The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Material and Methods
2.1. Genome Sequences
2.2. miR Prediction
2.3. Mutational Analysis of Potential miRNA Sites
2.4. Pathway Analysis
3. Results
3.1. Analysis of SARS-CoV-2 Viral Genome for miR Sequences with High Human Similarity and Functional Characterisation
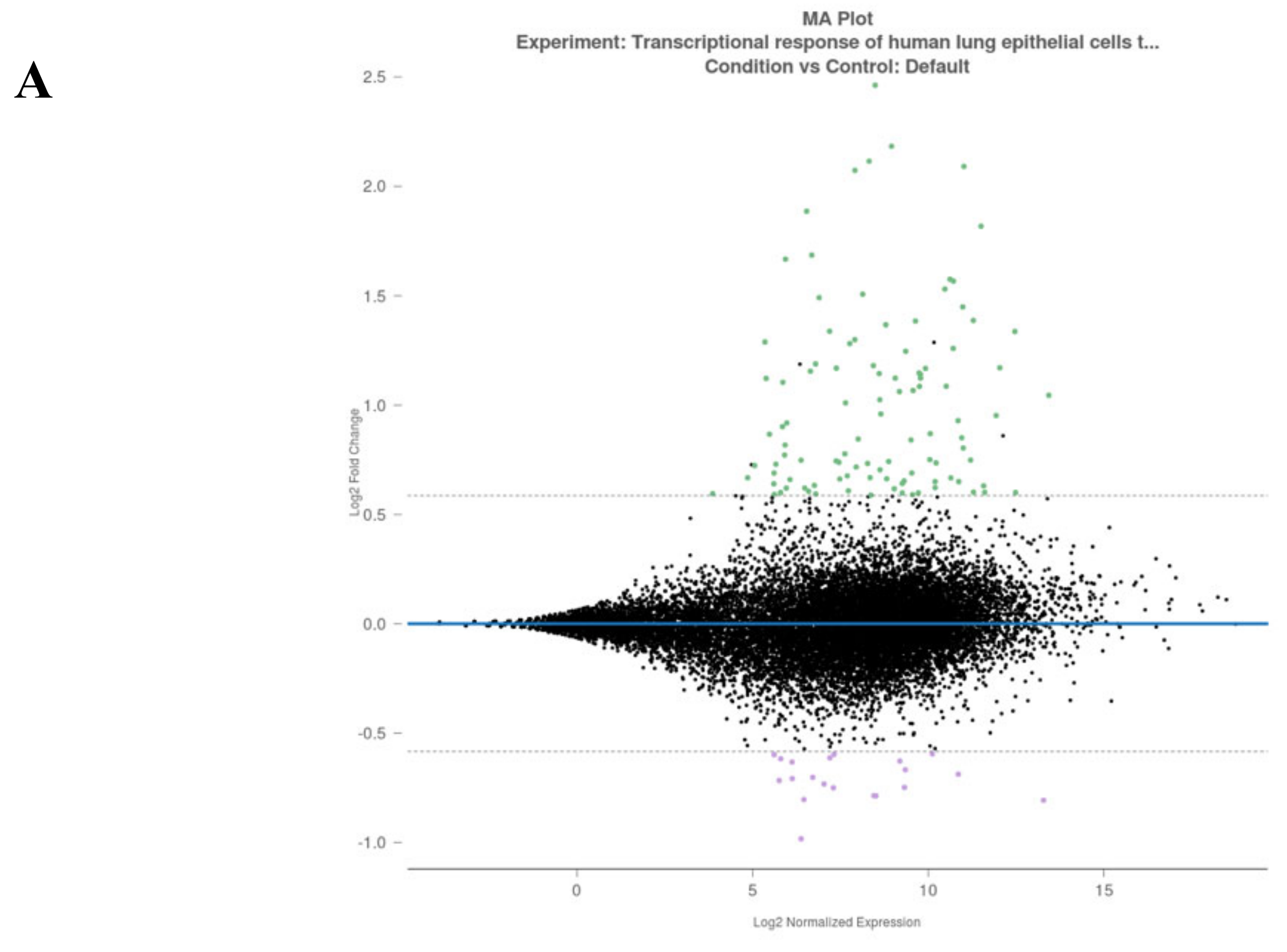
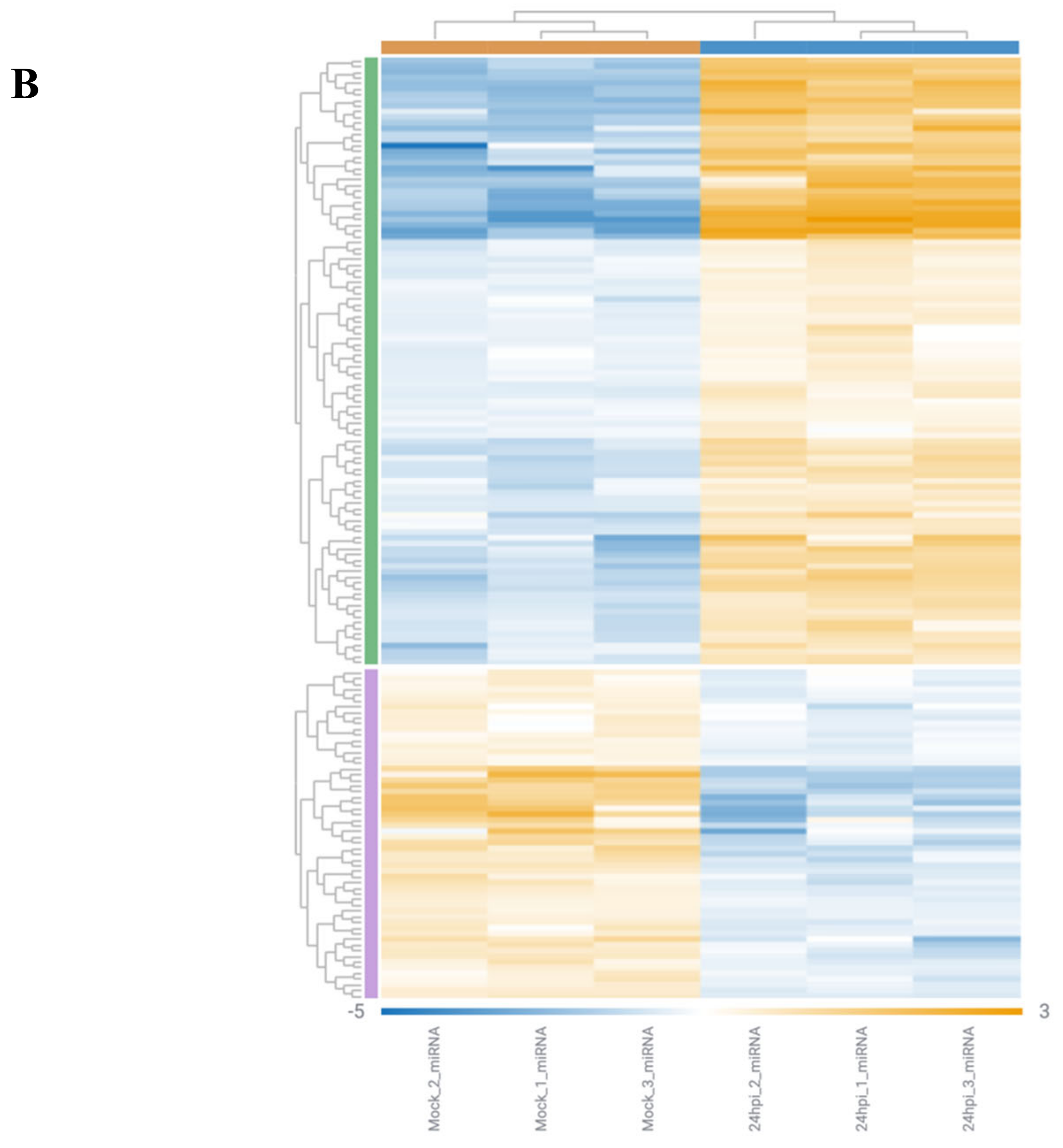
3.2. Analysis of Gene Alterations in NHEB Bronchial Epithelial and A549 Cells Due to SARS-CoV-2 Infection
4. Discussion
4.1. Biological Significance of Top Ranked miRs in Humans
4.1.1. miR-8066
4.1.2. miR-5197
4.1.3. miR-3611
4.1.4. miR-3934-3p
4.1.5. miR-1307-3p
4.1.6. miR-3691-3p
4.1.7. miR1468-5p
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Tsang, K.W.; Ooi, G.C.; Ho, P.L. Diagnosis and pharmacotherapy of severe acute respiratory syndrome: What have we learnt? Eur. Respir. J. 2004, 24, 1025–1032. [Google Scholar] [CrossRef] [Green Version]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Whisnant, A.W.; Kehl, T.; Bao, Q.; Materniak, M.; Kuzmak, J.; Löchelt, M.; Cullen, B.R. Identification of novel, highly expressed retroviral microRNAs in cells infected by bovine foamy virus. J. Virol. 2014, 88, 4679–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundhoff, A.; Sullivan, C.S. Virus-encoded microRNAs. Virology 2011, 411, 325–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwig, A.; Jongejan, A.; van Kampen, A.H.; Berkhout, B.; Das, A.T. Tat-dependent production of an HIV-1 TAR-encoded miRNA-like small RNA. Nucleic Acids Res. 2016, 44, 4340–4353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruscella, P.; Bottini, S.; Baudesson, C.; Pawlotsky, J.M.; Feray, C.; Trabucchi, M. Viruses and miRNAs: More Friends than Foes. Front. Microbiol. 2017, 8, 824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nightingale, K.; Dry, I.; Hopkins, J.; Dalziel, R. Regulation of Ov2 by virus encoded microRNAs. Vet. Res. Commun. 2019, 43, 99–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, B.R. MicroRNAs as mediators of viral evasion of the immune system. Nat. Immunol. 2013, 14, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Cullen, B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004, 78, 12868–12876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennasser, Y.; Yeung, M.L.; Jeang, K.-T. HIV-1 TAR RNA Subverts RNA Interference in Transfected Cells through Sequestration of TAR RNA-binding Protein, TRBP. J. Biol. Chem. 2006, 281, 27674–27678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harwig, A.; Das, A.T.; Berkhout, B. Retroviral microRNAs. Curr. Opin. Virol. 2014, 7, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Scheel, T.K.H.; Rice, C.M.; Darnell, R.B. Hepatitis C Virus RNA Functionally Sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef] [Green Version]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Sanghvi, V.R.; Steel, L.F. RNA silencing as a cellular defence against HIV-1 infection: Progress and issues. FASEB J. 2012, 26, 3937–3945. [Google Scholar] [CrossRef]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Zhu, Y.; Bisaro, D.M.; Parris, D.S. Herpes simplex virus type 1 suppresses RNA-induced gene silencing in mammalian cells. J. Virol. 2009, 83, 6652–6663. [Google Scholar] [CrossRef] [Green Version]
- Hussain, M.; Asgari, S. MicroRNA-like viral small RNA from Dengue virus 2 autoregulates its replication in mosquito cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2746–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data-from vision to reality. EuroSurveillance 2017, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlachos, I.S.; Konstantinos, Z.; Maria, D.; Paraskevopoulou, G.G.; Dimitra, K.; Thanasis, V.; Theodore, D.; Artemis, G.H. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/ (accessed on 18 April 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [Green Version]
