Immunological Effects and Viral Gene Expression Determine the Efficacy of Oncolytic Measles Vaccines Encoding IL-12 or IL-15 Agonists

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Viruses
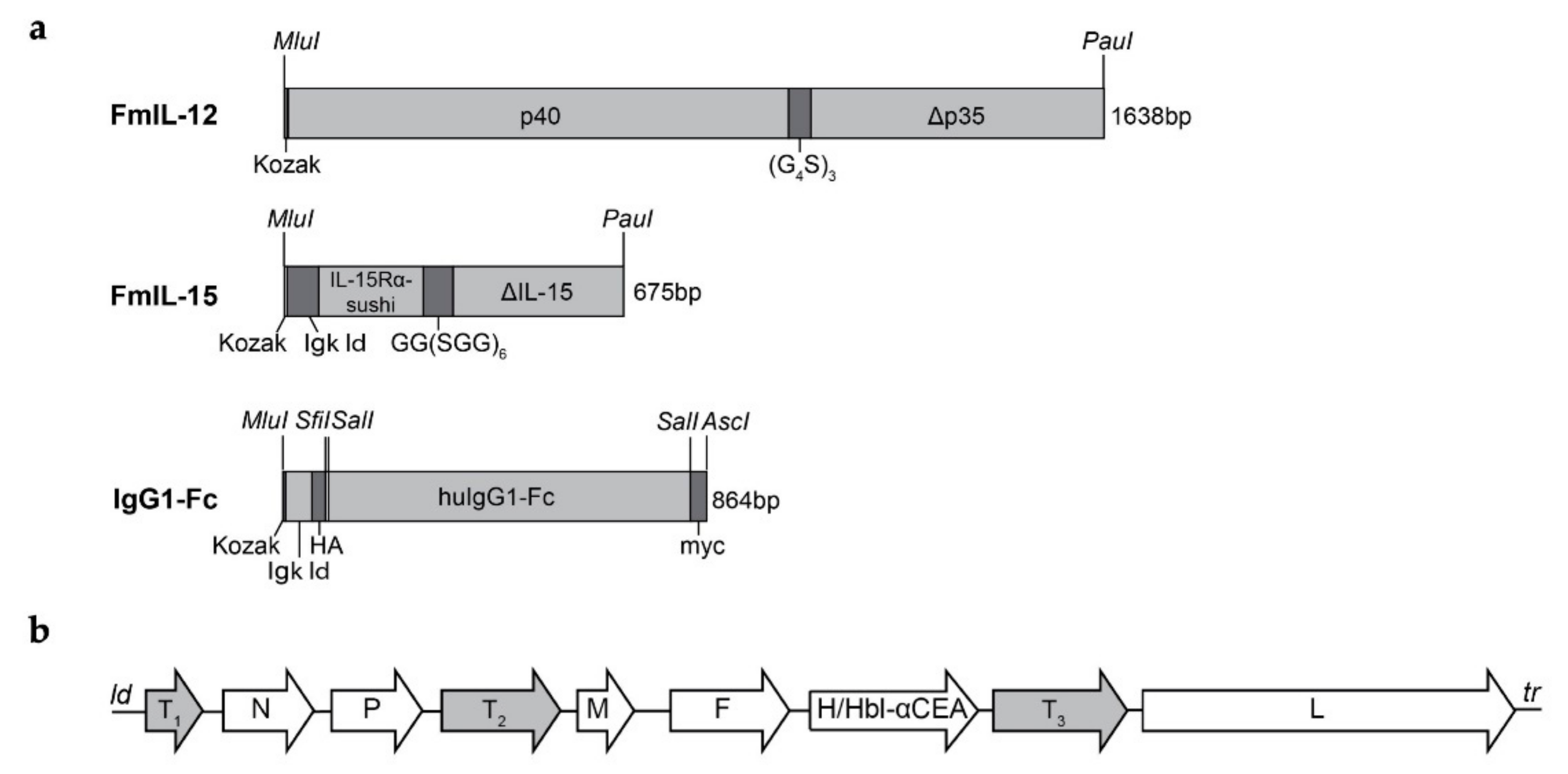
2.2. Cloning and Rescue of Recombinant MeVac
- FhIL-12 p40 for 5′ CCCGGGACGCGTGCCACCATGTGTCACCAGCAGT 3′
- FhIL-12 p40 rev 5′ AGATCCGCCGCCACCGCCACCACTGCAGGGCAC 3′
- FhIL-12 p35 for 5′ GGTGGCGGTGGCGGCGGATCTAGAAACCTCCCC 3′
- FhIL-12 p35 rev 5′ CCCGGGGCGCGCTCACTAGGAAGCATTCAGATAGCTC 3′
2.3. MeVac Titration, Propagation and Infection Experiments
2.4. Transgene Expression and Bioactivity
2.5. Characterization of MeVac Variants
2.6. Animal Experiments
2.7. Flow Cytometry
2.8. RT-qPCR
- mBcl2 for 5′–AGGCTGGGATGCCTTTGTGG–3′,
- mBcl2 rev 5′–ACTTGTGGCCCAGGTATGC–3′,
- mCish for 5′–CCTCGTCCTTCCAAGCTGTT–3′,
- mCish rev 5′–CCCAGTACCACCCCCTGTA–3′,
- mFasL for 5′–TCTGTGGCTACCGGTGGTAT–3′,
- mFasL rev 5′–GTACTGGGGTTGGCTCACG–3′,
- mGzmb for 5′–ACAAAGGCAGGGGAGATCAT–3′,
- mGzmb rev 5′–CGAATAAGGAAGCCCCCACA–3′,
- mL13A for 5′–GGCTGCCGAAGATGGCGGAG–3′,
- mL13A rev 5′–GCCTTCACAGCGTACGACCACC–3′.
- MV-N 241 for 5′-TTACCACTCGATCCAGACTTC-3′,
- MV-N 331+ rev 5′-CCTATTAGTGCCCCTGTTAGTTT-3′,
- FmIL-12p40 for 5′–CACTCCCCATTCCTACTTCT–3′,
- FmIL-12p35 rev 5′–CAGGATGCAGAGCTTCATTT–3′,
- FmIL-15 for 5′–CAACAGGGGACACAACCTGT–3′,
- FmIL-15 rev 5′–TGCACTTGAGGCTAGGTGTG–3′.
