1 | ND | ND | 1200 | |
| D. acuminata | VGO1391/7 | ES | 100 | ND | 0.01 | 0.3 | ND | ND | 0.1 | ND | ND | 2700 | |
| P. lima | CCMP2579 | RI, US | 7.3 | ND | 93 | ND | ND | ND | 0.01 | ND | 0.04 | 2300 | |
| P. lima | JHPL2 | NS, CA | 7.4 | ND | 92 | ND | ND | ND | 0.01 | ND | 0.07 | 18,000 | |
| P. lima | JHPL3 | NS, CA | 5.8 | ND | 94 | ND | ND | ND | 0.01 | ND | 0.07 | 12,000 | |
| P. lima | KP200 | PE, CA | 27 | ND | 73 | ND | ND | ND | 0.02 | ND | 0.03 | 13,000 | |
| P. lima | KP209 | NB, CA | 37 | ND | 60 | ND | ND | ND | 2.1 | ND | 0.03 | 4100 | |
| P. lima | IP197 | NS, CA | 44 | ND | 45 | ND | ND | ND | 11 | ND | 0.05 | 3100 | |
| P. lima | CCMI1036 | IE | 5.0 | ND | 95 | ND | ND | ND | ND | ND | 0.08 | 1900 | |
| P. lima | PL20V, Galicia | ES | 81 | ND | 17 | ND | ND | ND | 0.08 | ND | 0.02 | 1800 | |
| FDMT1-SPE (conc.) | CRM concentrate | IE/NO | 19 | 55 | 13 | 8.6 | 3.9 | 0.3 | 0.1 | 0.2 | 0.07 | 6500 | |
| CRM FDMT1 | CRM Extract | IE/NO | 25 | 59 | 8.8 | 5.1 | 2.5 | 0.2 | ND | ND | ND | 120 | |
| CRM DSP-Mus | CRM Extract | IE/NO | 34 | 30 | 32 | 1.2 | 0.9 | 0.03 | 1.3 | 0.03 | 0.02 | 330 | 1.63 |
| Mussel HP | Flødevigen | NO | 14 | 20 | 57 | 1.5 | 0.4 | 6.9 | ND | ND | ND | 7.3 | 0.04 |
| Whole mussel | Flødevigen | NO | 7.4 | 8.5 | 78 | 0.4 | ND | 5.4 | ND | ND | ND | 61 | 0.32 |
| Whole broth | Indian Pt. | NS, CA | 0.2 | ND | 98 | ND | ND | 1.4 | ND | ND | 0.02 | 93 | 0.52 |
| Whole mussel | Bonavista Bay | NF, CA | 0.2 | ND | 97 | ND | ND | 2.5 | ND | ND | 0.5 | 41 | 0.33 |
| Whole mussel | Ship Harbour | NS, CA | 92 | ND | 4.2 | 3.9 | ND | 0.2 | ND | ND | ND | 50 | 0.48 |
| Whole mussel c | Castlemaine Harbour | IE | 35 | 5.7 | 3.8 | 54 | 1.3 | 0.2 | ND | ND | ND | 10 | 0.09 |
| Whole mussel d | Bantry Middle | IE | 67 | 8.7 | 2.0 | 21 | 0.5 | 0.1 | 0.7 | ND | ND | 36 | 0.43 |
| SPATT (conc.) | Ship Harbour | NS, CA | 8.8 | ND | 91 | ND | ND | 0.2 | ND | ND | 0.1 | 37 | |
| Reported Results | |||||||
|---|---|---|---|---|---|---|---|
| Location | Date b | Species | 4 + 5 (%) | 1 | 2 | 1 Equiv. c | |
| 1 | Ardgroom | 02/06/20 | M. edulis | 5.5 | 2.68 | <LOD | 2.68 |
| 2 | Kilmakilloge | 02/06/20 | M. edulis | 6.7 | 2.61 | <LOD | 2.61 |
| 3 | Mine Head Ground | 03/06/20 | P. maximus | ND | 0.11 | 0.03 | 0.13 |
| 4 | Youghal Bay | 10/06/20 | S. solida | ND | 0.13 | <LOD | 0.13 |
| 5 | Valentia River | 11/06/20 | C. gigas | ND | 0.12 | <LOD | 0.12 |
| 6 | Castlemaine Harbour | 24/06/20 | C. edule | ND | 0.20 | <LOD | 0.20 |
| 7 | Tahilla | 30/06/20 | M. edulis | 20.2 | 0.69 | 0.02 | 0.70 |
| 8 | Bantry Middle | 30/06/20 | M. edulis | 16.8 | 1.21 | 0.08 | 1.26 |
| 9 | Gouleenacoush | 01/07/20 | M. edulis | 17.2 | 1.45 | <LOD | 1.45 |
| 10 | Adrigole | 01/07/20 | M. edulis | 14.1 | 0.66 | 0.07 | 0.70 |
| 11 | Tahilla | 13/07/20 | M. edulis | 39.2 | 0.33 | 0.04 | 0.35 |
| 12 | Castlemaine Harbour d | 13/07/20 | M. edulis | 58.0 | 0.09 | <LOQ | 0.09 |
| 13 | Castlemaine Harbour | 13/07/20 | M. edulis | 44.1 | 0.08 | <LOQ | 0.08 |