- Alexa, A.; Rahnenfuehrer, J. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2010, 2, 2010. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridgem, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2009, 38, D492–D496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 43, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 43, 1739–1740. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2017, 46, D661–D667. [Google Scholar] [CrossRef]
- Saçar-Demirci, M.D.; Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.J.; Wu, L.L.; Chen, D.F.; Li, Y.X.; Liu, Y.J.; Fan, Y.Q.; Du, S.H.; Huang, H.; Liu, N.; Caserta, S.; et al. Circulating Plasma microRNAs can differentiate Human Sepsis and Systemic Inflammatory Response Syndrome (SIRS). Sci. Rep. 2016, 6, 28006. [Google Scholar]
- Pluta, L.; Yousefi, B.; Damania, B.; Khan, A.A. Endosomal TLR-8 Senses microRNA-1294 Resulting in the Production of NFḱB Dependent Cytokines. Front. Immunol. 2019, 10, 2860. [Google Scholar] [CrossRef]
- Liao, B.; Zhou, M.X.; Zhou, F.K.; Luo, X.M.; Zhong, S.X.; Zhou, Y.F.; Qin, Y.S.; Li, P.P.; Qin, C. Exosome-Derived MiRNAs as Biomarkers of the Development and Progression of Intracranial Aneurysms. J. Atheroscler. Thromb. 2019, 27(6), 51102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivieri, F.; Rippo, M.R.; Procopio, A.D.; Fazioli, F. Circulating inflamma-miRs in aging and age-related diseases. Front. Genet. 2013, 4, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaschetto, L. A putative miRNA in the spike gene of SARS-CoV-2 has perfect sequence identity to both the forward and reverse complementary strands of hsa-mir-8055 involved in T-cell response to antigen. OSF Preprints 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Han, B.; Wang, J.; Liu, Q.; Kong, Y.; Jiang, D.; Jia, H. Differential expression profiles and functional analysis of circular RNAs in children with fulminant myocarditis. Epigenomics 2019, 11, 1129–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, D.; Pfefferle, S.; Drosten, C.; Stevermann, L.; Traggiai, E.; Lanzavecchia, A.; Becker, S. Studies on membrane topology, N-glycosylation and functionality of SARS-CoV membrane protein. Virol. J. 2009, 6, 79. [Google Scholar] [CrossRef] [Green Version]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.; Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Nicholls, J.M.; Chen, Y.G. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J. Biol. Chem. 2008, 283, 3272–3280. [Google Scholar] [CrossRef]
- Iwuchukwu, I.; Nguyen, D.; Beavers, M.; Tran, V.; Sulaiman, W.; Fannin, E.; Lasseigne, L.; Ramsay, E.; Wilson, J.; Bazan, N.G.; et al. MicroRNA Regulatory Network as Biomarkers of Late Seizure in Patients with Spontaneous Intracerebral Hemorrhage. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, Q.; Hu, L.; Chen, F.; Hu, Z.; Heist, R.S.; Su, L.; Amos, C.I.; Shen, H.; Christiani, D.C. Polymorphisms in MicroRNAs are associated with survival in non-small cell lung cancer. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2503–2511. [Google Scholar] [CrossRef] [Green Version]
- Schotte, D.; Akbari Moqadam, F.; Lange-Turenhout, E.A.; Chen, C.; van Ijcken, W.F.; Pieters, R.; den Boer, M.L. Discovery of new microRNAs by small RNAome deep sequencing in childhood acute lymphoblastic leukemia. Leukemia 2011, 25, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Li, X.P.; Gong, W.J.; Wu, N.Y.; Tang, J.; Yin, J.Y.; Li, X.; Zhang, W.; Zhou, H.H.; Liu, Z.Q. Age-related common miRNA polymorphism associated with severe toxicity in lung cancer patients treated with platinum-based chemotherapy. Clin. Exp. Pharmacol. Physiol. 2017, 44 (Suppl. S1), 21–29. [Google Scholar] [CrossRef]
- Michlewski, G.; Cáceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Filatov, A. Role of O-glycosylation and expression of CD43 and CD45 on the surfaces of effector T cells in human T cell leukemia virus type 1 cell-to-cell infection. J. Virol. 2012, 86, 2447–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, E.J.; Linstedt, A.D. Site-specific glycosylation of Ebola virus glycoprotein by human polypeptide GalNAc-transferase 1 induces cell adhesion defects. J. Biol. Chem. 2018, 293, 19866–19873. [Google Scholar] [CrossRef] [Green Version]