2.9. NanoString Gene Expression Profiling and Pathway Analysis
2.10. Statistical Analyses
3. Results
3.1. Oncolytic Measles Vaccines Encoding FmIL-12 or FmIL-15
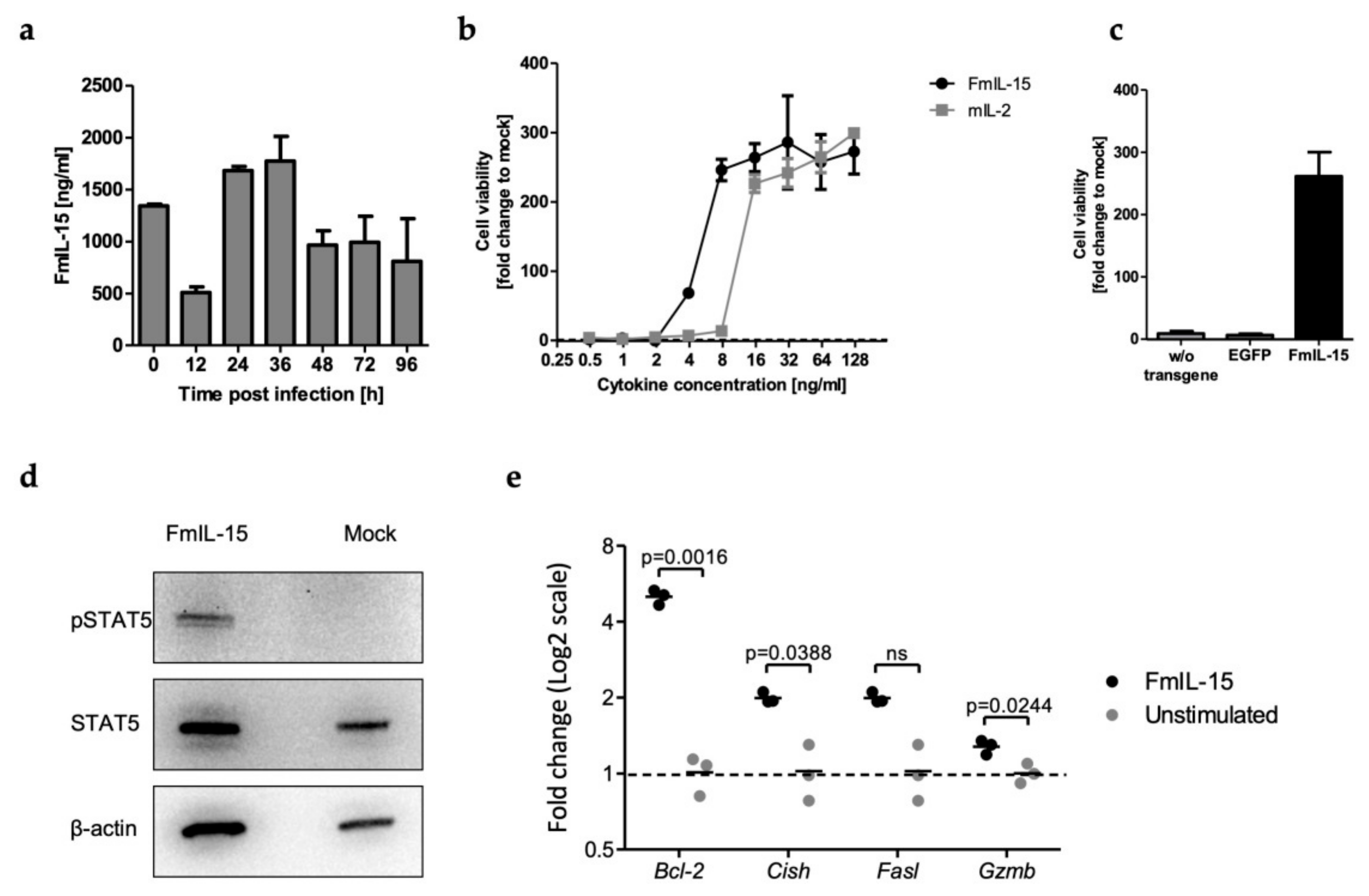
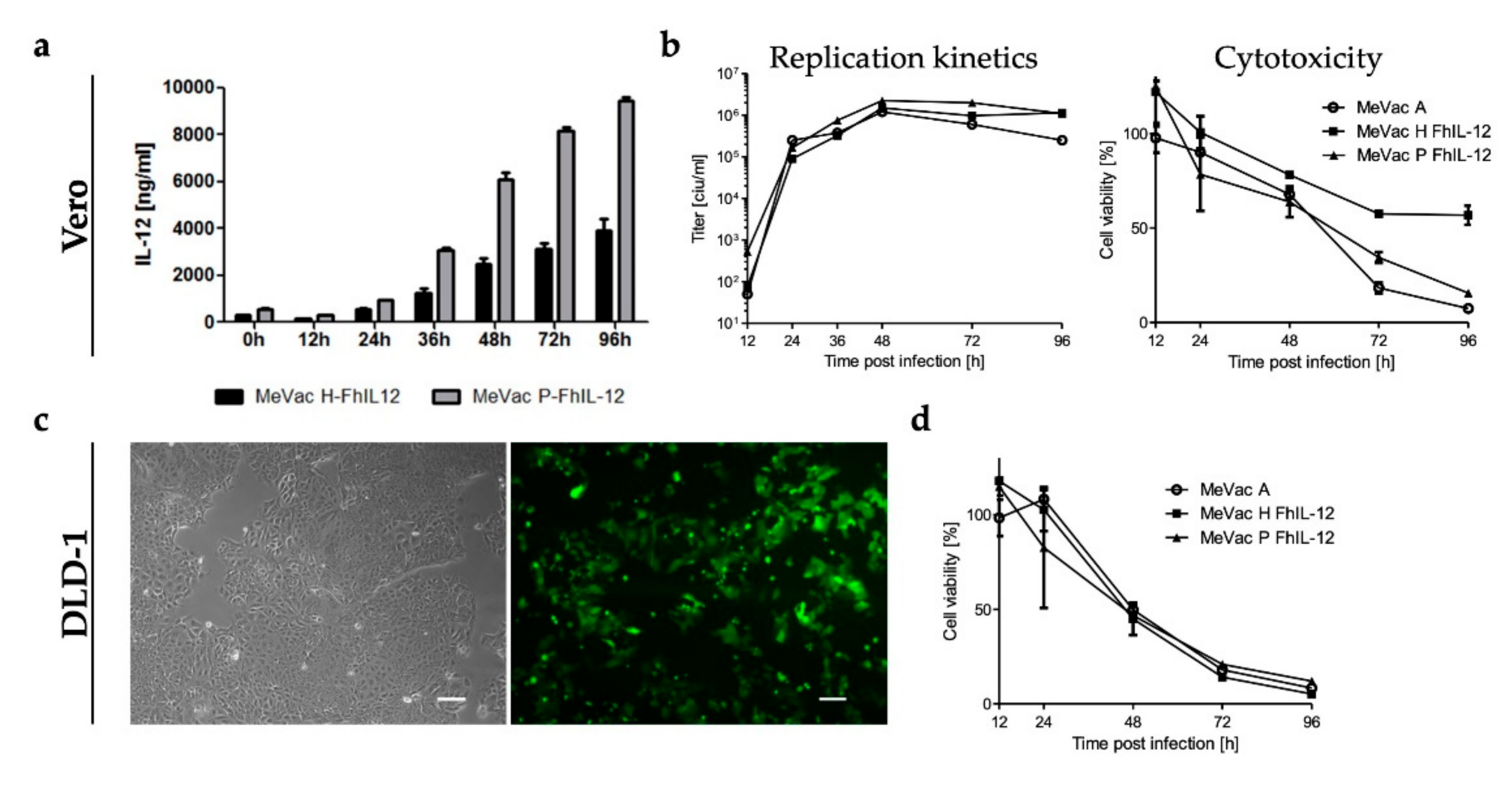
3.2. Secretion and Bioactivity of FmIL-15
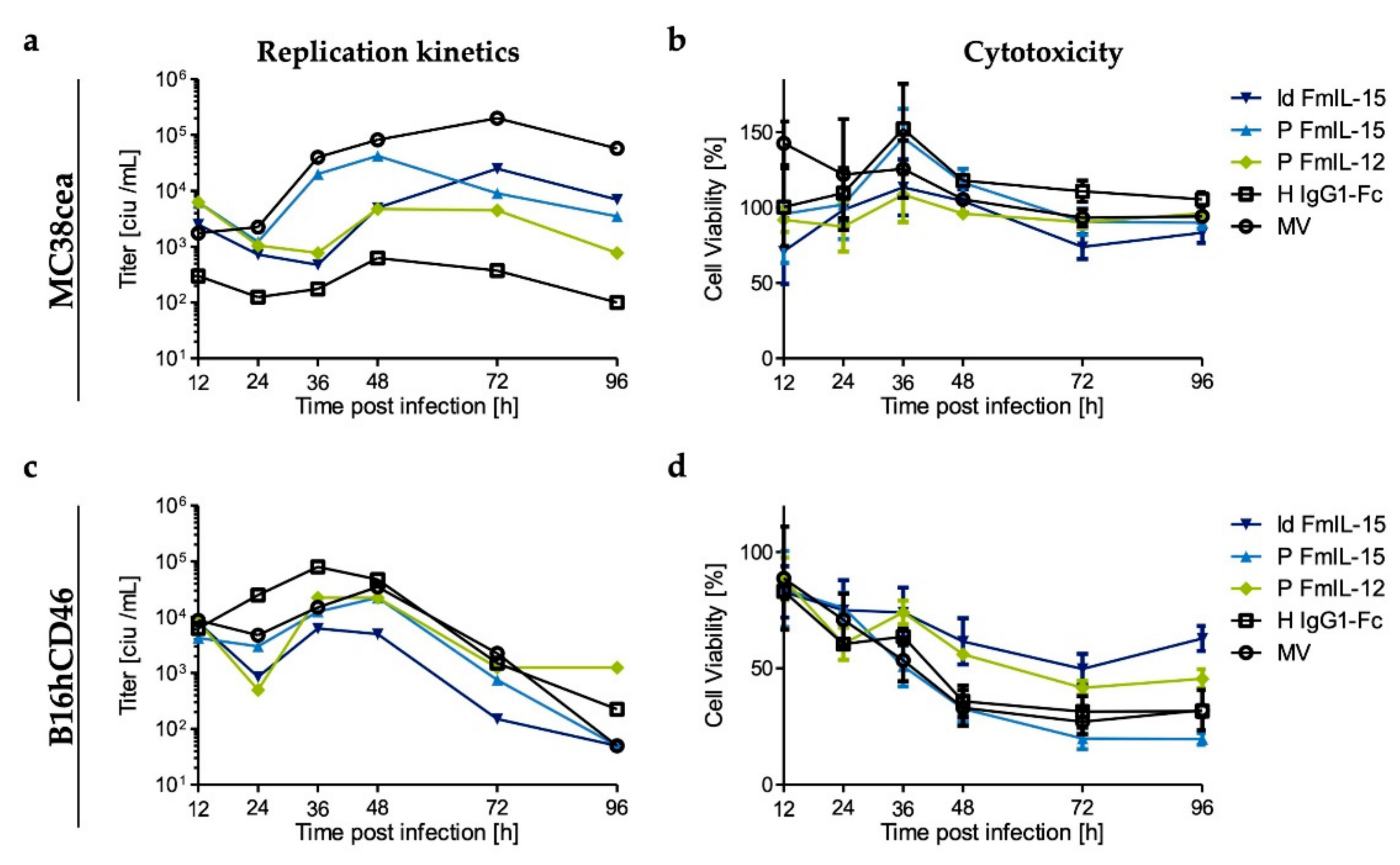
3.3. Replication and Cytotoxicity of Recombinant Measles Viruses in Murine Tumor Cell Lines
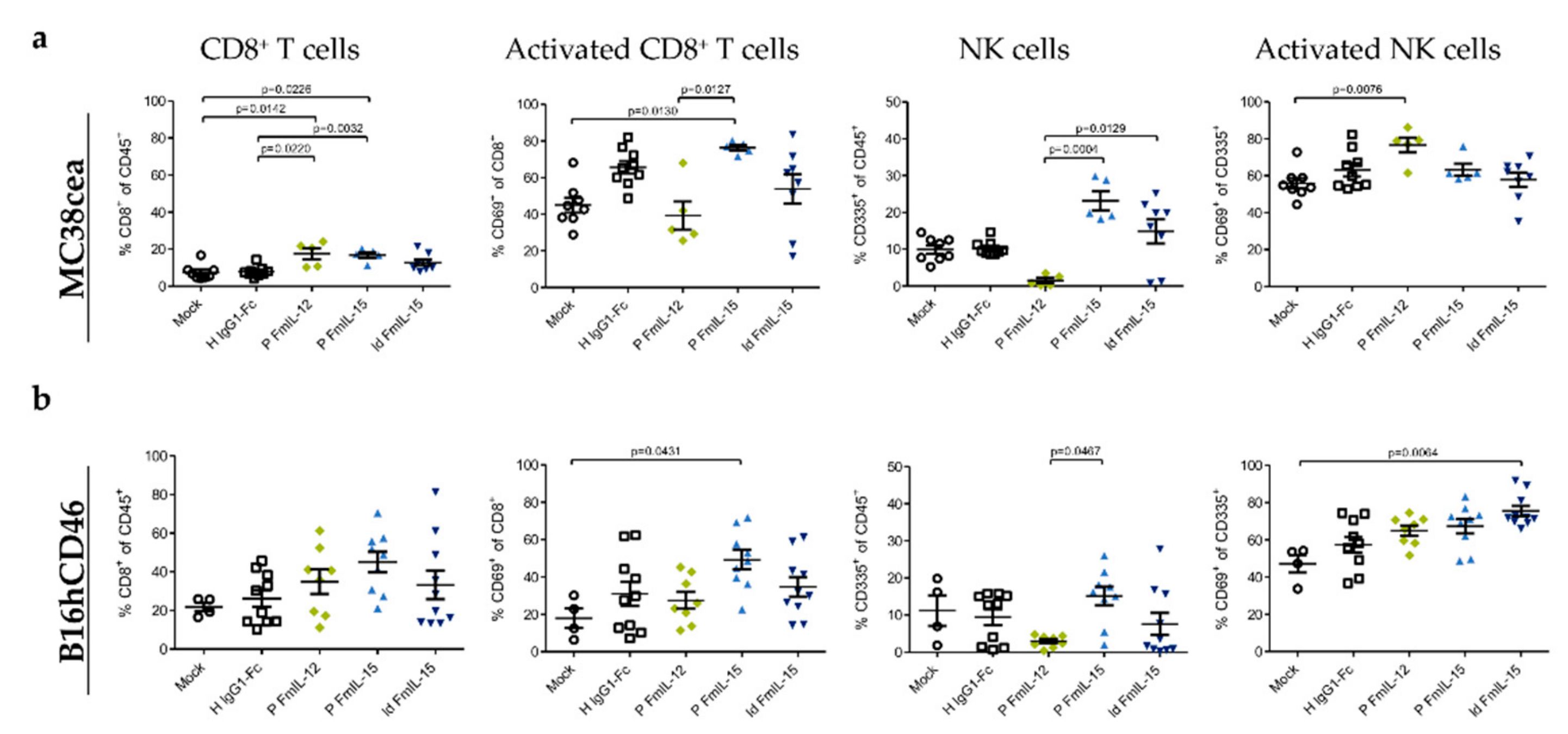
3.4. Immunovirotherapy with MeVac Encoding FmIL-15 Increases CD8+ T Cell and NK Cell Infiltration
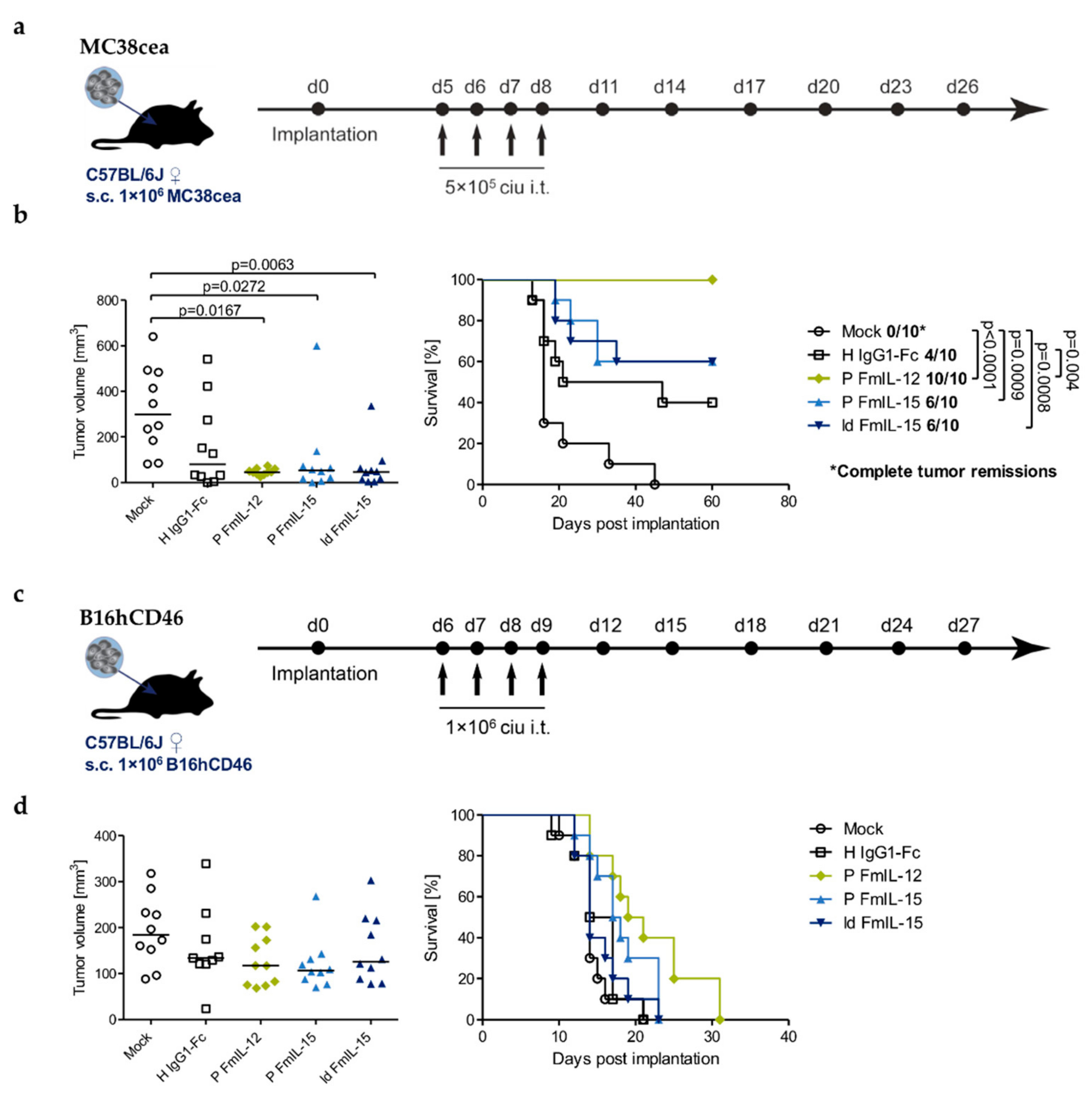
3.5. Therapeutic Efficacy in Syngeneic Mouse Models
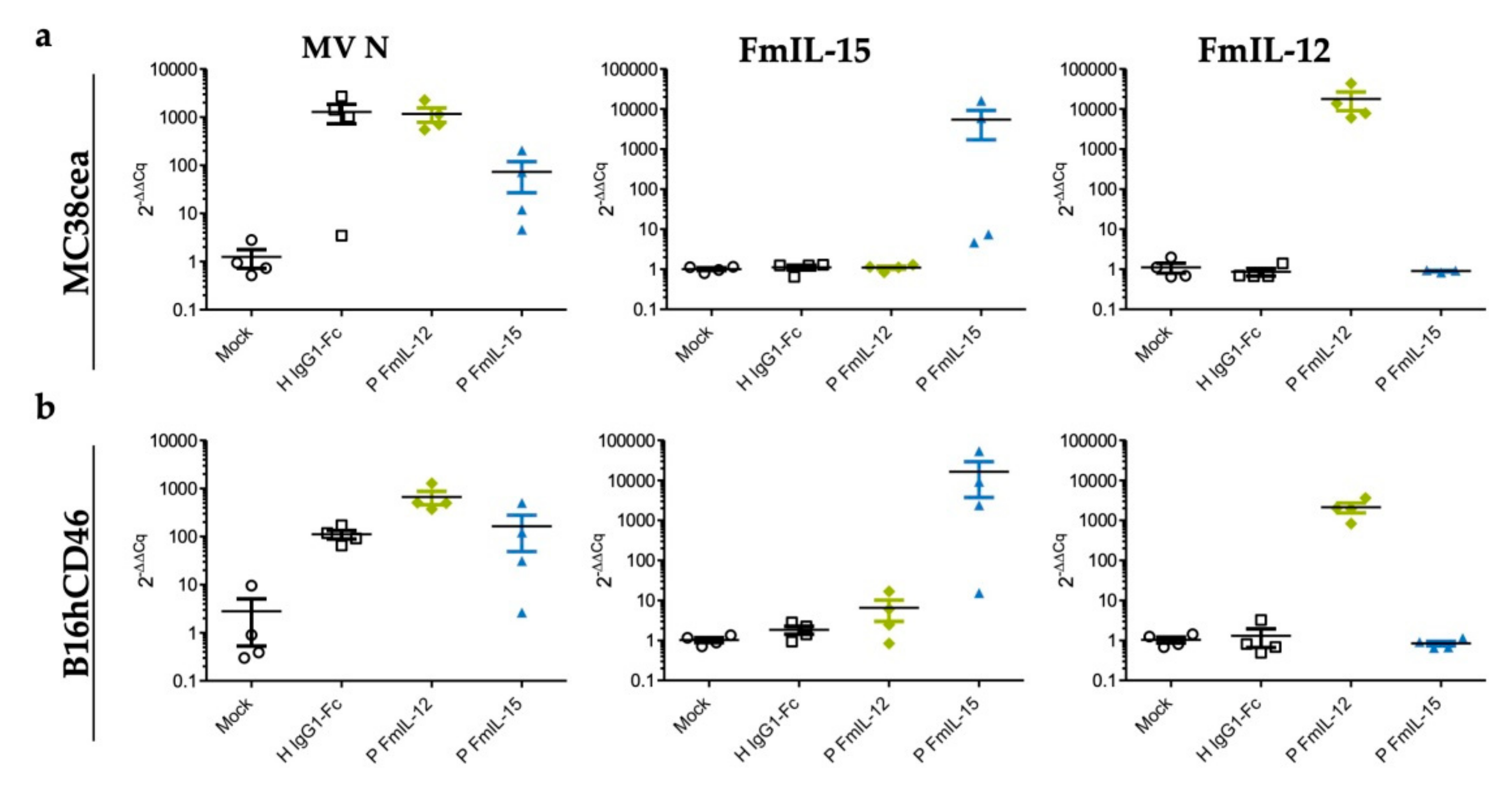
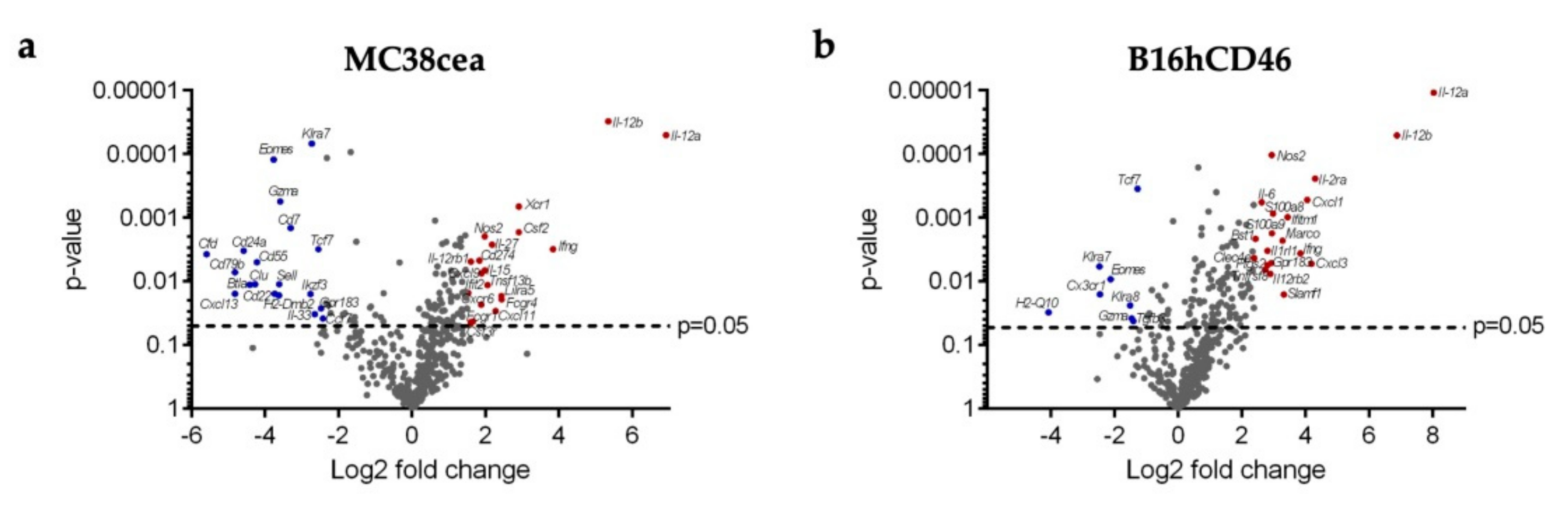
3.6. Differential Expression of Viral and Immune-Related Genes In Vivo
3.7. Oncolytic Measles Vaccines Encoding FhIL-12 for Treatment of Human Colorectal Cancer
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef]
- Robinson, S.; Galanis, E. Potential and clinical translation of oncolytic measles viruses. Expert Opin. Biol. Ther. 2017, 17, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aref, S.; Bailey, K.; Fielding, A. Measles to the rescue: A review of oncolytic measles virus. Viruses 2016, 8, 294. [Google Scholar] [CrossRef]
- Anderson, B.D.; Nakamura, T.; Russell, S.J.; Peng, K.W. High cd46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004, 64, 4919–4926. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Russell, S.J. Measles virus as an oncolytic vector platform. Curr. Gene Ther. 2008, 8, 162–175. [Google Scholar] [CrossRef]
- Leber, M.F.; Baertsch, M.A.; Anker, S.C.; Henkel, L.; Singh, H.M.; Bossow, S.; Engeland, C.E.; Barkley, R.; Hoyler, B.; Albert, J.; et al. Enhanced control of oncolytic measles virus using microrna target sites. Mol. Ther. Oncolyt. 2018, 9, 30–40. [Google Scholar] [CrossRef]
- Dingli, D.; Peng, K.W.; Harvey, M.E.; Greipp, P.R.; O’Connor, M.K.; Cattaneo, R.; Morris, J.C.; Russell, S.J. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood 2004, 103, 1641–1646. [Google Scholar] [CrossRef] [Green Version]
