Decanoyl-Arg-Val-Lys-Arg-Chloromethylketone: An Antiviral Compound That Acts against Flaviviruses through the Inhibition of Furin-Mediated prM Cleavage

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Virus
2.2. Reagents
2.3. Cell Viability Assay and Efficacy Study of dec-RVKR-cmk
2.4. Immunofluorescence Assay (IFA)
2.5. Plaque Assay
2.6. Time-of-Drug Addition Assay
2.7. Western Blot Analysis
2.8. RNA Extraction and Quantitative Real-Time PCR
2.9. Statistical Analysis
3. Results
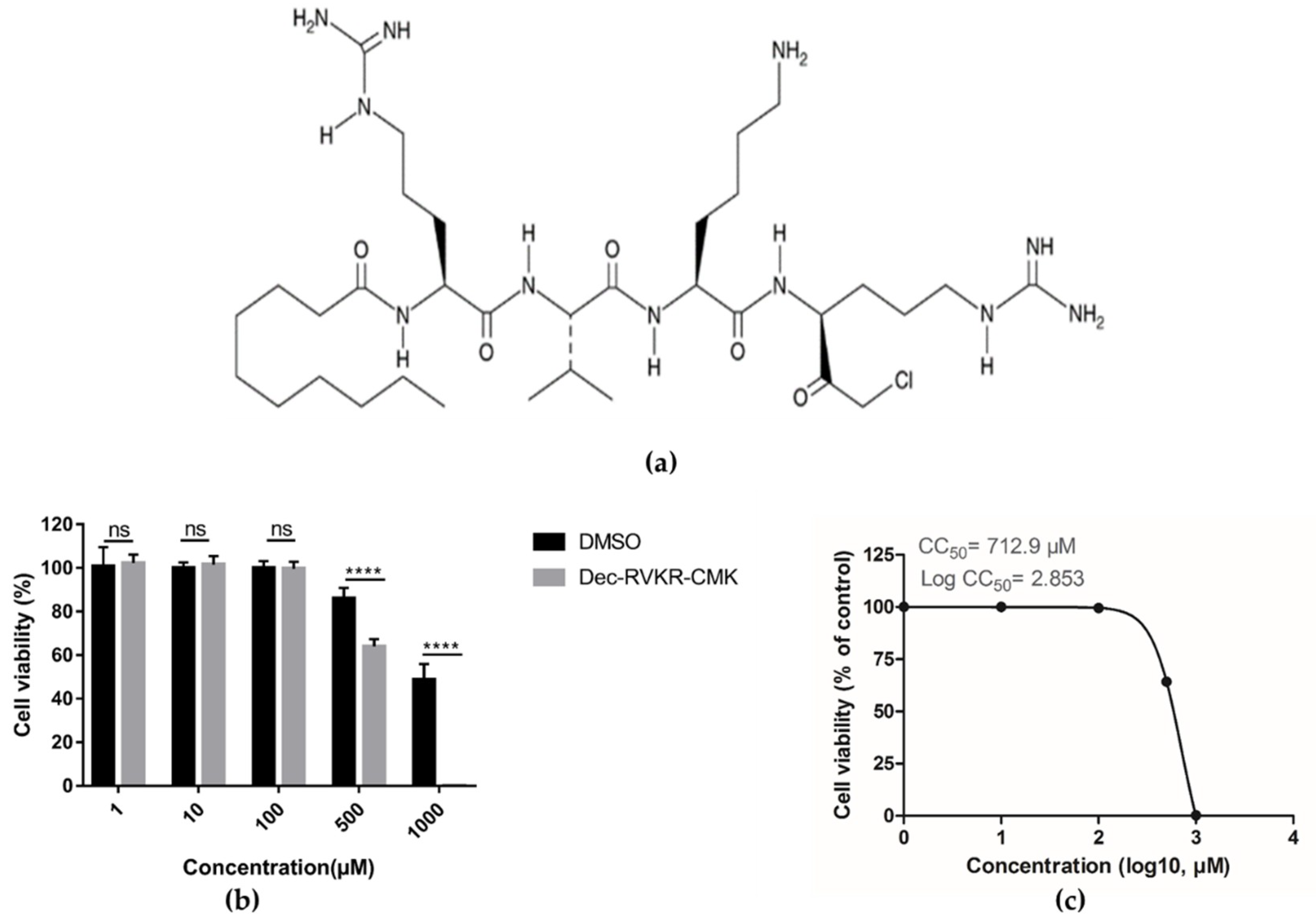
3.1. Cytotoxicity of dec-RVKR-cmk in Vero Cells
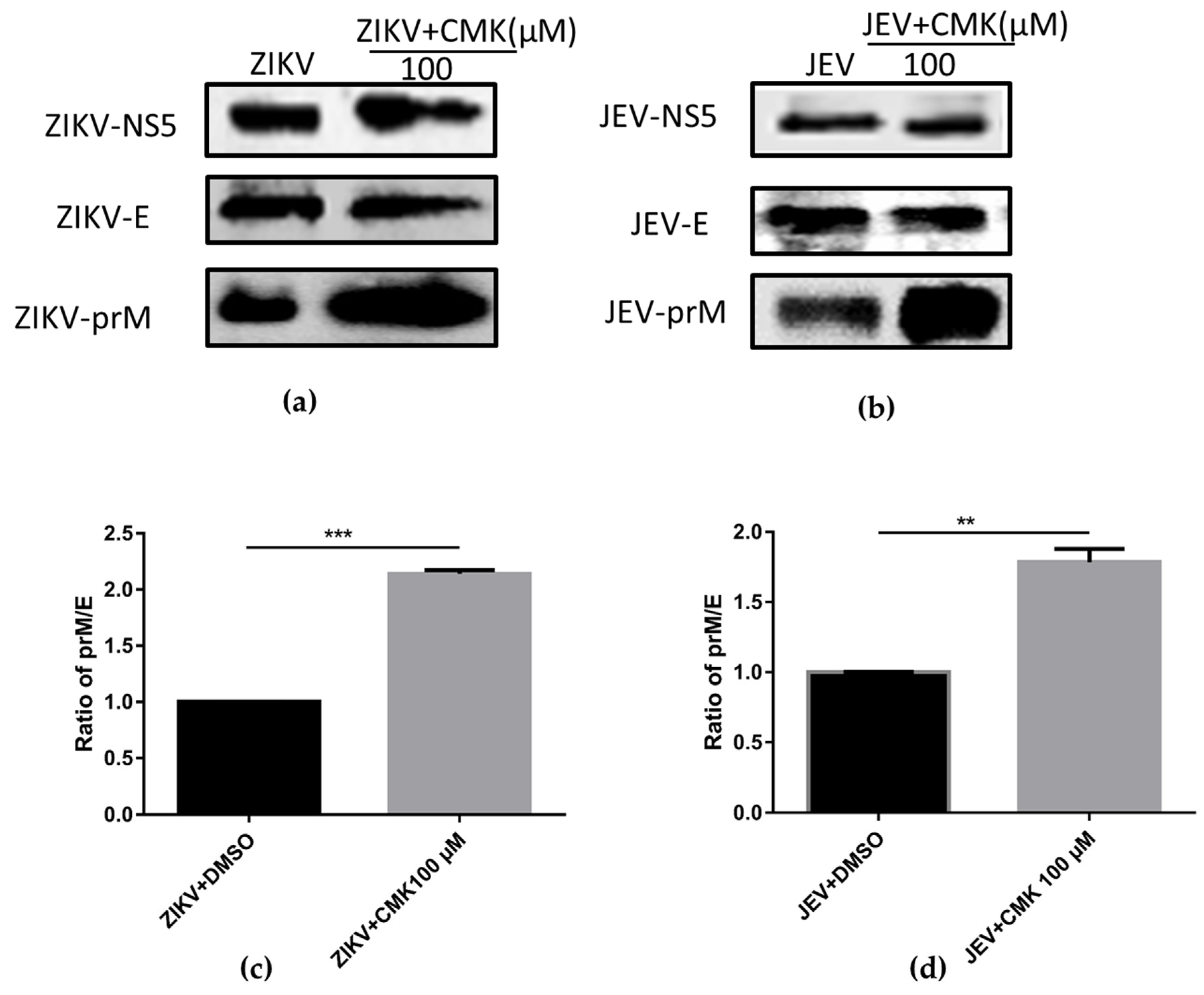
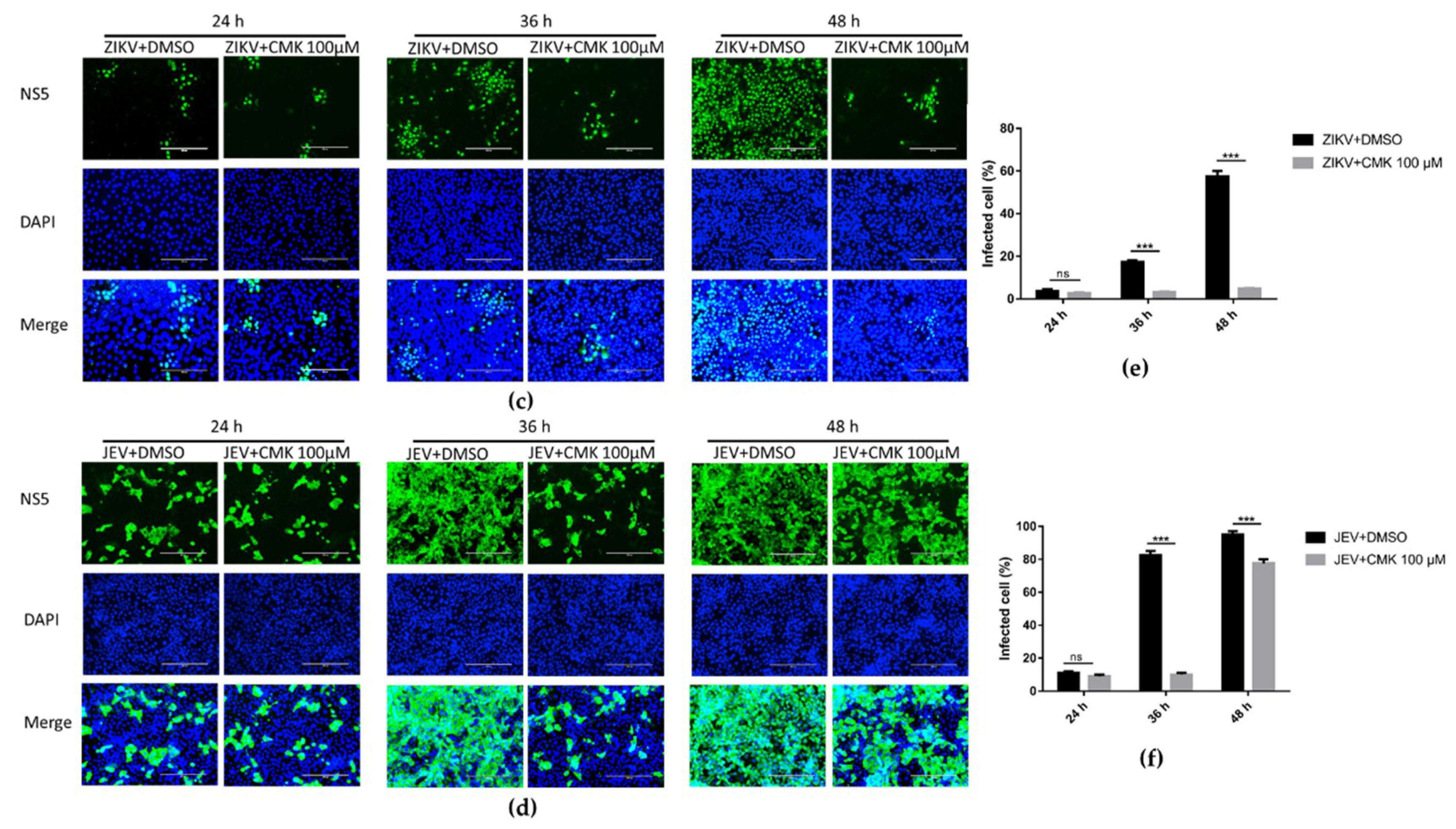
3.2. Dec-RVKR-cmk Inhibits prM Cleavage during ZIKV and JEV Infection
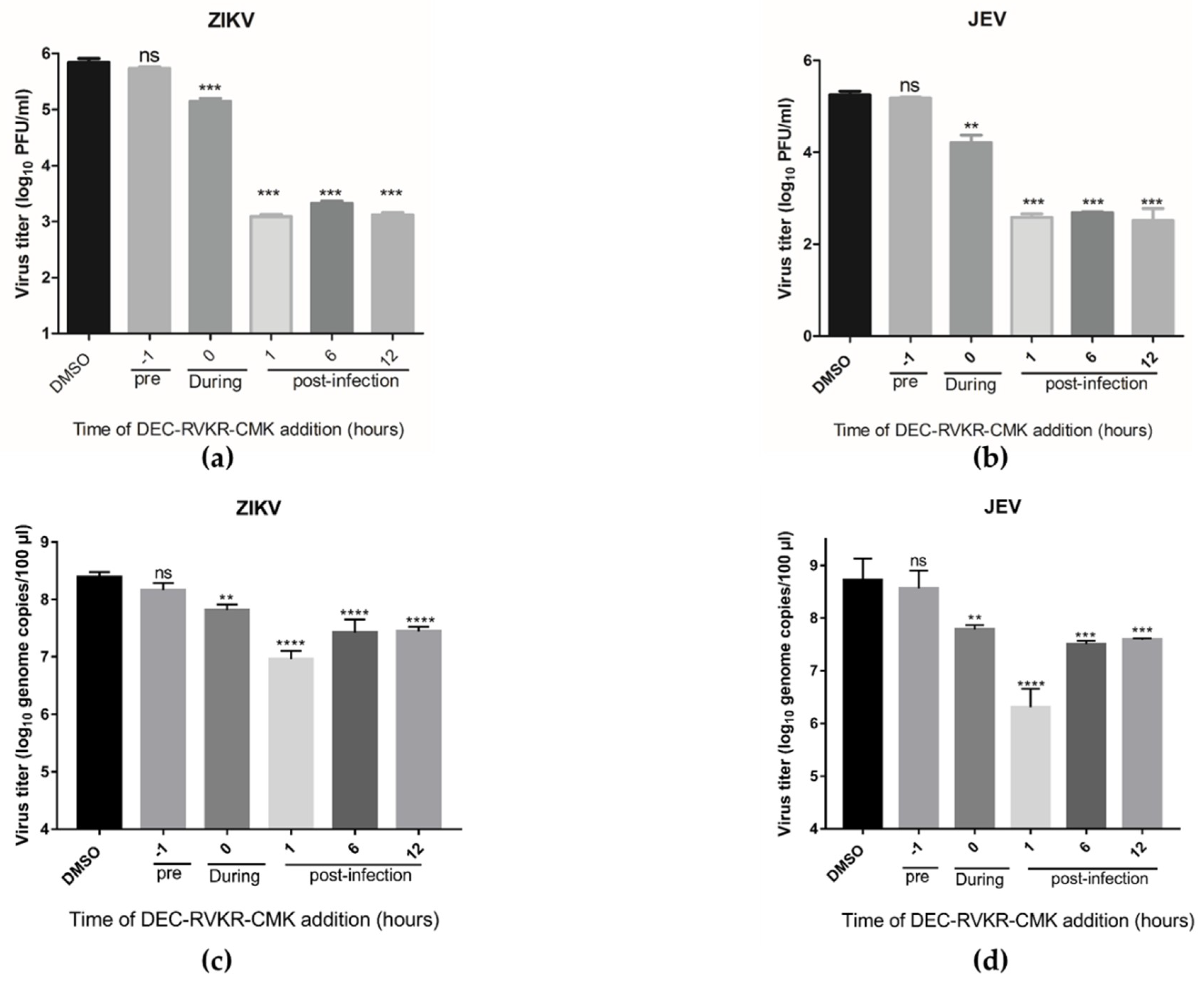
3.3. Dec-RVKR-cmk Inhibits ZIKV and JEV Infection at Different Times of Drug Administration
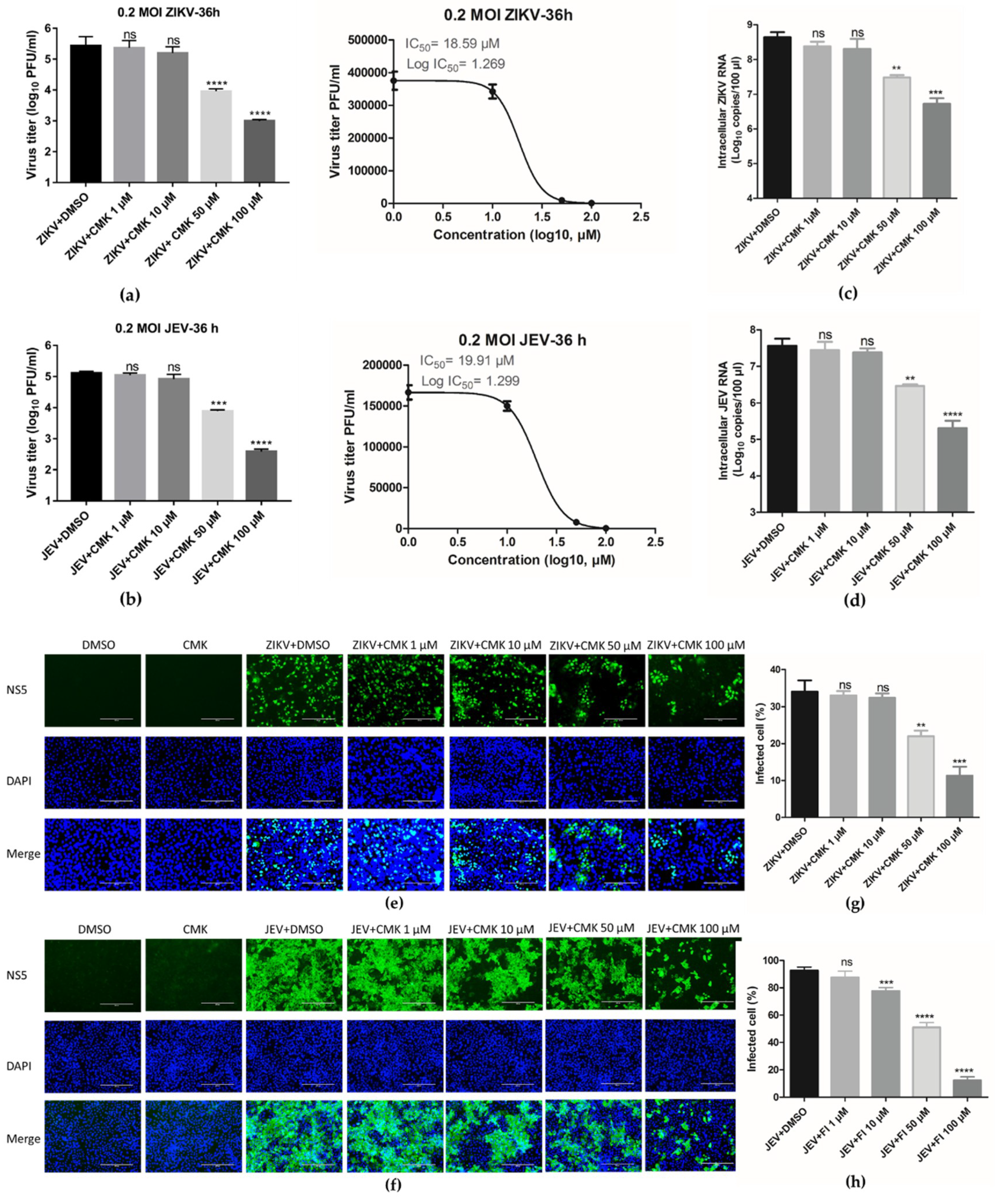
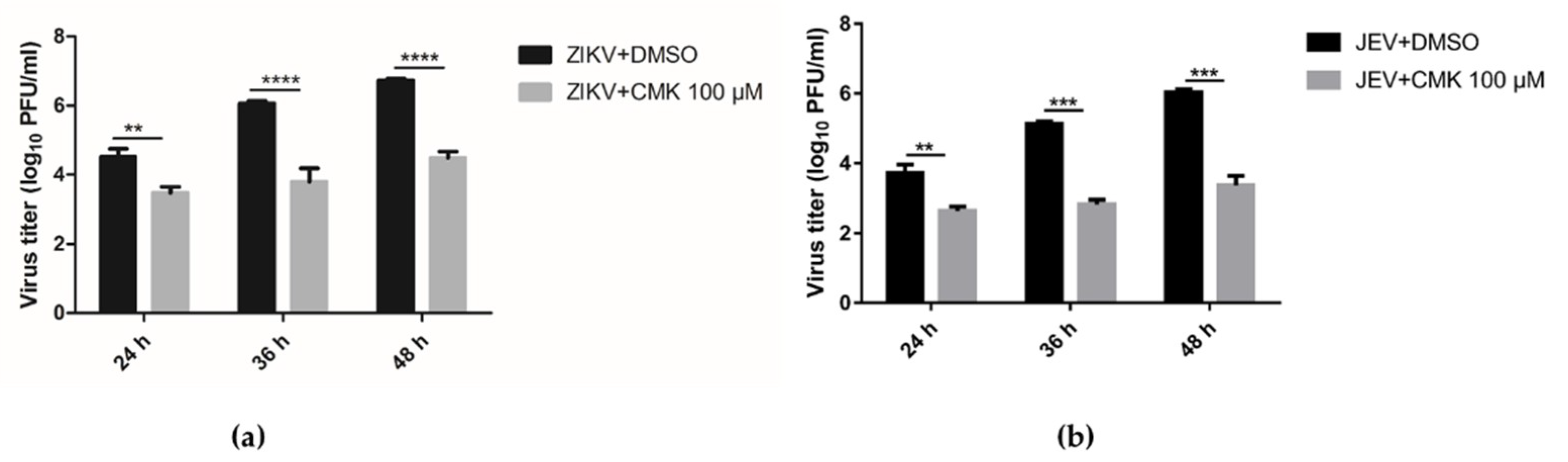
3.4. Dec-RVKR-cmk Suppresses ZIKV and JEV Propagation in a Dose-Dependent Manner
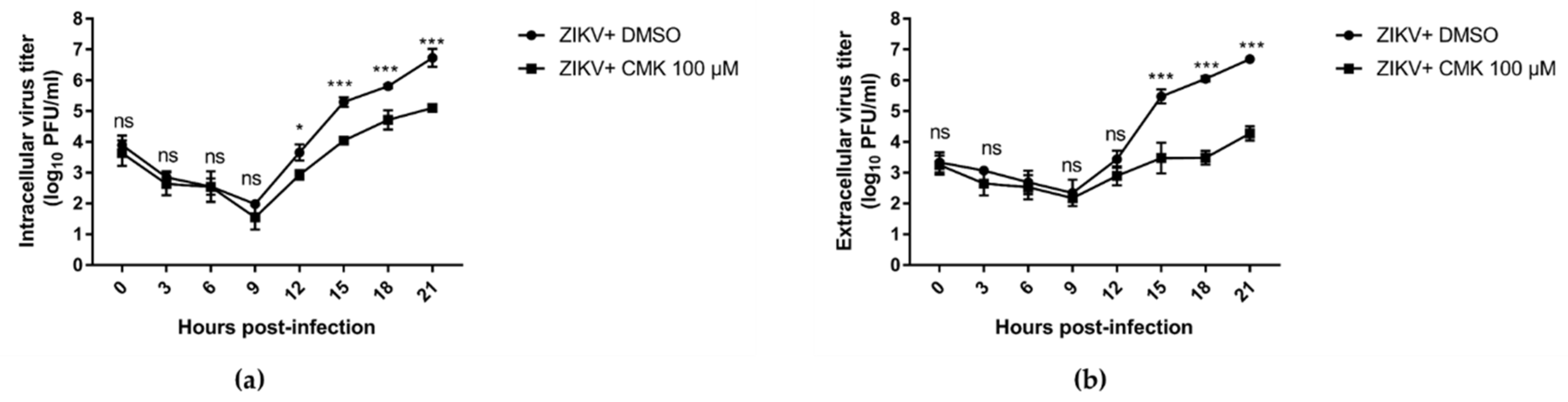
3.5. Dec-RVKR-cmk Inhibits ZIKV and JEV Propagation at Various Time Points
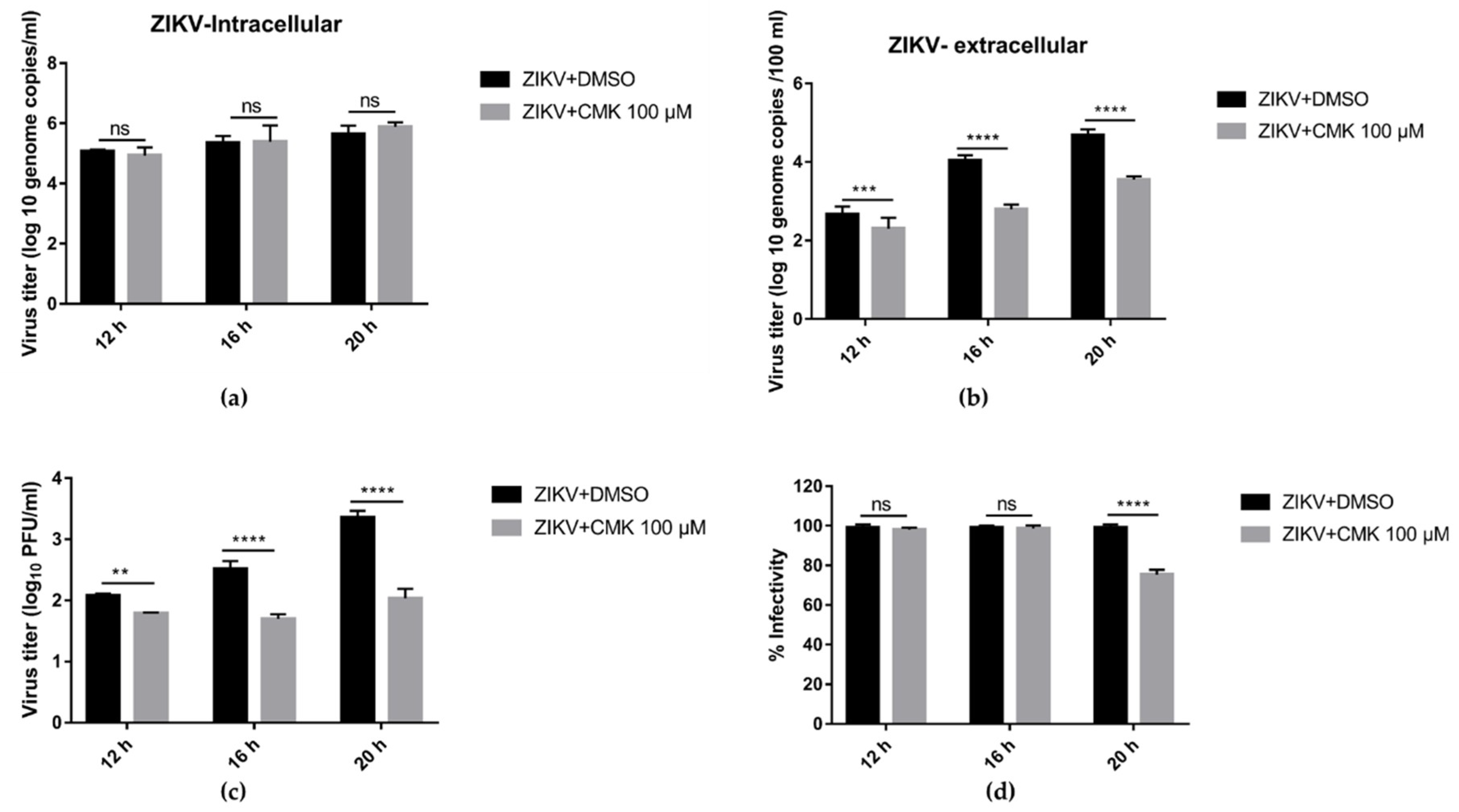
3.6. Dec-RVKR-cmk Inhibits ZIKV Release and Next Round Infectivity