- Lantéri, M.; Giordanengo, V.; Hiraoka, N.; Fuzibet, J.G.; Auberger, P.; Fukuda, M.; Baum, L.G.; Lefebvre, J.C. Altered T cell surface glycosylation in HIV-1 infection results in increased susceptibility to galectin-1-induced cell death. Glycobiology 2003, 13, 909–918. [Google Scholar] [CrossRef]
- Gaunitz, S.; Liu, J.; Nilsson, A.; Karlsson, N.; Holgersson, J. Avian influenza H5 hemagglutinin binds with high avidity to sialic acid on different O-linked core structures on mucin-type fusion proteins. Glycoconj. J. 2014, 31, 145–159. [Google Scholar] [CrossRef]
- Nordén, R.; Nyström, K.; Adamiak, B.; Halim, A.; Nilsson, J.; Larson, G.; Trybala, E.; Olofsson, S. Involvement of viral glycoprotein gC-1 in expression of the selectin ligand sialyl-Lewis X induced after infection with herpes simplex virus type 1. Apmis 2013, 121, 280–289. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Qiu, L.; Pan, R.; Bai, H.; Jiang, Y.; Wang, Z.; Bi, Y.; Chen, G.; Chang, G. Expression patterns of novel circular RNAs in chicken cells after avian leukosis virus subgroup J infection. Gene 2019, 15, 72–81. [Google Scholar] [CrossRef]
- Reid, D.J.; Pham, N.T. Emerging Therapeutic Options for the Management of COPD. Clin. Med. Insights Circ. Respir. Pulm. Med. 2013, 9, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Shen, Z.; Tang, W.; Guo, J.; Sun, S. miR-483-5p plays a protective role in chronic obstructive pulmonary disease. Int. J. Mol. Med. 2017, 40, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 4, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Martinez, P.; Séron, K.; Luo, G.; Allain, F.; Dubuisson, J.; Belouzard, S. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J. Virol. 2015, 89, 3846–3858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Jiang, J.; Liang, B.; Wei, F.; Huang, J.; Pan, P.; Su, J.; Zhou, B.; Zang, N.; Ye, L.; et al. Opiate use inhibits TLR9 signaling pathway in vivo: Possible role in pathogenesis of HIV-1 infection. Sci. Rep. 2017, 7, 13071. [Google Scholar] [CrossRef] [Green Version]
- Nyland, S.B.; Cao, C.; Bai, Y.; Loughran, T.P.; Ugen, K.E. Modulation of infection and type 1 cytokine expression parameters by morphine during in vitro coinfection with human T-cell leukemia virus type I and HIV-1. J. Acquir. Immune Defic. Syndr. 2003, 32, 406–416. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Douglas, S.D.; Lai, J.P.; Xiao, W.D.; Pleasure, D.E.; Ho, W.Z. Morphine enhances hepatitis C virus (HCV) replicon expression. Am. J. Pathol. 2003, 163, 1167–1175. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ye, L.; Peng, J.S.; Wang, C.Q.; Luo, G.X.; Zhang, T.; Wan, Q.; Ho, W.Z. Morphine inhibits intrahepatic interferon- alpha expression and enhances complete hepatitis C virus replication. J. Infect. Dis. 2007, 196, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Chuang, R.Y.; Suzuki, S.; Chuang, T.K.; Miyagi, T.; Chuang, L.F.; Doi, R.H. Opioids and the progression of simian AIDS. Front. Biosci. 2005, 10, 1666–1677. [Google Scholar] [CrossRef] [Green Version]
- Coussons-Read, M.E.; Daniels, M.; Gilmour, M.I. Morphine alters the immune response to influenza virus infection in Lewis rats. Adv. Exp. Med. Biol. 1998, 437, 73–82. [Google Scholar]
- Chung, K.F. Drugs to suppress cough. Expert Opin. Investig. Drugs 2005, 14, 19–27. [Google Scholar] [CrossRef]
- Hiew, M.S.Y.; Cheng, H.P.; Huang, C.J.; Chong, K.Y.; Cheong, S.K.; Choo, K.B.; Kamarul, T. Incomplete cellular reprogramming of colorectal cancer cells elicits an epithelial/mesenchymal hybrid phenotype. J. Biomed. Sci. 2018, 25, 57. [Google Scholar] [CrossRef] [PubMed]
- Yerukala Sathipati, S.; Ho, S.Y. Identifying the miRNA signature associated with survival time in patients with lung adenocarcinoma using miRNA expression profiles. Sci. Rep. 2017, 7, 7507. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Wen, Z.; Zheng, L. Downregulation of miR-3934-5p enhances A549 cell sensitivity to cisplatin by targeting TP53INP1. Exp. Ther. Med. 2019, 18, 1653–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slattery, M.L.; Trivellas, A.; Pellatt, A.J.; Mullany, L.E.; Stevens, J.R.; Wolff, R.K.; Herrick, J.S. Genetic variants in the TGF-β-signaling pathway influence expression of miRNAs in colon and rectal normal mucosa and tumor tissue. Oncotarget 2017, 8, 16765–16783. [Google Scholar] [CrossRef] [Green Version]