- Billeter, M.A.; Naim, H.Y.; Udem, S.A. Reverse genetics of measles virus and resulting multivalent recombinant vaccines: Applications of recombinant measles viruses. Curr. Top. Microbiol. Immunol. 2009, 329, 129–162. [Google Scholar]
- Dietz, L.; Engeland, C.E. Immunomodulation in oncolytic measles virotherapy. Methods Mol. Biol. 2020, 2058, 111–126. [Google Scholar] [PubMed]
- Grote, D.; Cattaneo, R.; Fielding, A.K. Neutrophils contribute to the measles virus-induced antitumor effect: Enhancement by granulocyte macrophage colony-stimulating factor expression. Cancer Res. 2003, 63, 6463–6468. [Google Scholar] [PubMed]
- Grossardt, C.; Engeland, C.E.; Bossow, S.; Halama, N.; Zaoui, K.; Leber, M.F.; Springfeld, C.; Jaeger, D.; von Kalle, C.; Ungerechts, G. Granulocyte-macrophage colony-stimulating factor-armed oncolytic measles virus is an effective therapeutic cancer vaccine. Hum. Gene Ther. 2013, 24, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Peng, K.W.; Dingli, D.; Kratzke, R.A.; Russell, S.J. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010, 17, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Iankov, I.D.; Allen, C.; Federspiel, M.J.; Myers, R.M.; Peng, K.W.; Ingle, J.N.; Russell, S.J.; Galanis, E. Expression of immunomodulatory neutrophil-activating protein of helicobacter pylori enhances the antitumor activity of oncolytic measles virus. Mol. Ther. 2012, 20, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. Ctla-4 and pd-l1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef]
- Speck, T.; Heidbuechel, J.P.W.; Veinalde, R.; Jaeger, D.; von Kalle, C.; Ball, C.R.; Ungerechts, G.; Engeland, C.E. Targeted bite expression by an oncolytic vector augments therapeutic efficacy against solid tumors. Clin. Cancer Res. 2018, 24, 2128–2137. [Google Scholar] [CrossRef] [PubMed]
- Veinalde, R.; Grossardt, C.; Hartmann, L.; Bourgeois-Daigneault, M.C.; Bell, J.C.; Jager, D.; von Kalle, C.; Ungerechts, G.; Engeland, C.E. Oncolytic measles virus encoding interleukin-12 mediates potent antitumor effects through t cell activation. Oncoimmunology 2017, 6, e1285992. [Google Scholar] [CrossRef]
- Seaman, W.E.; Sleisenger, M.; Eriksson, E.; Koo, G.C. Depletion of natural killer cells in mice by monoclonal antibody to nk-1.1. Reduction in host defense against malignancy without loss of cellular or humoral immunity. J. Immunol. 1987, 138, 4539–4544. [Google Scholar]
