The Segment Matters: Probable Reassortment of Tilapia Lake Virus (TiLV) Complicates Phylogenetic Analysis and Inference of Geographical Origin of New Isolate from Bangladesh

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Site Description and Sample Collection
2.2. RNA Extraction and Detection of TiLV by RT-PCR
2.3. Total RNA Sequencing and TiLV Genome Assembly
2.4. Phylogenetic Analyses
2.5. Quartet Tree Analysis to Detect Reassortment
2.6. Data Availability
3. Results and Discussion
3.1. Detection of TiLV Nucleic Acids in Bangladesh Samples
3.2. Bangladesh TiLV Genome and Similarity with Other TiLV Isolates
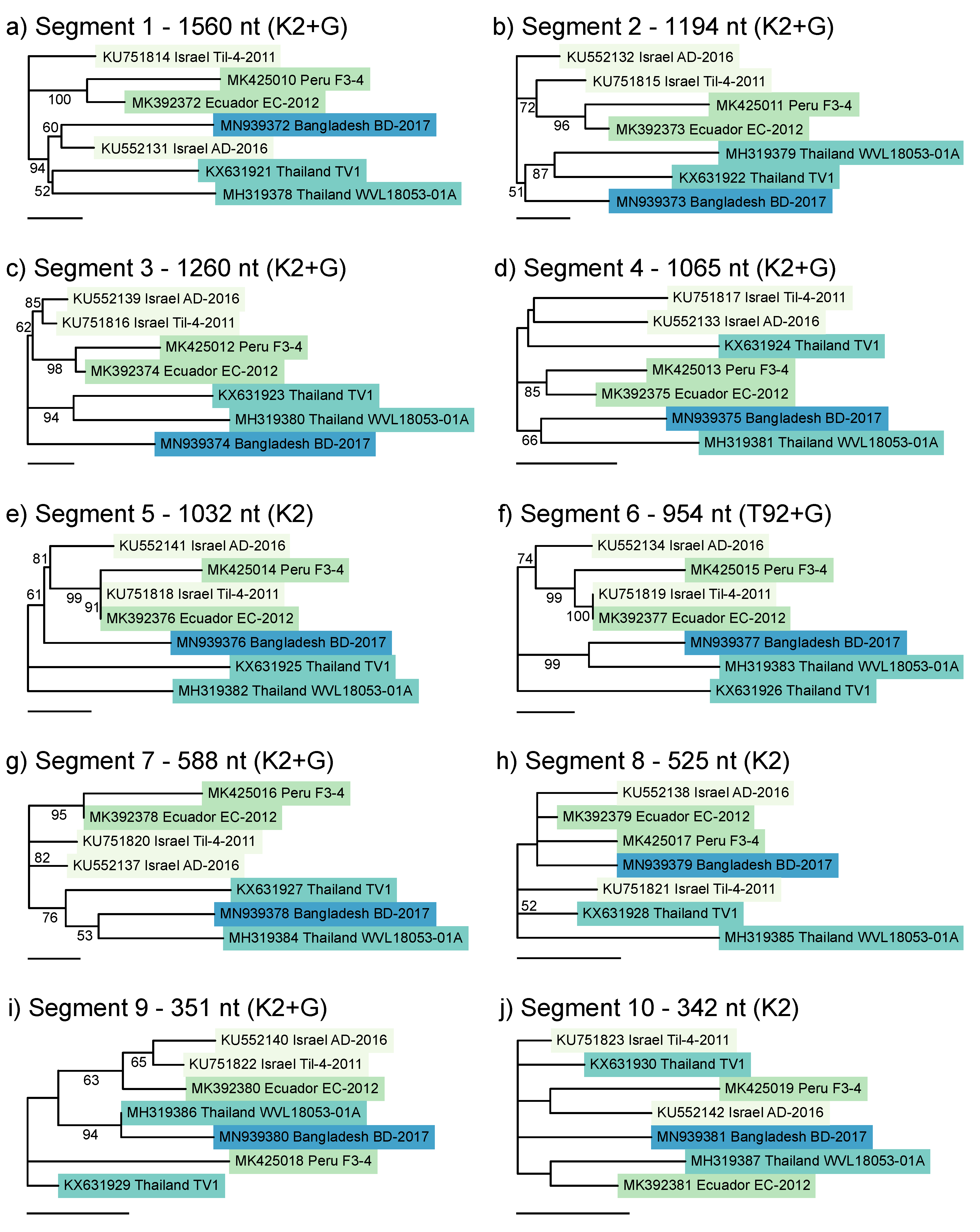
3.3. Possible Reassortment of TiLV Segments
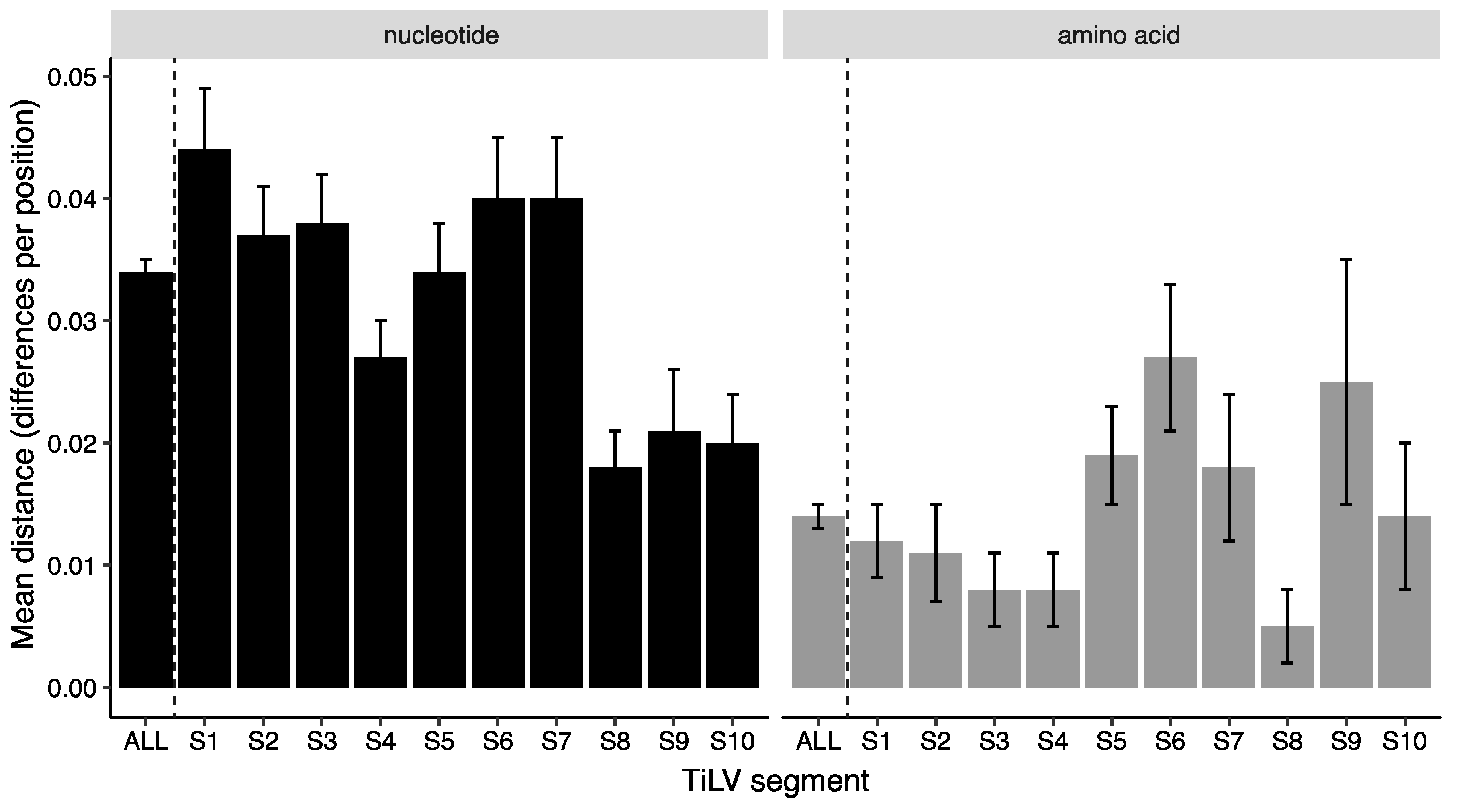
3.4. Amino Acid Substitutions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- FAO Outbreaks of Tilapia lake virus (TiLV) threaten the livelihoods and food security of millions of people dependent on tilapia farming. Glob. Inf. Early Warn. Syst. Food Agric. 2017, 338, 1–6.
- DoF Yearbook of fisheries statistics of Bangladesh, 2017-18. Fish. Resour. Surv. Syst. (FRSS), Dep. Fish. Minist. Fish. Livestock, Bangladesh 2018, 35, 121.