- Chinnapaiyan, S.; Dutta, R.K.; Nair, M.; Chand, H.S.; Rahman, I.; Unwalla, H.J. TGF-β1 increases viral burden and promotes HIV-1 latency in primary differentiated human bronchial epithelial cells. Sci. Rep. 2019, 9, 12552. [Google Scholar] [CrossRef] [Green Version]
- Lang, J.; Yang, N.; Deng, J.; Liu, K.; Yang, P.; Zhang, G.; Jiang, C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE 2011, 6, e23710. [Google Scholar] [CrossRef]
- Milewska, A.; Zarebski, M.; Nowak, P.; Stozek, K.; Potempa, J.; Pyrc, K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 2014, 88, 13221–13230. [Google Scholar] [CrossRef] [Green Version]
- Tamhankar, M.; Gerhardt, D.M.; Bennett, R.S.; Murphy, N.; Jahrling, P.B.; Patterson, J.L. Heparan sulfate is an important mediator of Ebola virus infection in polarized epithelial cells. Virol. J. 2018, 15, 135. [Google Scholar] [CrossRef] [Green Version]
- Kalia, M.; Chandra, V.; Rahman, S.A.; Sehgal, D.; Jameel, S. Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J. Virol. 2009, 83, 12714–12724. [Google Scholar] [CrossRef] [Green Version]
- Robinson-McCarthy, L.R.; McCarthy, K.R.; Raaben, M.; Piccinotti, S.; Nieuwenhuis, J.; Stubbs, S.H.; Bakkers, M.J.G.; Whelan, S.P.J. Reconstruction of the cell entry pathway of an extinct virus. PLoS Pathog. 2018, 14, e1007123. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Koetzner, C.A.; Payne, A.F.; Nierode, G.J.; Yu, Y.; Wang, R.; Barr, E.; Dordick, J.S.; Kramer, L.D.; Zhang, F.; et al. Glycosaminoglycan Compositional Analysis of Relevant Tissues in Zika Virus Pathogenesis and in Vitro Evaluation of Heparin as an Antiviral against Zika Virus Infection. Biochemistry 2019, 58, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Huan, C.C.; Wang, Y.; Ni, B.; Wang, R.; Huang, L.; Ren, X.F.; Tong, G.Z.; Ding, C.; Fan, H.J.; Mao, X. Porcine epidemic diarrhea virus uses cell-surface heparan sulfate as an attachment factor. Arch. Virol. 2015, 160, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Anindita, P.D.; Ito, N.; Sugiyama, M.; Carr, M.; Fukuhara, H.; Ose, T.; Maenaka, K.; Takada, A.; Hall, W.W.; et al. The Role of Heparan Sulfate Proteoglycans as an Attachment Factor for Rabies Virus Entry and Infection. J. Infect. Dis. 2018, 217, 1740–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, C.; Dhondt, K.P.; Châlons, M.; Mély, S.; Raoul, H.; Negre, D.; Cosset, F.L.; Gerlier, D.; Vivès, R.R.; Horvat, B. Heparan sulfate-dependent enhancement of henipavirus infection. Mbio 2015, 6, e02427. [Google Scholar] [CrossRef] [Green Version]
- Picchianti-Diamanti, A.; Rosado, M.M.; D’Amelio, R. Infectious Agents and Inflammation: The Role of Microbiota in Autoimmune Arthritis. Front. Microbiol. 2018, 8, 2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demignot, S.; Beilsteinm, F.; Morel, E. Triglyceride-rich lipoproteins and cytosolic lipid droplets in enterocytes: Key players in intestinal physiology and metabolic disorders. Biochimie 2014, 96, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Herzlich, B.C.; Schiano, T.D.; Moussa, Z.; Zimbalist, E.; Panagopoulos, G.; Ast, A.; Nawabi, I. Decreased intrinsic factor secretion in AIDS: Relation to parietal cell acid secretory capacity and vitamin B12 malabsorption. Am. J. Gastroenterol. 1992, 87, 1781–1788. [Google Scholar]
- Ehrenpreis, E.D.; Carlson, S.J.; Boorstein, H.L.; Craig, R.M. Malabsorption and deficiency of vitamin B12 in HIV-infected patients with chronic diarrhea. Dig. Dis. Sci. 1994, 39, 2159–2162. [Google Scholar] [CrossRef]
- Sultana, S.; Li, H.; Puche, A.; Jones, O.; Bryant, J.L.; Royal, W. Quantitation of parvalbumin+ neurons and human immunodeficiency virus type 1 (HIV-1) regulatory gene expression in the HIV-1 transgenic rat: Effects of vitamin A deficiency and morphine. J. Neurovirol. 2010, 16, 33–40. [Google Scholar] [CrossRef] [Green Version]