- Carson, W.E.; Giri, J.G.; Lindemann, M.J.; Linett, M.L.; Ahdieh, M.; Paxton, R.; Anderson, D.; Eisenmann, J.; Grabstein, K.; Caligiuri, M.A. Interleukin (il) 15 is a novel cytokine that activates human natural killer cells via components of the il-2 receptor. J. Exp. Med. 1994, 180, 1395–1403. [Google Scholar] [CrossRef]
- Marks-Konczalik, J.; Dubois, S.; Losi, J.M.; Sabzevari, H.; Yamada, N.; Feigenbaum, L.; Waldmann, T.A.; Tagaya, Y. Il-2-induced activation-induced cell death is inhibited in il-15 transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 11445–11450. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.S.; Kallas, E.G.; Thomas, E.K.; Looney, J.; Campbell, M.; Evans, T.G. Effects of interleukin-15 on in vitro human t cell proliferation and activation. J. Interferon Cytokine Res. 2000, 20, 119–123. [Google Scholar] [CrossRef]
- Zhang, X.; Sun, S.; Hwang, I.; Tough, D.F.; Sprent, J. Potent and selective stimulation of memory-phenotype cd8+ t cells in vivo by il-15. Immunity 1998, 8, 591–599. [Google Scholar] [CrossRef]
- Perna, S.K.; de Angelis, B.; Pagliara, D.; Hasan, S.T.; Zhang, L.; Mahendravada, A.; Heslop, H.E.; Brenner, M.K.; Rooney, C.M.; Dotti, G.; et al. Interleukin 15 provides relief to ctls from regulatory t cell-mediated inhibition: Implications for adoptive t cell-based therapies for lymphoma. Clin. Cancer Res. 2013, 19, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Burkett, P.R.; Koka, R.; Chien, M.; Chai, S.; Boone, D.L.; Ma, A. Coordinate expression and trans presentation of interleukin (il)-15ralpha and il-15 supports natural killer cell and memory cd8+ t cell homeostasis. J. Exp. Med. 2004, 200, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Steel, J.C.; Waldmann, T.A.; Morris, J.C. Interleukin-15 biology and its therapeutic implications in cancer. Trends Pharmacol. Sci. 2012, 33, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, G.C.; Radvanyi, L. The il-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014, 25, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Dubois, S.; Mariner, J.; Waldmann, T.A.; Tagaya, Y. Il-15ralpha recycles and presents il-15 in trans to neighboring cells. Immunity 2002, 17, 537–547. [Google Scholar] [CrossRef]