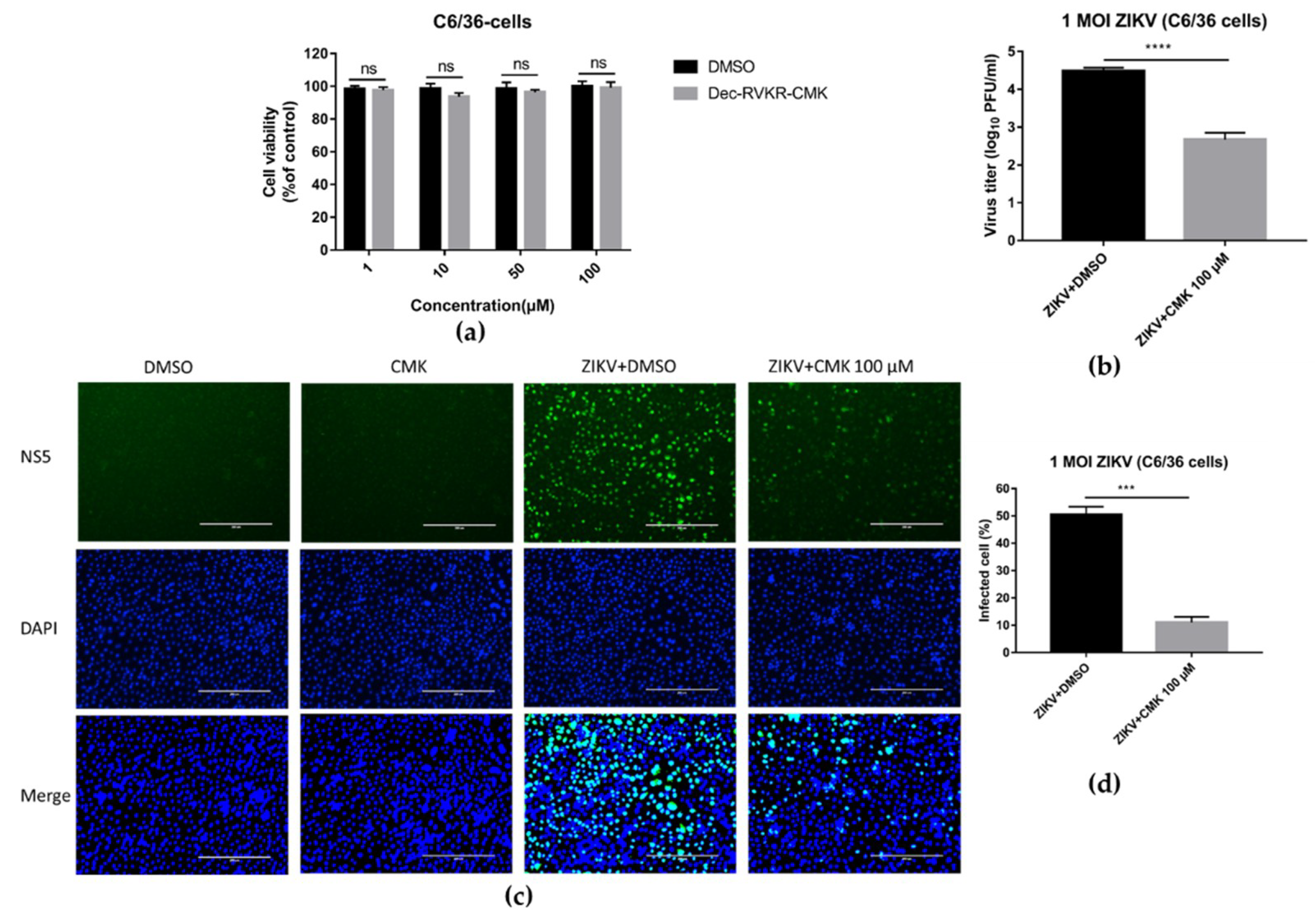
3.7. Dec-RVKR-cmk Inhibits ZIKV and JEV Infection in Mosquito Cells
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Panayiotou, C.; Lindqvist, R.; Kurhade, C.; Vonderstein, K.; Pasto, J.; Edlund, K.; Upadhyay, A.S.; Overby, A.K. Viperin Restricts Zika Virus and Tick-Borne Encephalitis Virus Replication by Targeting NS3 for Proteasomal Degradation. J. Virol. 2018, 92, e02054-17. [Google Scholar] [CrossRef] [PubMed]
- Gould, E.; Solomon, T. Pathogenic flaviviruses. Lancet 2008, 371, 500–509. [Google Scholar] [CrossRef]
- Puerta-Guardo, H.; Glasner, D.R.; Espinosa, D.A.; Biering, S.B.; Patana, M.; Ratnasiri, K.; Wang, C.; Beatty, P.R.; Harris, E. Flavivirus NS1 Triggers Tissue-Specific Vascular Endothelial Dysfunction Reflecting Disease Tropism. Cell Rep. 2019, 26, 1598–1613. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Li, B.; Linhardt, R. Pathogenesis and inhibition of flaviviruses from a carbohydrate perspective. Pharmaceuticals 2017, 10, 44. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K. Flaviviruses and flavivirus vaccines. Vaccine 2012, 30, 4301–4306. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Nambala, P.; Su, W.