- West, C.E.; Sijtsma, S.R.; Kouwenhoven, B.; Rombout, J.H.; van der Zijpp, A.J. Epithelia-damaging virus infections affect vitamin A status in chickens. J. Nutr. 1992, 122, 333–339. [Google Scholar] [CrossRef]
- Daneshkhah, A.; Eshein, A.; Subramanian, H.; Roy, H.K.; Backman, V. The Role of Vitamin D in Suppressing Cytokine Storm in COVID-19 Patients and Associated Mortality. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Nagai, A.; Matsumiya, H.; Hayashi, M.; Yasui, S.; Okamoto, H.; Konno, K. Effects of nicotinamide and niacin on bleomycin-induced acute injury and subsequent fibrosis in hamster lungs. Exp. Lung Res. 1994, 20, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Rivero, M.; Zhang, R.; Heilmann-Heimbach, S.; Mueller, A.; Bagci, S.; Dresbach, T.; Schröder, L.; Holdenrieder, S.; Reutter, H.M.; Kipfmueller, F. Circulating microRNAs are associated with Pulmonary Hypertension and Development of Chronic Lung Disease in Congenital Diaphragmatic Hernia. Sci. Rep. 2018, 8, 10735. [Google Scholar] [CrossRef] [PubMed]
- Ruan, D.T.; Gao, S.; Shelat, H.; King, B.; Geng, Y.J. Differential expression of microRNA and arachidonic acid metabolism in aspirin-treated human cardiac and peri-cardiac fat-derived mesenchymal stem cells. Vascul. Pharmacol. 2020, 127, 106651. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.S.; White, K.; MacLean, M.R.; Baker, A.H. MicroRNAs in pulmonary arterial remodeling. Cell Mol. Life Sci. 2013, 70, 4479–4494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandre-Alcázar, M.A.; Michiels-Corsten, M.; Vicencio, A.G.; Reiss, I.; Ryu, J.; de Krijger, R.R.; Haddad, G.G.; Tibboel, D.; Seeger, W.; Eickelberg, O.; et al. TGF-beta signaling is dynamically regulated during the alveolarization of rodents and human lungs. Dev. Dyn. 2008, 237, 259–269. [Google Scholar] [CrossRef]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef] [Green Version]
- Morty, R.E.; Königshoff, M.; Eickelberg, O. Transforming growth factor-beta signaling across ages: From distorted lung development to chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 607–613. [Google Scholar] [CrossRef]
- Lal, C.V.; Olave, N.; Travers, C.; Rezonzew, G.; Dolma, K.; Simpson, A.; Halloran, B.; Aghai, Z.; Das, P.; Sharma, N.; et al. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. JCI Insight 2018, 3, 93994. [Google Scholar] [CrossRef]
- Lin, R.; Rahtu-Korpela, L.; Magga, J.; Ulvila, J.; Swan, J.; Kemppi, A.; Pakanen, L.; Porvari, K.; Huikuri, H.; Junttila, J.; et al. miR-1468-3p promotes aging-related cardiac fibrosis. Mol. Ther. Nucl. Acids 2020. [Google Scholar] [CrossRef] [PubMed]
- Erener, S.; Marwaha, A.; Tan, R.; Panagiotopoulos, C.; Kieffer, T.J. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes. JCI Insight 2017, 2, e89656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, K.; Zhi, T.; Xu, W.; Xu, X.; Wu, W.; Yu, T.; Nie, E.; Zhou, X.; Bao, Z.; Jin, X.; et al. MicroRNA-1468-5p inhibits glioma cell proliferation and induces cell cycle arrest by targeting RRM1. Am. J. Cancer Res. 2017, 7, 784–800. [Google Scholar] [PubMed]
- Liu, F.; Zhao, H.; Gong, L.; Yao, L.; Li, Y.; Zhang, W. MicroRNA-129-3p functions as a tumor suppressor in serous ovarian cancer by targeting BZW1. Int. J. Clin. Exp. Pathol. 2018, 11, 5901–5908. [Google Scholar]
- Ludwig, N.; Fehlmann, T.; Kern, F.; Gogol, M.; Maetzler, W.; Deutscher, S.; Gurlit, S.; Schulte, C.; von Thaler, A.K.; Deuschle, C.; et al. Machine Learning to Detect Alzheimer’s Disease from Circulating Non-coding RNAs. Genom. Proteom. Bioinform. 2019, 17, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, B.; Han, S.; Lu, L.; Chen, Y.; Chu, X.; Wang, R.; Chen, L. MicroRNA-129 in Human Cancers: From Tumorigenesis to Clinical Treatment. Cell Physiol. Biochem. 2016, 39, 2186–2202. [Google Scholar] [CrossRef]
