Recombination Analysis of Non-Poliovirus Members of the Enterovirus C Species: Restriction of Recombination Events to Members of the Same 3DPol Cluster

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Processing and Full Length Sequencing
2.2. Database Construction
2.3. Detecting Phylogenetic Violation
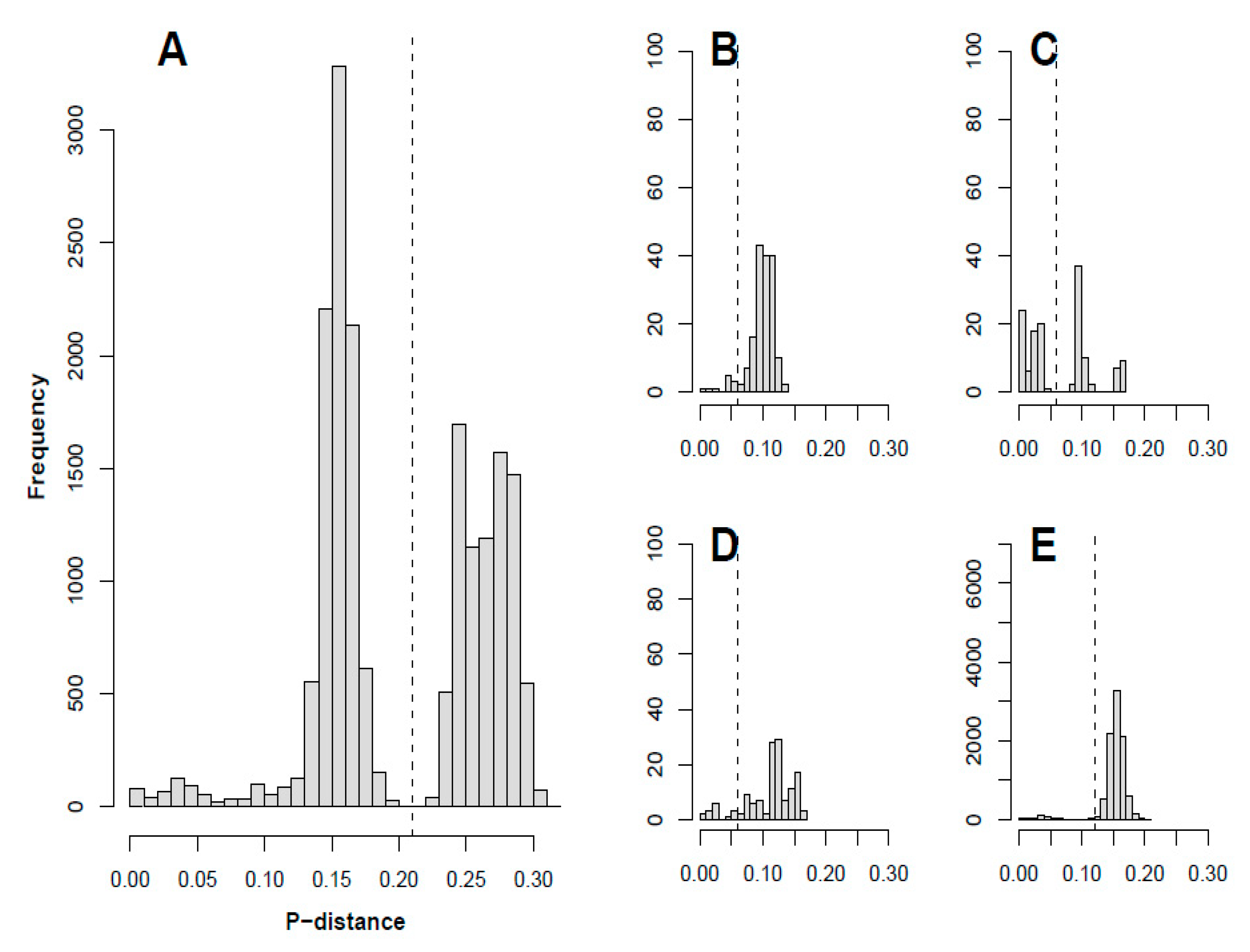
2.4. Plotting p-Distance and Determining Recombinant Forms
3. Results
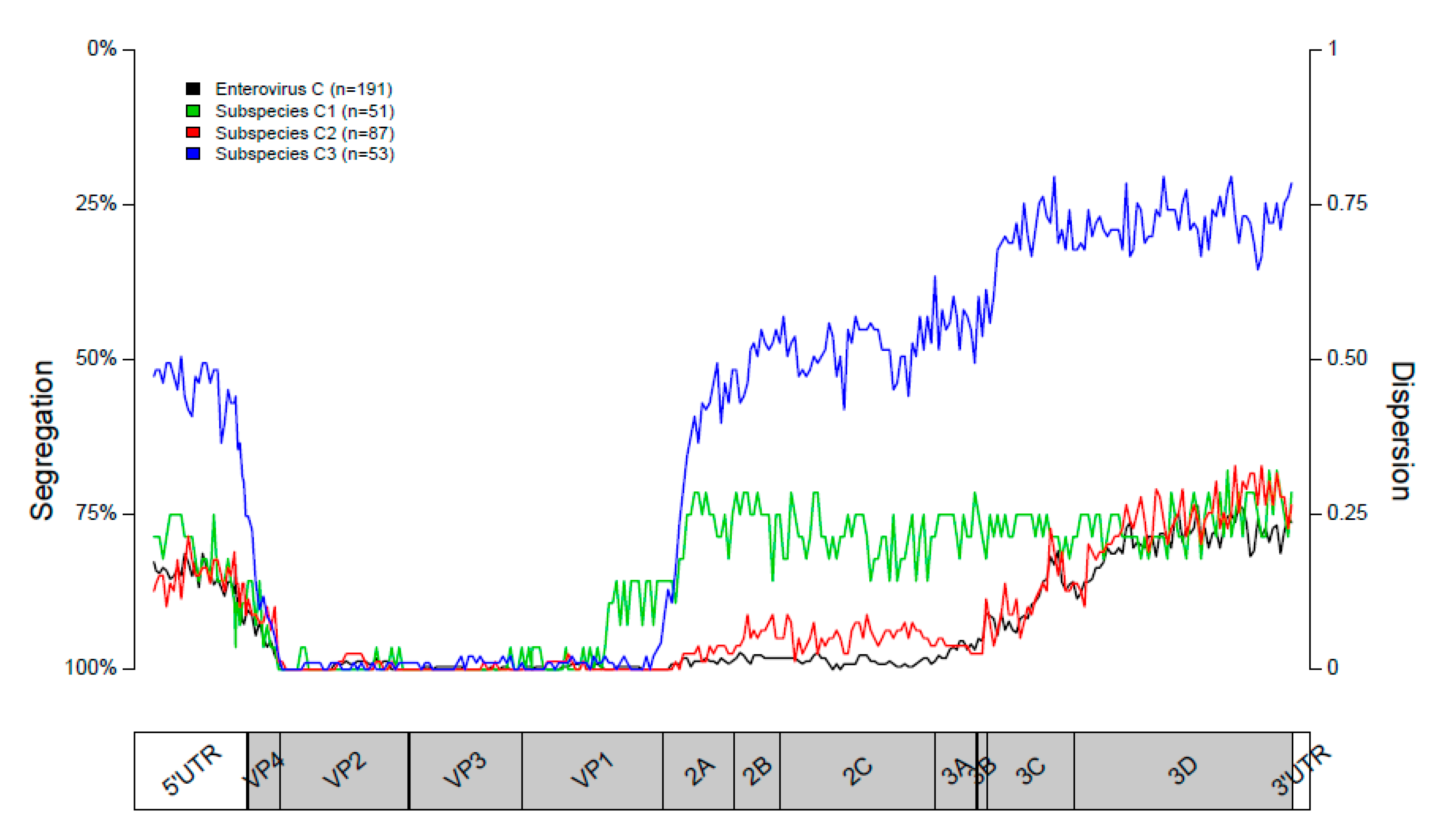
3.1. Segregation of Sequences in the VP1 Genomic Region
3.2. Segregation of Sequences in the 2C Genomic Region
3.3. Segregation of Sequences in the 3DPol Genomic Region
3.4. Segregation of Sequences Across the Genome
3.5. Recombinant Forms
3.6. Clustering Associated with Site of Origin
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Suresh, S.; Rawlinson, W.D.; Andrews, P.I.; Stelzer-Braid, S. Global epidemiology of nonpolio enteroviruses causing severe neurological complications: A systematic review and meta-analysis. Rev. Med. Virol. 2019, e2082. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.L.; Lau, J.S.Y.; Goh, S.M.; Wilson, H.L.; Catton, M.; Korman, T.M. Myocarditis caused by human parechovirus in adult. Emerg. Infect Dis. 2017, 23, 1571–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaidis, M.; Mimouli, K.; Kyriakopoulou, Z.; Tsimpidis, M.; Tsakogiannis, D.; Markoulatos, P.; Amoutzias, G.D. Large-scale genomic analysis reveals recurrent patterns of intertypic recombination in human enteroviruses. Virology 2019, 526, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Welch, J. Frequency and dynamics of recombination within different species of human enteroviruses. J. Virol. 2006, 80, 483–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakopoulou, Z.; Pliaka, V.; Amoutzias, G.D.; Markoulatos, P. Recombination among human non-polio enteroviruses: Implications for epidemiology and evolution. Virus Genes 2015, 50, 177–188. [Google Scholar] [CrossRef]
- Runckel, C.; Westesson, O.; Andino, R.; DeRisi, J.L. Identification and manipulation of the molecular determinants influencing poliovirus recombination. PLoS Pathog. 2013, 9, e1003164. [Google Scholar] [CrossRef]
- Smura, T.; Blomqvist, S.; Vuorinen, T.; Ivanova, O.; Samoilovich, E.; Al-Hello, H.; Savolainen-Kopra, C.; Hovi, T.; Roivainen, M. Recombination in the evolution of enterovirus C species sub-group that contains types CVA-21, CVA-24, EV-C95, EV-C96 and EV-C99. PLoS ONE 2014, 9, e94579. [Google Scholar] [CrossRef]