-C. Role of Zika Virus prM protein in viral pathogenicity and use in vaccine development. Front. Microbiol. 2018, 9, 1797. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, I.C.; Kartenbeck, J.; Mezzacasa, A.; Allison, S.L.; Heinz, F.X.; Helenius, A. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis virus. J. Virol. 2003, 77, 4370–4382. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Westaway, E.G. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 2001, 75, 10787–10799. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lok, S.-M.; Yu, I.-M.; Zhang, Y.; Kuhn, R.J.; Chen, J.; Rossmann, M.G. The flavivirus precursor membrane-envelope protein complex: Structure and maturation. Science 2008, 319, 1830–1834. [Google Scholar] [CrossRef]
- Yu, I.-M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Elshuber, S.; Mandl, C.W. Resuscitating mutations in a furin cleavage-deficient mutant of the flavivirus tick-borne encephalitis virus. J. Virol. 2005, 79, 11813–11823. [Google Scholar] [CrossRef] [PubMed]
- Yoshii, K.; Igarashi, M.; Ichii, O.; Yokozawa, K.; Ito, K.; Kariwa, H.; Takashima, I. A conserved region in the prM protein is a critical determinant in the assembly of flavivirus particles. J. Gen. Virol. 2012, 93, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Garten, W.; Hallenberger, S.; Ortmann, D.; Schafer, W.; Vey, M.; Angliker, H.; Shaw, E.; Klenk, H.D. Processing of viral glycoproteins by the subtilisin-like endoprotease furin and its inhibition by specific peptidylchloroalkylketones. Biochimie 1994, 76, 217–225. [Google Scholar] [CrossRef]
- Becker, G.L.; Sielaff, F.; Than, M.E.; Lindberg, I.; Routhier, S.; Day, R.; Lu, Y.; Garten, W.; Steinmetzer, T. Potent inhibitors of furin and furin-like proprotein convertases containing decarboxylated P1 arginine mimetics. J. Med. Chem. 2010, 53, 1067–1075. [Google Scholar] [CrossRef]
- Pang, Y.J.; Tan, X.J.; Li, D.M.; Zheng, Z.H.; Lei, R.X.; Peng, X.M. Therapeutic potential of furin inhibitors for the chronic infection of hepatitis B virus. Liver Int. 2013, 33, 1230–1238. [Google Scholar] [CrossRef]
- Hallenberger, S.; Bosch, V.; Angliker, H.; Shaw, E.; Klenk, H.-D.; Garten, W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gpl60. Nature 1992, 360, 358. [Google Scholar] [CrossRef]
- Ozden, S.; Lucas-Hourani, M.; Ceccaldi, P.-E.; Basak, A.; Valentine, M.; Benjannet, S.; Hamelin, J.; Jacob, Y.; Mamchaoui, K.; Mouly, V. Inhibition of Chikungunya virus infection in cultured human muscle cells by furin inhibitors impairment of the maturation of the E2 surface glycoprotein. J. Biol. Chem. 2008, 283, 21899–21908. [Google Scholar] [CrossRef]