- Tang, X.; Tang, J.; Liu, X.; Zeng, L.; Cheng, C.; Luo, Y.; Li, L.; Qin, S.L.; Sang, Y.; Deng, L.M.; et al. Downregulation of miR-129-2 by promoter hypermethylation regulates breast cancer cell proliferation and apoptosis. Oncol. Rep. 2016, 35, 2963–2969. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ruan, A.; Wang, X.; Han, W.; Wang, R.; Lou, N.; Ruan, H.; Qiu, B.; Yang, H.; Zhang, X. miR-129-3p, as a diagnostic and prognostic biomarker for renal cell carcinoma, attenuates cell migration and invasion via downregulating multiple metastasis-related genes. J. Cancer Res. Clin. Oncol. 2014, 140, 1295–1304. [Google Scholar] [CrossRef]
- Wong, K.Y.; Yim, R.L.; Kwong, Y.L.; Leung, C.Y.; Hui, P.K.; Cheung, F.; Liang, R.; Jin, D.Y.; Chim, C.S. Epigenetic inactivation of the MIR129-2 in hematological malignancies. J. Hematol. Oncol. 2013, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Qian, J.; Li, C.; Kwok, L.; Cheng, F.; Liu, P.; Perdomo, C.; Kotton, D.; Vaziri, C.; Anderlind, C.; et al. miR-129 regulates cell proliferation by downregulating Cdk6 expression. Cell Cycle 2010, 9, 1809–1818. [Google Scholar] [CrossRef] [Green Version]
- Katayama, Y.; Maeda, M.; Miyaguchi, K.; Nemoto, S.; Yasen, M.; Tanaka, S.; Mizushima, H.; Fukuoka, Y.; Arii, S.; Tanaka, H. Identification of pathogenesis-related microRNAs in hepatocellular carcinoma by expression profiling. Oncol. Lett. 2012, 4, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Sannigrahi, M.K.; Sharma, R.; Singh, V.; Panda, N.K.; Rattan, V.; Khullar, M. DNA methylation regulated microRNAs in HPV-16-induced head and neck squamous cell carcinoma (HNSCC). Mol. Cell Biochem. 2018, 448, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Dreher, A.; Rossing, M.; Kaczkowski, B.; Andersen, D.K.; Larsen, T.J.; Christophersen, M.K.; Nielsen, F.C.; Norrild, B. Differential expression of cellular microRNAs in HPV 11, -16, and -45 transfected cells. Biochem. Biophys. Res. Commun. 2011, 412, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–106. [Google Scholar] [PubMed]
- Liu, Z.; Wang, J.; Xu, Y.; Guo, M.; Mi, K.; Xu, R.; Pei, Y.; Zhang, Q.; Luan, X.; Hu, Z.; et al. Implications of the virus-encoded miRNA and host miRNA in the pathogenicity of SARS-CoV-2. arXiv 2020, arXiv:2004.04874. [Google Scholar]
- Rakhmetullina, A.; Ivashchenko, A.; Akimniyazova, A.; Aisina, D.; Pyrkova, A. The miRNA Complexes Against Coronaviruses COVID-19, SARS-CoV, And MERS-CoV. Res. Sq. 2020. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhong, L. Genomics functional analysis and drug screening of SARS-CoV-2. Genes Dis. 2020. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRs | Score | E-Value | Alignment | Wuhan | Italy | UK | Valencia | Turkey | Vero E6 |
|---|---|---|---|---|---|---|---|---|---|
| NC_045512.2 | MT066156.1 | hCoV-19/England/20136087804/2020|EPI_ISL_420910 | MT198652.2 | hCoV-19/Turkey/GLAB-CoV008/2020 | hCoV-19/Turkey/ERAGEM-001/2020 | ||||
| hsa-miR-8066 | 80 | 1.6–2.8 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-5197-3p | 79 | 1.6–2.8 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-3611 | 77 | 2.8–3.8 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-3934-3p | 76 | 3.4–5.0 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-1468-5p | 71 | 4.7–8.8 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-1307-3p | 72 | 4.3–6.3 |  | √ | √ | √ | √ | √ | |
| hsa-miR-3691-3p | 74 | 5.0–9.5 |  | √ | √ | √ | √ | √ | |
| hsa-miR-3120-5p | 73 | 6.0–7.2 |  | √ | √ | √ | √ | √ | |
| hsa-miR-3914 | 73 | 6.0–8.5 |  | √ | √ | √ | √ | √ | |
| hsa-miR-3672 | 72 | 7.3–9.8 |  | X | X | X | X | ||
| hsa-miR-7107-3p | 73 | 6.0–6.2 |  | √ | √ | √ | √ | √ | |
| hsa-miR-1287-5p | 73 | 6.0–8.3 |  | √ | √ | √ | √ | √ | |
| hsa-miR-129-2-3p | 73 | 6.0–7.7 |  | √ | √ | √ | |||
| hsa-miR-378c | 71 | 8.8–9.3 |  | √ | √ | √ | |||
| hsa-miR-10397-5p | 72 | 6.9–10.0 |  | √ | √ | √ | |||
| hsa-miR-584-3p | 72 | 7.3–9.8 |  | √ | √ | ||||
| hsa-miR-3085-3p | 71 | 8.8–9.9 |  | √ | √ | √ | |||
| hsa-miR-3191-3p | 70 | 7.4–8.5 |  | √ | √ | ||||
| hsa-miR-148b-3p | 72 | 8.2–9.8 |  | √ | √ | ||||
| hsa-miR-3529-3p | 69 | 9.0 |  | √ | |||||
| hsa-miR-3682-5p | 68 | 9.0 |  | √ | √ |
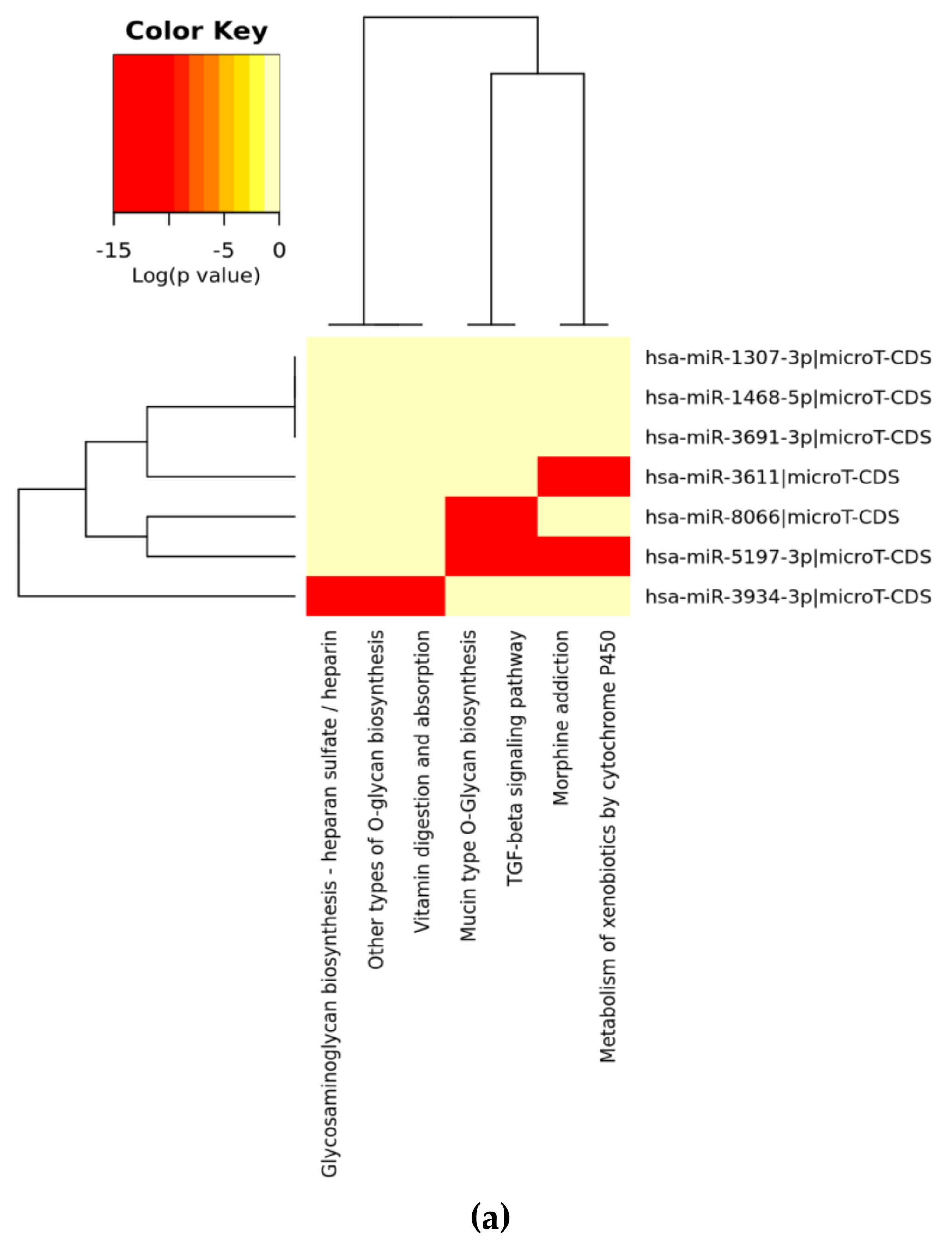
| KEGG Pathway (A) | p-Value | #genes | #miRNAs |