- Bessaud, M.; Joffret, M.L.; Holmblat, B.; Razafindratsimandresy, R.; Delpeyroux, F. Genetic relationship between cocirculating Human enteroviruses species C. PLoS ONE 2011, 6, e24823. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Kroes, A.C.M.; Lukashev, A.; Muir, P.; Odoom, J.; Roivainen, M.; et al. Evolutionary dynamics and temporal/geographical correlates of recombination in the human enterovirus echovirus types 9, 11, and 30. J. Virol. 2010, 84, 9292–9300. [Google Scholar] [CrossRef] [Green Version]
- Cabrerizo, M.; Trallero, G.; Simmonds, P. Recombination and evolutionary dynamics of human echovirus 6. J. Med. Virol. 2014, 86, 857–864. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Lashkevich, V.A.; Ivanova, O.E.; Koroleva, G.A.; Hinkkanen, A.E.; Ilonen, J. Recombination in circulating human enterovirus B: Independent evolution of structural and non-structural genome regions. J. Gen. Virol. 2005, 86 Pt 12, 3281–3290. [Google Scholar] [CrossRef]
- McWilliam Leitch, E.C.; Bendig, J.; Cabrerizo, M.; Cardosa, J.; Hyypia, T.; Ivanova, O.E.; Kelly, A.; Kroes, A.C.M.; Lukashev, A.; MacAdam, A.; et al. Transmission networks and population turnover of echovirus 30. J. Virol. 2009, 83, 2109–2118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Koike, S.; Kroes, A.C.M.; Lukashev, A.; Perera, D.; Roivainen, M.; et al. The association of recombination events in the founding and emergence of subgenogroup evolutionary lineages of human enterovirus 71. J. Virol. 2012, 86, 2676–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puenpa, J.; Vongpunsawad, S.; Osterback, R.; Waris, M.; Eriksson, E.; Albert, J.; Kroes, A.C.M.; Lukashev, A.; Perera, D.; Roivainen, M. Molecular epidemiology and the evolution of human coxsackievirus A6. J. Gen. Virol. 2016, 97, 3225–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, A.N.; Shumilina, E.Y.; Belalov, I.S.; Ivanova, O.E.; Eremeeva, T.P.; Reznik, V.I.; Trotsenko, O.E.; Drexler, I.F.; Drostenet, C. Recombination strategies and evolutionary dynamics of the Human enterovirus A global gene pool. J. Gen. Virol. 2014, 95 Pt 4, 868–873. [Google Scholar] [CrossRef]
- van der Sanden, S.; van Eek, J.; Martin, D.P.; van der Avoort, H.; Vennema, H.; Koopmans, M. Detection of recombination breakpoints in the genomes of human enterovirus 71 strains isolated in the Netherlands in epidemic and non-epidemic years, 1963–2010. Infect Genet Evol. 2011, 11, 886–894. [Google Scholar] [CrossRef]
- Adeniji, J.A.; Faleye, T.O. Enterovirus, C. strains circulating in Nigeria and their contribution to the emergence of recombinant circulating vaccine-derived polioviruses. Arch. Virol. 2015, 160, 675–683. [Google Scholar] [CrossRef]