- Tong, Y.; Tong, S.; Zhao, X.; Wang, J.; Jun, J.; Park, J.; Wands, J.; Li, J. Initiation of duck hepatitis B virus infection requires cleavage by a furin-like protease. J. Virol. 2010, 84, 4569–4578. [Google Scholar] [CrossRef]
- Day, P.M.; Schiller, J.T. The role of furin in papillomavirus infection. Future Microbiol. 2009, 4, 1255–1262. [Google Scholar] [CrossRef] [Green Version]
- Couture, F.; Kwiatkowska, A.; Dory, Y.L.; Day, R. Therapeutic uses of furin and its inhibitors: A patent review. Expert Opin. Ther. Pat. 2015, 25, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Creemers, J.W.; Khatib, A.M. Knock-out mouse models of proprotein convertases: Unique functions or redundancy? Front. Biosci. J. Virtual Libr. 2008, 13, 4960–4971. [Google Scholar] [CrossRef] [PubMed]
- Couture, F.; D’Anjou, F.; Day, R. On the cutting edge of proprotein convertase pharmacology: From molecular concepts to clinical applications. Biomol. Concepts 2011, 2, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Roebroek, A.J.; Taylor, N.A.; Louagie, E.; Pauli, I.; Smeijers, L.; Snellinx, A.; Lauwers, A.; Van de Ven, W.J.; Hartmann, D.; Creemers, J.W. Limited redundancy of the proprotein convertase furin in mouse liver. J. Biol. Chem. 2004, 279, 53442–53450. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ye, J.; Ashraf, U.; Li, Y.; Wei, S.; Wan, S.; Zohaib, A.; Song, Y.; Chen, H.; Cao, S. MicroRNA-33a-5p modulates Japanese Encephalitis Virus replication by targeting eukaryotic translation elongation factor 1A1. J. Virol. 2016, 90, 3722–3734. [Google Scholar] [CrossRef]
- Mukherjee, S.; Lin, T.Y.; Dowd, K.A.; Manhart, C.J.; Pierson, T.C. The infectivity of prM-containing partially mature West Nile virus does not require the activity of cellular furin-like proteases. J. Virol. 2011, 85, 12067–12072. [Google Scholar] [CrossRef]
- Kovanich, D.; Saisawang, C.; Sittipaisankul, P.; Ramphan, S.; Kalpongnukul, N.; Somparn, P.; Pisitkun, T.; Smith, D.R. Analysis of the Zika and Japanese encephalitis virus NS5 interactomes. J. Proteome Res. 2019. [Google Scholar] [CrossRef]
- Becker, G.L.; Lu, Y.; Hardes, K.; Strehlow, B.; Levesque, C.; Lindberg, I.; Sandvig, K.; Bakowsky, U.; Day, R.; Garten, W.; et al. Highly potent inhibitors of proprotein convertase furin as potential drugs for treatment of infectious diseases. J. Biol. Chem. 2012, 287, 21992–22003. [Google Scholar] [CrossRef]