| Mucin type O-Glycan biosynthesis | 2.52 × 10−2 | 7 | 3 |
| TGF-beta signaling pathway | 4.96 × 10−1 | 12 | 4 |
| Morphine addiction | 0.0001128919 | 14 | 5 |
| Metabolism of xenobiotics by cytochrome P450 | 0.0002215491 | 5 | 2 |
| Other types of O-glycan biosynthesis | 0.0003646344 | 1 | 1 |
| Vitamin digestion and absorption | 0.001008222 | 2 | 1 |
| Glycosaminoglycan biosynthesis—heparan sulfate/heparin | 0.00385809 | 1 | 1 |
| GABAergic synapse | 0.01342039 | 13 | 4 |
| Cytokine-cytokine receptor interaction | 0.02096334 | 9 | 1 |
| Signaling pathways regulating pluripotency of stem cells | 0.180299 | 9 | 1 |
| Amphetamine addiction | 0.2150865 | 7 | 1 |
| Axon guidance | 0.2239648 | 22 | 3 |
| Hippo signaling pathway | 0.2278356 | 7 | 1 |
| Prolactin signaling pathway | 0.2284669 | 5 | 1 |
| mRNA surveillance pathway | 0.2795597 | 1 | 1 |
| Glycosphingolipid biosynthesis—lacto and neolacto series | 0.3157068 | 1 | 1 |
| Bile secretion | 0.4120997 | 1 | 1 |
| Circadian entrainment | 0.4608082 | 9 | 1 |
| N-Glycan biosynthesis | 0.488078 | 2 | 1 |
| Mismatch repair | 0.6174557 | 1 | 1 |
| Drug metabolism—cytochrome P450 | 0.7063987 | 6 | 1 |
| Glutamatergic synapse | 0.7319762 | 6 | 1 |
| Glycosaminoglycan degradation | 0.7395672 | 2 | 1 |
| Antigen processing and presentation | 0.7591685 | 1 | 1 |
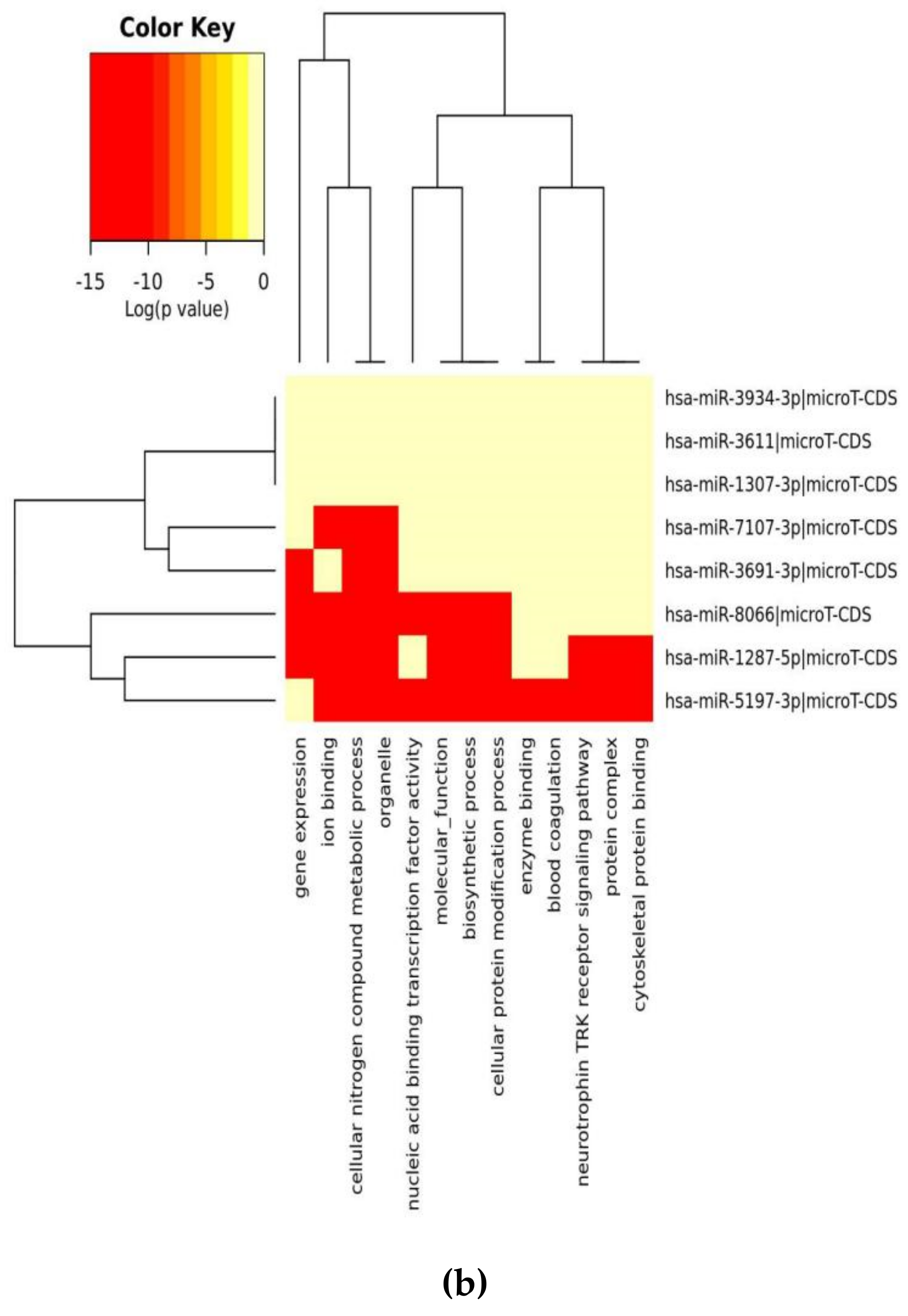
| GO Category (B) | p-Value | #genes | #miRNAs |
| organelle | 1 × 10−38 | 848 | 6 |
| cellular nitrogen compound metabolic process | 1 × 10−12 | 414 | 7 |
| ion binding | 8 × 10−8 | 495 | 7 |
| biosynthetic process | 3 × 10−7 | 351 | 7 |
| nucleic acid binding transcription factor activity | 4 × 10−2 | 115 | 6 |
| cellular protein modification process | 2 × 101 | 205 | 7 |
| molecular_function | 5 × 103 | 1303 | 7 |
| cellular_component | 1 × 105 | 1312 | 7 |
| enzyme binding | 2 × 105 | 119 | 5 |
| gene expression | 3 × 105 | 54 | 6 |
| protein binding transcription factor activity | 1 × 106 | 52 | 5 |
| blood coagulation | 0.000176061599974 | 44 | 6 |
| protein complex | 0.00115693944276 | 290 | 5 |
| post-translational protein modification | 0.00185490003064 | 19 | 5 |
| neurotrophin TRK receptor signaling pathway | 0.00197464302174 | 24 | 5 |
| synaptic transmission | 0.00204631087649 | 42 | 5 |
| cellular protein metabolic process | 0.00275618650845 | 39 | 5 |
| small molecule metabolic process | 0.00275618650845 | 170 | 7 |
| cytoskeletal protein binding | 0.00396272124679 | 68 | 4 |
| cell-cell signaling | 0.00396272124679 | 60 | 5 |
| transcription, DNA-templated | 0.00450420995446 | 208 | 6 |
| symbiosis, encompassing mutualism through parasitism | 0.0140041886634 | 41 | 5 |
| catabolic process | 0.0141620388146 | 142 | 6 |
| Fc-epsilon receptor signaling pathway | 0.0222360628043 | 15 | 6 |
| cellular component assembly | 0.02375306792 | 99 | 5 |
| transcription initiation from RNA polymerase II promoter | 0.0250016205995 | 24 | 5 |
| nucleoplasm | 0.0335128910566 | 92 | 6 |
| platelet activation | 0.0350801245107 | 20 | 5 |
| positive regulation of telomere maintenance via telomerase | 0.0370638891992 | 3 | 3 |
| RNA polymerase II core promoter proximal region sequence-specific DNA binding transcription factor activity involved in positive regulation of transcription | 0.0448871926331 | 32 | 5 |
| O-glycan processing | 0.0449415771561 | 8 | 5 |
| miRs | Alignment | Wuhan/China | Italy | Spain | France | England | USA | India |
|---|---|---|---|---|---|---|---|---|
| n = 28 | n = 44 | n = 133 | n = 104 | n = 104 | n = 104 | n = 34 | ||
| hsa-miR-8066 | ccaaaagaucacauug | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-5197-3p | auucgaagacccagucccuacuu | 0 | 0 | 0 | 0 | 0 | 0.9% | 0 |
| hsa-miR-3611 | ugagaagcaagaaauucuu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-3934-3p | ucagguuggacagcugg | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-1307-3p | accgaggccacgcggagu | 3.5% | 2.2% | 8.27% | 1.92% | 2.88% | 2.88% | 38.23% |
| hsa-miR-3691-3p | gagauguugacacagacuuugu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-1468-5p | cucaguuugccuguuu | 0 | 0 | 2.25% | 0.96% | 0 | 0 | 8.83% |