- Arita, M.; Zhu, S.L.; Yoshida, H.; Yoneyama, T.; Miyamura, T.; Shimizu, H. A Sabin 3-derived poliovirus recombinant contained a sequence homologous with indigenous human enterovirus species C in the viral polymerase coding region. J. Virol. 2005, 79, 12650–12657. [Google Scholar] [CrossRef] [Green Version]
- Combelas, N.; Holmblat, B.; Joffret, M.L.; Colbere-Garapin, F.; Delpeyroux, F. Recombination between poliovirus and coxsackie A viruses of species C: A model of viral genetic plasticity and emergence. Viruses 2011, 3, 1460–1484. [Google Scholar] [CrossRef] [Green Version]
- Mbaeyi, C.; Alleman, M.M.; Ehrhardt, D.; Wiesen, E.; Burns, C.C.; Liu, H.; Ewetola, R.; Seakamela, L.; Mdodo, R.; Ndoutabe, M.; et al. Update on vaccine-derived poliovirus outbreaks—Democratic Republic of the Congo and Horn of Africa, 2017–2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 225–230. [Google Scholar] [CrossRef]
- Mbaeyi, C.; Wadood, Z.M.; Moran, T.; Mjourn, A.F.; Stehling-Ariza, T.; Nikulin, J.; Al Safadi, M.; Iber, J.; Zomahoun, L.; Abourshaid, N.; et al. Strategic Response to an Outbreak of Circulating Vaccine-Derived Poliovirus Type 2—Syria, 2017–2018. MMWR Morb. Mortal. Wkly Rep. 2018, 67, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Adamu, U.S.; Archer, W.R.; Braka, F.; Damisa, E.; Siddique, A.; Baig, S.; Higgins, J.; Etapelong Sume, G.; Banda, R.; Kipkoech Korir, C.; et al. Progress Toward Poliomyelitis Eradication—Nigeria, January 2018–May 2019. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 642–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessaud, M.; Joffret, M.L.; Blondel, B.; Delpeyroux, F. Exchanges of genomic domains between poliovirus and other cocirculating species C enteroviruses reveal a high degree of plasticity. Sci. Rep. 2016, 6, 38831. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, L.; Van Der Sanden, S.M.G.; Calis, J.C.J.; Bruning, A.H.L.; Wang, S.; Wildenbeest, J.G.; Rebers, S.P.H.; Phiri, K.S.; Westerhius, B.M.; Van Hensbroek, M.B.; et al. High frequency of Polio-like Enterovirus C strains with differential clustering of CVA-13 and EV-C99 subgenotypes in a cohort of Malawian children. Arch. Virol. 2018, 163, 2645–2653. [Google Scholar] [CrossRef] [Green Version]
- Cabrerizo, M.; Trallero, G.; Echevarria, J.E.; Moreno-Docon, A.; Pena, M.J.; Perez-Ruiz, M.; Avellón, A. Molecular characterization of enteroviruses associated with neurological infections in Spain, 2008. J. Med. Virol. 2013, 85, 1975–1977. [Google Scholar] [CrossRef]