- Peng, M.; Watanabe, S.; Chan, K.W.K.; He, Q.; Zhao, Y.; Zhang, Z.; Lai, X.; Luo, D.; Vasudevan, S.G.; Li, G. Luteolin restricts dengue virus replication through inhibition of the proprotein convertase furin. Antivir. Res. 2017, 143, 176–185. [Google Scholar] [CrossRef]
- Thomas, G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef] [Green Version]
- Decroly, E.; Vandenbranden, M.; Ruysschaert, J.M.; Cogniaux, J.; Jacob, G.S.; Howard, S.C.; Marshall, G.; Kompelli, A.; Basak, A.; Jean, F.; et al. The convertases furin and PC1 can both cleave the human immunodeficiency virus (HIV)-1 envelope glycoprotein gp160 into gp120 (HIV-1 SU) and gp41 (HIV-I TM). J. Biol. Chem. 1994, 269, 12240–12247. [Google Scholar] [PubMed]
- Bolt, G.; Pedersen, L.O.; Birkeslund, H.H. Cleavage of the respiratory syncytial virus fusion protein is required for its surface expression: Role of furin. Virus Res. 2000, 68, 25–33. [Google Scholar] [CrossRef]
- Molloy, S.S.; Anderson, E.D.; Jean, F.; Thomas, G. Bi-cycling the furin pathway: From TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 1999, 9, 28–35. [Google Scholar] [CrossRef]