- Hernandez, R.; Belton, B.; Reardon, T.; Hu, C.; Zhang, X.; Ahmed, A. The “quiet revolution” in the aquaculture value chain in Bangladesh. Aquaculture 2018, 493, 456–468. [Google Scholar] [CrossRef]
- Tran, K.; Verdegem, M.; Wolkenfelt, B.; Mohan, C.; Verreth, J. The relation between farming practices and tilapia production in small-scale fish farms in Bangladesh. CGIAR Res. Progr. Fish Agri-Food Syst. Progr. Rep. 2019. FISH-2019-06. [Google Scholar]
- Uddin, M.T.; Goswami, A.; Rahman, M.S.; Dhar, A.R. How can governance improve efficiency and effectiveness of value chains? An analysis of pangas and tilapia stakeholders in Bangladesh. Aquaculture 2019, 510, 206–215. [Google Scholar] [CrossRef]
- Eyngor, M.; Zamostiano, R.; Tsofack, J.E.K.; Berkowitz, A.; Bercovier, H.; Tinman, S.; Lev, M.; Hurvitz, A.; Galeotti, M.; Bacharach, E.; et al. Identification of a novel RNA virus lethal to tilapia. J. Clin. Microbiol. 2014, 52, 4137–4146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacharach, E.; Mishra, N.; Briese, T.; Zody, M.C.; Kembou Tsofack, J.E.; Zamostiano, R.; Berkowitz, A.; Ng, J.; Nitido, A.; Corvelo, A.; et al. Characterization of a Novel Orthomyxo-like Virus Causing Mass Die-Offs of Tilapia. MBio 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, V.; Chakraborty, H.J.; Rout, A.K.; Balabantaray, S.; Behera, B.K.; Das, B.K. Structural characterization of open reading frame-encoded functional genes from tilapia lake virus (TiLV). Mol. Biotechnol. 2019, 61, 945–957. [Google Scholar] [CrossRef]
- ICTV International Committee on Taxonomy of Viruses. Available online: https://talk.ictvonline.org/ (accessed on 7 February 2020).
- Dong, H.T.; Ataguba, G.A.; Khunrae, P.; Rattanarojpong, T.; Senapin, S. Evidence of TiLV infection in tilapia hatcheries in Thailand from 2012 to 2017 reveals probable global spread of the disease. Aquaculture 2017, 479, 579–583. [Google Scholar] [CrossRef]
- Fathi, M.; Dickson, C.; Dickson, M.; Leschen, W.; Baily, J.; Muir, F.; Ulrich, K.; Weidmann, M. Identification of Tilapia Lake Virus in Egypt in Nile tilapia affected by “summer mortality” syndrome. Aquaculture 2017, 473, 430–432. [Google Scholar] [CrossRef]
- Dong, H.T.; Siriroob, S.; Meemetta, W.; Santimanawong, W.; Gangnonngiw, W.; Pirarat, N.; Khunrae, P.; Rattanarojpong, T.; Vanichviriyakit, R.; Senapin, S. Emergence of tilapia lake virus in Thailand and an alternative semi-nested RT-PCR for detection. Aquaculture 2017, 476, 111–118. [Google Scholar] [CrossRef]
- Mugimba, K.K.; Chengula, A.A.; Wamala, S.; Mwega, E.D.; Kasanga, C.J.; Byarugaba, D.K.; Mdegela, R.H.; Tal, S.; Bornstein, B.; Dishon, A.; et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. J. Fish Dis. 2018, 41, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Pulido, L.L.H.; Mora, C.M.; Hung, A.L.; Dong, H.T.; Senapin, S. Tilapia lake virus (TiLV) from Peru is genetically close to the Israeli isolates. Aquaculture 2019, 510, 61–65. [Google Scholar] [CrossRef]
- Senapin, S.; Shyam, K.U.; Meemetta, W.; Rattanarojpong, T.; Dong, H.T. Inapparent infection cases of tilapia lake virus (TiLV) in farmed tilapia. Aquaculture 2018, 487, 51–55. [Google Scholar] [CrossRef]