- Bubba, L.; Martinelli, M.; Pellegrinelli, L.; Primache, V.; Tanzi, E.; Pariani, E.; Sandro, B. A 4-year Study on Epidemiologic and Molecular Characteristics of Human Parechoviruses and Enteroviruses Circulating in Children Younger Than 5 Years in Northern Italy. Pediatr. Infect. Dis. J. 2017, 36, 13–19. [Google Scholar] [CrossRef]
- Cristanziano, V.D.; Bottcher, S.; Diedrich, S.; Timmen-Wego, M.; Knops, E.; Lubke, N.; Kaiser, R.; Pfister, H.; Kabore, Y.; D’Alfonso, R. Detection and characterization of enteroviruses and parechoviruses in healthy people living in the South of Cote d’Ivoire. J. Clin. Virol. 2015, 71, 40–43. [Google Scholar] [CrossRef]
- Abedi, G.R.; Watson, J.T.; Nix, W.A.; Oberste, M.S.; Gerber, S.I. Enterovirus and Parechovirus Surveillance—United States, 2014–2016. MMWR Morb. Mortal Wkly. Rep. 2018, 67, 515–518. [Google Scholar] [CrossRef]
- Abedi, G.R.; Watson, J.T.; Pham, H.; Nix, W.A.; Oberste, M.S.; Gerber, S.I. Enterovirus and human parechovirus surveillance—United States, 2009–2013. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 940–943. [Google Scholar] [CrossRef]
- Jeong, E.J.; Lee, J.H.; Kim, M.S.; Bae, G.R.; Jung, C.; Lee, K.; Choi, S.-M.; Kim, D.-K.; Lee, D.-S.; Kim, W.-D.; et al. Molecular characterization of enteroviruses detected in Gyeong-Ju and Po-Hang provinces of Korea in 2003. Arch. Virol. 2010, 155, 1707–1712. [Google Scholar] [CrossRef]
- Kowada, K.; Takeuchi, K.; Hirano, E.; Toho, M.; Sada, K. Development of a multiplex real-time PCR assay for detection of human enteric viruses other than norovirus using samples collected from gastroenteritis patients in Fukui Prefecture, Japan. J. Med. Virol. 2018, 90, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Shukla, D.; Srivastava, S.; Idris, M.Z.; Dhole, T.N. High frequency of enterovirus serotype circulation in a densely populated area of India. J. Infect. Dev. Ctries. 2013, 7, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilsker, M.; Moosa, Y.; Nooij, S.; Fonseca, V.; Ghysens, Y.; Dumon, K.; Pauwels, R.; Alcantra, L.C.; Eynden, E.V.; Vandamme, A.-M.; et al. Genome Detective: An automated system for virus identification from high-throughput sequencing data. Bioinformatics 2019, 35, 871–873. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Lam, T.T.; Zhu, H.; Guan, Y. Two methods for mapping and visualizing associated data on phylogeny using Ggtree. Mol. Biol. Evol. 2018, 35, 3041–3043. [Google Scholar] [CrossRef]
- Team RC. R: A Language and Environment for Statistical Computing. Vienna, Austria, 2019. Available online: https://www.R-project.org/ (accessed on 30 April 2020).
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 114. [Google Scholar] [CrossRef] [Green Version]