- Dubois, S.; Patel, H.J.; Zhang, M.; Waldmann, T.A.; Muller, J.R. Preassociation of il-15 with il-15r alpha-igg1-fc enhances its activity on proliferation of nk and cd8+/cd44high t cells and its antitumor action. J. Immunol. 2008, 180, 2099–2106. [Google Scholar] [CrossRef]
- Rubinstein, M.P.; Kovar, M.; Purton, J.F.; Cho, J.H.; Boyman, O.; Surh, C.D.; Sprent, J. Converting il-15 to a superagonist by binding to soluble il-15r alpha. Proc. Natl. Acad. Sci. USA 2006, 103, 9166–9171. [Google Scholar] [CrossRef]
- Mortier, E.; Quemener, A.; Vusio, P.; Lorenzen, I.; Boublik, Y.; Grotzinger, J.; Plet, A.; Jacques, Y. Soluble interleukin-15 receptor alpha (il-15r alpha)-sushi as a selective and potent agonist of il-15 action through il-15r beta/gamma. Hyperagonist il-15 x il-15r alpha fusion proteins. J. Biol. Chem. 2006, 281, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Peng, K.W.; Harvey, M.; Greiner, S.; Lorimer, I.A.; James, C.D.; Russell, S.J. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 2005, 23, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Kantor, J.A.; Salgaller, M.; Hand, P.H.; Fernsten, P.D.; Schlom, J. Transduction and expression of the human carcinoembryonic antigen gene in a murine colon carcinoma cell line. Cancer Res. 1991, 51, 3657–3662. [Google Scholar] [PubMed]
- Radecke, F.; Spielhofer, P.; Schneider, H.; Kaelin, K.; Huber, M.; Dotsch, C.; Christiansen, G.; Billeter, M.A. Rescue of measles viruses from cloned DNA. EMBO J. 1995, 14, 5773–5784. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Staeheli, P.; Schneider, U. Rna polymerase ii-controlled expression of antigenomic rna enhances the rescue efficacies of two different members of the mononegavirales independently of the site of viral genome replication. J. Virol. 2006, 80, 5708–5715. [Google Scholar] [CrossRef] [PubMed]
- Tosic, V.; Thomas, D.L.; Kranz, D.M.; Liu, J.; McFadden, G.; Shisler, J.L.; MacNeill, A.L.; Roy, E.J. Myxoma virus expressing a fusion protein of interleukin-15 (il15) and il15 receptor alpha has enhanced antitumor activity. PLoS ONE 2014, 9, e109801. [Google Scholar] [CrossRef] [PubMed]