- Presser, L.D.; Haskett, A.; Waris, G. Hepatitis C virus-induced furin and thrombospondin-1 activate TGF-beta1: Role of TGF-beta1 in HCV replication. Virology 2011, 412, 284–296. [Google Scholar] [CrossRef] [PubMed]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imran, M.; Saleemi, M.K.; Chen, Z.; Wang, X.; Zhou, D.; Li, Y.; Zhao, Z.; Zheng, B.; Li, Q.; Cao, S.; et al. Decanoyl-Arg-Val-Lys-Arg-Chloromethylketone: An Antiviral Compound That Acts against Flaviviruses through the Inhibition of Furin-Mediated prM Cleavage. Viruses 2019, 11, 1011. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111011
Imran M, Saleemi MK, Chen Z, Wang X, Zhou D, Li Y, Zhao Z, Zheng B, Li Q, Cao S, et al. Decanoyl-Arg-Val-Lys-Arg-Chloromethylketone: An Antiviral Compound That Acts against Flaviviruses through the Inhibition of Furin-Mediated prM Cleavage. Viruses. 2019; 11(11):1011. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111011
Chicago/Turabian StyleImran, Muhammad, Muhammad Kashif Saleemi, Zheng Chen, Xugang Wang, Dengyuan Zhou, Yunchuan Li, Zikai Zhao, Bohan Zheng, Qiuyan Li, Shengbo Cao, and et al. 2019. "Decanoyl-Arg-Val-Lys-Arg-Chloromethylketone: An Antiviral Compound That Acts against Flaviviruses through the Inhibition of Furin-Mediated prM Cleavage" Viruses 11, no. 11: 1011. https://0-doi-org.brum.beds.ac.uk/10.3390/v11111011