- Benschop, K.S.; de Vries, M.; Minnaar, R.P.; Stanway, G.; van der Hoek, L.; Wolthers, K.C.; Simmond, P. Comprehensive full-length sequence analyses of human parechoviruses: Diversity and recombination. J. Gen. Virol. 2010, 91 Pt 1, 145–154. [Google Scholar] [CrossRef]
- Calvert, J.; Chieochansin, T.; Benschop, K.S.; McWilliam Leitch, E.C.; Drexler, J.F.; Grywna, K.; da Costa Ribeiro, H., Jr.; Drosten, C.; Harvala, H.; Poovorawan, Y.; et al. Recombination dynamics of human parechoviruses: Investigation of type-specific differences in frequency and epidemiological correlates. J. Gen Virol. 2010, 91 Pt 5, 1229–1238. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Drexler, J.F.; Kotova, V.O.; Amjaga, E.N.; Reznik, V.I.; Gmyl, A.P.; da Costa Riberiro, H.J.; Drosten, C.; Harvala, H.; Poovorawan, Y.; et al. Novel Serotypes 105 and 116 are members of distinct subgroups of human enterovirus C. J. Gen Virol. 2012, 93 Pt 11, 2357–2362. [Google Scholar] [CrossRef]
- Simmonds, P. Recombination and selection in the evolution of picornaviruses and other Mammalian positive-stranded RNA viruses. J. Virol. 2006, 80, 11124–11140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, P. SSE: A nucleotide and amino acid sequence analysis platform. BMC Res. Notes. 2012, 5, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokarz, R.; Haq, S.; Sameroff, S.; Howie, S.R.; Lipkin, W.I. Genomic analysis of coxsackieviruses A1, A19, A22, enteroviruses 113 and 104: Viruses representing two clades with distinct tropism within enterovirus C. J. Gen. Virol. 2013, 94 Pt 9, 1995–2004. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brouwer, L.; Benschop, K.S.M.; Nguyen, D.; Kamau, E.; Pajkrt, D.; Simmonds, P.; Wolthers, K.C. Recombination Analysis of Non-Poliovirus Members of the Enterovirus C Species: Restriction of Recombination Events to Members of the Same 3DPol Cluster. Viruses 2020, 12, 706. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070706
Brouwer L, Benschop KSM, Nguyen D, Kamau E, Pajkrt D, Simmonds P, Wolthers KC. Recombination Analysis of Non-Poliovirus Members of the Enterovirus C Species: Restriction of Recombination Events to Members of the Same 3DPol Cluster. Viruses. 2020; 12(7):706. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070706
Chicago/Turabian StyleBrouwer, Lieke, Kimberley S.M. Benschop, Dung Nguyen, Everlyn Kamau, Dasja Pajkrt, Peter Simmonds, and Katja C. Wolthers. 2020. "Recombination Analysis of Non-Poliovirus Members of the Enterovirus C Species: Restriction of Recombination Events to Members of the Same 3DPol Cluster" Viruses 12, no. 7: 706. https://0-doi-org.brum.beds.ac.uk/10.3390/v12070706