- Calain, P.; Roux, L. The rule of six, a basic feature for efficient replication of sendai virus defective interfering rna. J. Virol. 1993, 67, 4822–4830. [Google Scholar]
- Heidbuechel, J.P.W.; Engeland, C.E. Paramyxoviruses for tumor-targeted immunomodulation: Design and evaluation ex vivo. J. Vis. Exp. 2019, 143, e58651. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, G.J.; Rao, P.K.; Gately, M.K.; Mulligan, R.C. Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat. Biotechnol. 1997, 15, 35–40. [Google Scholar] [CrossRef]
- Großardt, C. Engineering Targeted and Cytokine-Armed Oncolytic Measles Viruses. Ph.D. Thesis, Ruperto-Carola University of Heidelberg, Heidelberg, Germany, 2013. [Google Scholar]
- Vincent, S.; Tigaud, I.; Schneider, H.; Buchholz, C.J.; Yanagi, Y.; Gerlier, D. Restriction of measles virus rna synthesis by a mouse host cell line: Trans-complementation by polymerase components or a human cellular factor(s). J. Virol. 2002, 76, 6121–6130. [Google Scholar] [CrossRef]
- Cattaneo, R.; Rebmann, G.; Baczko, K.; Ter Meulen, V.; Billeter, M.A. Altered ratios of measles virus transcripts in diseased human brains. Virology 1987, 160, 523–526. [Google Scholar] [CrossRef]
- Robinson, T.O.; Schluns, K.S. The potential and promise of il-15 in immuno-oncogenic therapies. Immunol. Lett. 2017, 190, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Stoklasek, T.A.; Schluns, K.S.; Lefrancois, L. Combined il-15/il-15ralpha immunotherapy maximizes il-15 activity in vivo. J. Immunol. 2006, 177, 6072–6080. [Google Scholar] [CrossRef] [PubMed]
- Ugen, K.E.; Kutzler, M.A.; Marrero, B.; Westover, J.; Coppola, D.; Weiner, D.B.; Heller, R. Regression of subcutaneous b16 melanoma tumors after intratumoral delivery of an il-15-expressing plasmid followed by in vivo electroporation. Cancer Gene Ther. 2006, 13, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Kishida, T.; Asada, H.; Satoh, E.; Tanaka, S.; Shinya, M.; Hirai, H.; Iwai, M.; Tahara, H.; Imanishi, J.; Mazda, O. In vivo electroporation-mediated transfer of interleukin-12 and interleukin-18 genes induces significant antitumor