- Ferguson, H.W.; Kabuusu, R.; Beltran, S.; Reyes, E.; Lince, J.A.; del Pozo, J. Syncytial hepatitis of farmed tilapia, Oreochromis niloticus (L.): A case report. J. Fish Dis. 2014, 37, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Kembou Tsofack, J.E.; Zamostiano, R.; Watted, S.; Berkowitz, A.; Rosenbluth, E.; Mishra, N.; Briese, T.; Lipkin, W.I.; Kabuusu, R.M.; Ferguson, H.; et al. Detection of tilapia lake virus in clinical samples by culturing and nested reverse transcription-PCR. Clin. Vet. Microbiol. 2017, 55, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, P.; Fathi, M.A.; Fischer, A.; Mohan, C.; Schieck, E.; Mishra, N.; Heinimann, A.; Frey, J.; Wieland, B.; Jores, J. Detection of Tilapia Lake Virus in Egyptian fish farms experiencing high mortalities in 2015. J. Fish Dis. 2017, 1–4. [Google Scholar] [CrossRef]
- Behera, B.K.; Pradhan, P.K.; Swaminathan, T.R.; Sood, N.; Paria, P.; Das, A.; Verma, D.K.; Kumar, R.; Yadav, M.K.; Dev, A.K.; et al. Emergence of Tilapia Lake Virus associated with mortalities of farmed Nile Tilapia Oreochromis niloticus (Linnaeus 1758) in India. Aquaculture 2018, 484, 168–174. [Google Scholar] [CrossRef]
- Surachetpong, W.; Janetanakit, T.; Nonthabenjawan, N.; Tattiyapong, P.; Sirikanchana, K.; Amonsin, A. Outbreaks of tilapia lake virus infection, Thailand, 2015–2016. Emerg. Infect. Dis. 2017, 23, 1031–1033. [Google Scholar] [CrossRef] [Green Version]
- Amal, M.N.A.; Koh, C.B.; Nurliyana, M.; Suhaiba, M.; Nor-Amalina, Z.; Santha, S.; Diyana-Nadhirah, K.P.; Yusof, M.T.; Ina-Salwany, M.Y.; Zamri-Saad, M. A case of natural co-infection of Tilapia Lake Virus and Aeromonas veronii in a Malaysian red hybrid tilapia (Oreochromis niloticus × O. mossambicus) farm experiencing high mortality. Aquaculture 2018, 485, 12–16. [Google Scholar] [CrossRef]
- Skornik, R.; Eyngor, M.; Behar, A.; Markovich, M.P.; Wajsbrot, N.; Klement, E.; Davidovich, N. Tilapia Lake Virus disease: phylogenetic analysis reveal that two distinct clades are circulating in Israel simultaneously. Transbound. Emerg. Dis. 2019, 67. [Google Scholar] [CrossRef]
- Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 7 February 2020).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. RnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience 2019, 8, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. CAP3: A DNA Sequence Assembly Program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- Al-Hussinee, L.; Subramaniam, K.; Ahasan, M.S.; Keleher, B.; Waltzek, T.B. Complete genome sequence of a tilapia lake virus isolate. Genome Announc. 2018, 6, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, K.; Ferguson, H.W.; Kabuusu, R.; Waltzek, T.B. Genome sequence of tilapia lake virus associated with syncytial hepatitis of tilapia in an Ecuadorian aquaculture facility. Microbiol. Resour. Announc. 2019, 8, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 2004, 5, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y. A phylogenetic approach to detecting reassortments in viruses with segmented genomes. Gene 2010, 464, 11–16. [Google Scholar] [CrossRef]
- Jaemwimol, P.; Rawiwan, P.; Tattiyapong, P.; Saengnual, P.; Kamlangdee, A.; Surachetpong, W. Susceptibility of important warm water fish species to tilapia lake virus (TiLV) infection. Aquaculture 2018, 497, 462–468. [Google Scholar] [CrossRef]
- Abdullah, A.; Ramly, R.; Mohammad Ridzwan, M.S.; Sudirwan, F.; Abas, A.; Ahmad, K.; Murni, M.; Kua, B.C. First detection of tilapia lake virus (TiLV) in wild river carp (Barbonymus schwanenfeldii) at Timah Tasoh Lake, Malaysia. J. Fish Dis. 2018, 41, 1459–1462. [Google Scholar] [CrossRef]
- McDonald, S.M.; Nelson, M.I.; Turner, P.E.; Patton, J.T. Reassortment in segmented RNA viruses: Mechanisms and outcomes. Nat. Rev. Microbiol. 2016, 14, 448–460. [Google Scholar] [CrossRef] [Green Version]