| hsa-miR-3120-5p | uguagaggaggcaaagacag | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-3914 | caucucacuugcugguuccu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-3672 | ugagucucauggaaaaca | 0 | 0 | 0.75% | 0.96% | 0 | 0 | 0 |
| hsa-miR-378c | acugggcauugauuuagaugagugg | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-7107-3p | ccaaaaagagaaagucaaca | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-1287-5p | acucaaaccacugaaacagc | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-10397-5p | uucuucaccugaugcugu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-584-3p | gccugguuugccuggcac | 0 | 0 | 0.75% | 0 | 0 | 0 | 0 |
| hsa-miR-3085-3p | ucuggcuguuauggcc | 0 | 0 | 0 | 0 | 0 | 0.96% | 0 |
| hsa-miR-3191-3p | cugucuauccaguugcgucacca | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-3529-3p | uggcagacgggcgauuuuguu | 0 | 0 | 0 | 0 | 0 | 0 | 2.94% |
| hsa-miR-3682-5p | auagcacaaguagauguag | 0 | 0 | 0 | 0 | 0 | 0.96% | 0 |
| hsa-miR-148b-3p | aaguucuaugaugcacag | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-129-2-3p | ugauuuuuguggaaagggcu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| WikiPathways | p-Adj |
| Photodynamic therapy-induced NF-kB survival signaling | 0 |
| IL-18 signaling pathway | 8.6 × 10−9 |
| miRNAs involvement in the immune response in sepsis | 2.4 × 10−8 |
| Cytokines and Inflammatory Response | 9.9 × 10−7 |
| Lung fibrosis | 2.5 × 10−6 |
| BioPlanet | p-Adj |
| Oncostatin M | 0 |
| Interleukin-1 regulation of extracellular matrix | 0 |
| Interleukin-5 regulation of apoptosis | 0 |
| TNF-alpha effects on cytokine activity, cell motility, and apoptosis | 0 |
| Immune system signaling by interferons, interleukins, prolactin, and growth hormones | 0 |
| KEGG | p-Adj |
| IL-17 signaling pathway | 1.3 × 10−9 |
| TNF signaling pathway | 1.6 × 10−9 |
| Legionellosis | 3.5 × 10−9 |
| Rheumatoid arthritis | 5.4 × 10−9 |
| Cytokine-cytokine receptor interaction | 6.6 × 10−9 |
| PANTHER | p-Adj |
| Plasminogen activating cascade | 0.00156 |
| Toll receptor signaling pathway | 0.00911 |
| CCKR signaling map ST | 0.02550 |
| Apoptosis signaling pathway | 0.10282 |
| Blood coagulation | 0.10433 |
| REACTOME | p-Adj |
| Interferon alpha/beta signaling | 1.6 × 10−9 |
| Interleukin-10 signaling | 2.5 × 10−9 |
| Interleukin-4 and Interleukin-13 signaling | 2.4 × 10−7 |
| Formation of the cornified envelope | 1.3 × 10−5 |
| Chemokine receptors bind chemokines | 0.00047 |
| Small Molecule Pathway DB | p-Adj |
| CD40L Signalling Pathway | 0.25268 |
| NF-kB Signaling Pathway | 0.25268 |
| Toll-Like Receptor Pathway 2 | 0.25268 |
| Capecitabine Metabolism Pathway | 0.25268 |
| Capecitabine Action Pathway | 0.25268 |
| BIOCYC | p-Adj |
| vitamin D3 biosynthesis | 0.03597 |
| guanosine nucleotides degradation | 0.03597 |
| retinoate biosynthesis II | 0.03597 |
| guanosine nucleotides degradation III | 0.03597 |
| adenosine nucleotides degradation II | 0.03597 |
| Pathway Interaction DB | p-Adj |
| Validated transcriptional targets of AP1 family members Fra1 and Fra2 | 3.8 × 10−5 |
| IL23-mediated signaling events | 0.00050 |
| CD40/CD40L signaling | 0.02539 |
| Glucocorticoid receptor regulatory network | 0.02603 |
| LPA receptor mediated events | 0.04171 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arisan, E.D.; Dart, A.; Grant, G.H.; Arisan, S.; Cuhadaroglu, S.; Lange, S.; Uysal-Onganer, P. The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities. Viruses 2020, 12, 614. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060614
Arisan ED, Dart A, Grant GH, Arisan S, Cuhadaroglu S, Lange S, Uysal-Onganer P. The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities. Viruses. 2020; 12(6):614. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060614
Chicago/Turabian StyleArisan, Elif Damla, Alwyn Dart, Guy H. Grant, Serdar Arisan, Songul Cuhadaroglu, Sigrun Lange, and Pinar Uysal-Onganer. 2020. "The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities" Viruses 12, no. 6: 614. https://0-doi-org.brum.beds.ac.uk/10.3390/v12060614