effects against melanoma in mice. Gene Ther. 2001, 8, 1234–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Yao, Z.; Dubois, S.; Ju, W.; Muller, J.R.; Waldmann, T.A. Interleukin-15 combined with an anti-cd40 antibody provides enhanced therapeutic efficacy for murine models of colon cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 7513–7518. [Google Scholar] [CrossRef] [PubMed]
- Lasek, W.; Basak, G.; Switaj, T.; Jakubowska, A.B.; Wysocki, P.J.; Mackiewicz, A.; Drela, N.; Jalili, A.; Kaminski, R.; Kozar, K.; et al. Complete tumour regressions induced by vaccination with il-12 gene-transduced tumour cells in combination with il-15 in a melanoma model in mice. Cancer Immunol. Immunother. 2004, 53, 363–372. [Google Scholar]
- Di Carlo, E.; Comes, A.; Basso, S.; de Ambrosis, A.; Meazza, R.; Musiani, P.; Moelling, K.; Albini, A.; Ferrini, S. The combined action of il-15 and il-12 gene transfer can induce tumor cell rejection without t and nk cell involvement. J. Immunol. 2000, 165, 3111–3118. [Google Scholar] [CrossRef]
- Kimura, K.; Nishimura, H.; Matsuzaki, T.; Yokokura, T.; Nimura, Y.; Yoshikai, Y. Synergistic effect of interleukin-15 and interleukin-12 on antitumor activity in a murine malignant pleurisy model. Cancer Immunol. Immunother. 2000, 49, 71–77. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Q.; Yu, X.; Zhao, L. Rescue of nonlytic newcastle disease virus (ndv) expressing il-15 for cancer immunotherapy. Virus Res. 2017, 233, 35–41. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Q.; Mei, Y.; Liu, Y.; Zhao, L. Newcastle disease virus co-expressing interleukin 7 and interleukin 15 modified tumor cells as a vaccine for cancer immunotherapy. Cancer Sci. 2018, 109, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, K.B.; Barra, N.G.; Davies, E.; Ashkar, A.A.; Lichty, B.D. Expressing human interleukin-15 from oncolytic vesicular stomatitis virus improves survival in a murine metastatic colon adenocarcinoma model through the enhancement of anti-tumor immunity. Cancer Gene Ther. 2012, 19, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Kowalsky, S.J.; Liu, Z.; Feist, M.; Berkey, S.E.; Ma, C.; Ravindranathan, R.; Dai, E.; Roy, E.J.; Guo, Z.S.; Bartlett, D.L. Superagonist il-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with pd-1 blockade. Mol. Ther. 2018, 26, 2476–2486. [Google Scholar] [CrossRef] [PubMed]