- Lowen, A.C. It’s in the mix: Reassortment of segmented viral genomes. PLoS Pathog. 2018, 14, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Smith, G.J.D.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Segment | Reported Length (nt) from Bacharach et al. [7] | BD-2017 Length (nt) | BD-2017 Coverage (Mean ± SD) | Trimmed Length with Full Coding Region Refs (nt) | Partial Segments with Shorter Refs (nt) |
|---|---|---|---|---|---|
| 1 | 1641 | 1620 | 90 ± 29 | 1560 | 1560 |
| 2 | 1471 | 1448 | 91 ± 24 | 1194 | 819 |
| 3 | 1371 | 1353 | 204 ± 44 | 1260 | 234 |
| 4 | 1250 | 1226 | 46 ± 18 | 1065 | 261 |
| 5 | 1099 | 1083 | 198 ± 61 | 1032 | |
| 6 | 1044 | 1024 | 177 ± 50 | 954 | |
| 7 | 777 | 758 | 92 ± 31 | 588 | |
| 8 | 657 | 637 | 289 ± 72 | 525 | |
| 9 | 548 | 531 | 341 ± 146 | 351 | 249 |
| 10 | 465 | 443 | 164 ± 44 | 342 | |
| Total | 10323 | 10123 | 150 ± 96 | 8871 |
| Pattern (Segs 1–7) | Number of Quartets | Notes |
|---|---|---|
| 1111111 | 17 | Identical pattern for all segments—no reassortment |
| 1111221 | 5 | 1 = Ecuador/Peru together and/or both Israel together, |
| 2211221 | 4 | 2 = Ecuador with Israel Til-4-2011 |
| xx11111 | 2 | Segs 1–2 poorly resolved, 3–7 well supported |
| unresolved | 7 | Most/all segments poorly resolved |
| Segment | Bootstrap Support (%) | Pair 1 | Pair 2 |
|---|---|---|---|
| Pattern 1111221 | |||
| 1 | 100 | Ecuador + Peru | Israel AD-2016 + Israel Til-4-2011 |
| 2 | 98 | Ecuador + Peru | Israel AD-2016 + Israel Til-4-2011 |
| 3 | 99 | Ecuador + Peru | Israel AD-2016 + Israel Til-4-2011 |
| 4 | 95 | Ecuador + Peru | Israel AD-2016 + Israel Til-4-2011 |
| 5 | 74 | Ecuador + Israel Til-4-2011 | Israel AD-2016 + Peru |
| 6 | 97 | Ecuador + Israel Til-4-2011 | Israel AD-2016 + Peru |
| 7 | 98 | Ecuador + Peru | Israel AD-2016 + Israel Til-4-2011 |
| Pattern 2211221 | |||
| 1 | 95 | Ecuador + Israel Til-4-2011 | Israel AD-2016 + Thailand WVL |
| 2 | 93 | Ecuador + Israel Til-4-2011 | Israel AD-2016 + Thailand WVL |
| 3 | 92 | Ecuador + Thailand WVL | Israel AD-2016 + Israel Til-4-2011 |
| 4 | 70 | Ecuador + Thailand WVL | Israel AD-2016 + Israel Til-4-2011 |
| 5 | 100 | Ecuador + Israel Til-4-2011 | Israel AD-2016 + Thailand WVL |
| 6 | 100 | Ecuador + Israel Til-4-2011 | Israel AD-2016 + Thailand WVL |
| 7 | 42 | Ecuador + Thailand WVL | Israel AD-2016 + Israel Til-4-2011 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaput, D.L.; Bass, D.; Alam, M.M.; Al Hasan, N.; Stentiford, G.D.; van Aerle, R.; Moore, K.; Bignell, J.P.; Haque, M.M.; Tyler, C.R. The Segment Matters: Probable Reassortment of Tilapia Lake Virus (TiLV) Complicates Phylogenetic Analysis and Inference of Geographical Origin of New Isolate from Bangladesh. Viruses 2020, 12, 258. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030258
Chaput DL, Bass D, Alam MM, Al Hasan N, Stentiford GD, van Aerle R, Moore K, Bignell JP, Haque MM, Tyler CR. The Segment Matters: Probable Reassortment of Tilapia Lake Virus (TiLV) Complicates Phylogenetic Analysis and Inference of Geographical Origin of New Isolate from Bangladesh. Viruses. 2020; 12(3):258. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030258
Chicago/Turabian StyleChaput, Dominique L., David Bass, Md. Mehedi Alam, Neaz Al Hasan, Grant D. Stentiford, Ronny van Aerle, Karen Moore, John P. Bignell, Mohammad Mahfujul Haque, and Charles R. Tyler. 2020. "The Segment Matters: Probable Reassortment of Tilapia Lake Virus (TiLV) Complicates Phylogenetic Analysis and Inference of Geographical Origin of New Isolate from Bangladesh" Viruses 12, no. 3: 258. https://0-doi-org.brum.beds.ac.uk/10.3390/v12030258