| 14 | Bantry North | 13/07/20 | M. edulis | 40.5 | 0.22 | 0.06 | 0.26 |
| 15 | Bantry Middle | 13/07/20 | M. edulis | 21.4 | 0.40 | 0.05 | 0.43 |
| 16 | South of Smalls Ground | 04/08/20 | P. maximus | ND | 0.10 | 0.03 | 0.12 |
| 17 | Bantry North d | 31/08/20 | M. edulis | 5.7 | 0.35 | 0.73 | 0.79 |
| 18 | Bantry South | 31/08/20 | M. edulis | 4.9 | 0.84 | 0.76 | 1.30 |
| 19 | Ardgroom | 31/08/20 | M. edulis | 26.0 | 0.07 | 0.12 | 0.14 |
| Non-Hydrolyzed | Hydrolyzed | |||||||
|---|---|---|---|---|---|---|---|---|
| Location | Species | Date b | 1 | 3 | 4 | 1 | 3 | 4 |
| Sumner | M. edulis | 04/01/17 | 0.151 | 0.008 | 0.011 | 0.226 | <LOQ | 0.013 |
| Akaroa harbour | M. edulis | 05/07/17 | 0.420 | 0.010 | 0.010 | 0.826 | 0.012 | <LOQ |
| Akaroa harbour | M. edulis | 12/07/17 | 0.268 | 0.005 | 0.004 | 0.590 | <LOQ | <LOQ |
| Akaroa harbour | M. edulis | 31/01/17 | 0.412 | 0.018 | 0.006 | 0.833 | 0.022 | <LOQ |
| Akaroa harbour | M. edulis | 07/08/17 | 0.514 | 0.025 | 0.0010 | 0.978 | 0.032 | <LOQ |
| Motukiekie Beach | P. canaliculus | 25/02/19 | 0.025 | ND | ND | 0.201 | ND | ND |
| Cape Foulwind | P. canaliculus | 04/03/19 | 0.021 | ND | ND | 0.191 | ND | ND |
| Cape Foulwind | P. canaliculus | 18/03/19 | 0.020 | ND | ND | 0.220 | ND | ND |
| Motukiekie Beach | M. edulis | 01/04/19 | 0.037 | ND | ND | 0.220 | ND | ND |
| Sumner | P. subtriangulata | 02/06/20 | ND | <LOQ | ND | ND | 0.025 | ND |
| Sumner | P. canaliculus | 09/06/20 | <LOQ | 0.009 | ND | 0.012 | 0.072 | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkins, A.L.; Rundberget, T.; Sandvik, M.; Rise, F.; Knudsen, B.K.; Kilcoyne, J.; Reguera, B.; Rial, P.; Wright, E.J.; Giddings, S.D.; et al. Identification of 24-O-β-d-Glycosides and 7-Deoxy-Analogues of Okadaic Acid and Dinophysistoxin-1 and -2 in Extracts from Dinophysis Blooms, Dinophysis and Prorocentrum Cultures, and Shellfish in Europe, North America and Australasia. Toxins 2021, 13, 510. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080510
Wilkins AL, Rundberget T, Sandvik M, Rise F, Knudsen BK, Kilcoyne J, Reguera B, Rial P, Wright EJ, Giddings SD, et al. Identification of 24-O-β-d-Glycosides and 7-Deoxy-Analogues of Okadaic Acid and Dinophysistoxin-1 and -2 in Extracts from Dinophysis Blooms, Dinophysis and Prorocentrum Cultures, and Shellfish in Europe, North America and Australasia. Toxins. 2021; 13(8):510. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080510
Chicago/Turabian StyleWilkins, Alistair L., Thomas Rundberget, Morten Sandvik, Frode Rise, Brent K. Knudsen, Jane Kilcoyne, Beatriz Reguera, Pilar Rial, Elliott J. Wright, Sabrina D. Giddings, and et al. 2021. "Identification of 24-O-β-d-Glycosides and 7-Deoxy-Analogues of Okadaic Acid and Dinophysistoxin-1 and -2 in Extracts from Dinophysis Blooms, Dinophysis and Prorocentrum Cultures, and Shellfish in Europe, North America and Australasia" Toxins 13, no. 8: 510. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins13080510