- Bankamp, B.; Takeda, M.; Zhang, Y.; Xu, W.; Rota, P.A. Genetic characterization of measles vaccine strains. J. Infect. Dis. 2011, 204 (Suppl. 1), S533–S548. [Google Scholar] [CrossRef] [PubMed]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L.; et al. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Boisgerault, N.; Guillerme, J.B.; Pouliquen, D.; Mesel-Lemoine, M.; Achard, C.; Combredet, C.; Fonteneau, J.F.; Tangy, F.; Gregoire, M. Natural oncolytic activity of live-attenuated measles virus against human lung and colorectal adenocarcinomas. BioMed Res. Int. 2013, 2013, 387362. [Google Scholar] [CrossRef] [PubMed]
- Amagai, Y.; Fujiyuki, T.; Yoneda, M.; Shoji, K.; Furukawa, Y.; Sato, H.; Kai, C. Oncolytic activity of a recombinant measles virus, blind to signaling lymphocyte activation molecule, against colorectal cancer cells. Sci. Rep. 2016, 6, 24572. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Backhaus, P.S.; Veinalde, R.; Hartmann, L.; Dunder, J.E.; Jeworowski, L.M.; Albert, J.; Hoyler, B.; Poth, T.; Jäger, D.; Ungerechts, G.; et al. Immunological Effects and Viral Gene Expression Determine the Efficacy of Oncolytic Measles Vaccines Encoding IL-12 or IL-15 Agonists. Viruses 2019, 11, 914. https://0-doi-org.brum.beds.ac.uk/10.3390/v11100914
Backhaus PS, Veinalde R, Hartmann L, Dunder JE, Jeworowski LM, Albert J, Hoyler B, Poth T, Jäger D, Ungerechts G, et al. Immunological Effects and Viral Gene Expression Determine the Efficacy of Oncolytic Measles Vaccines Encoding IL-12 or IL-15 Agonists. Viruses. 2019; 11(10):914. https://0-doi-org.brum.beds.ac.uk/10.3390/v11100914
Chicago/Turabian StyleBackhaus, Paul S., Rūta Veinalde, Laura Hartmann, Jessica E. Dunder, Lara M. Jeworowski, Jessica Albert, Birgit Hoyler, Tanja Poth, Dirk Jäger, Guy Ungerechts, and et al. 2019. "Immunological Effects and Viral Gene Expression Determine the Efficacy of Oncolytic Measles Vaccines Encoding IL-12 or IL-15 Agonists" Viruses 11, no. 10: 914. https://0-doi-org.brum.beds.ac.uk/10.3390/v11100914