The Phage-Encoded N-Acetyltransferase Rac Mediates Inactivation of Pseudomonas aeruginosa Transcription by Cleavage of the RNA Polymerase Alpha Subunit

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Manipulations and Bacteriophages
2.2. Protein Expression and Purification
2.3. Mass Spectrometry Identifications
2.4. Pulse Labeling with 3H-Uridine
2.5. In Vitro Transcription
2.6. Western Immunoblot Analyses
2.7. Acetyltransferase Substrate Profiling
3. Results
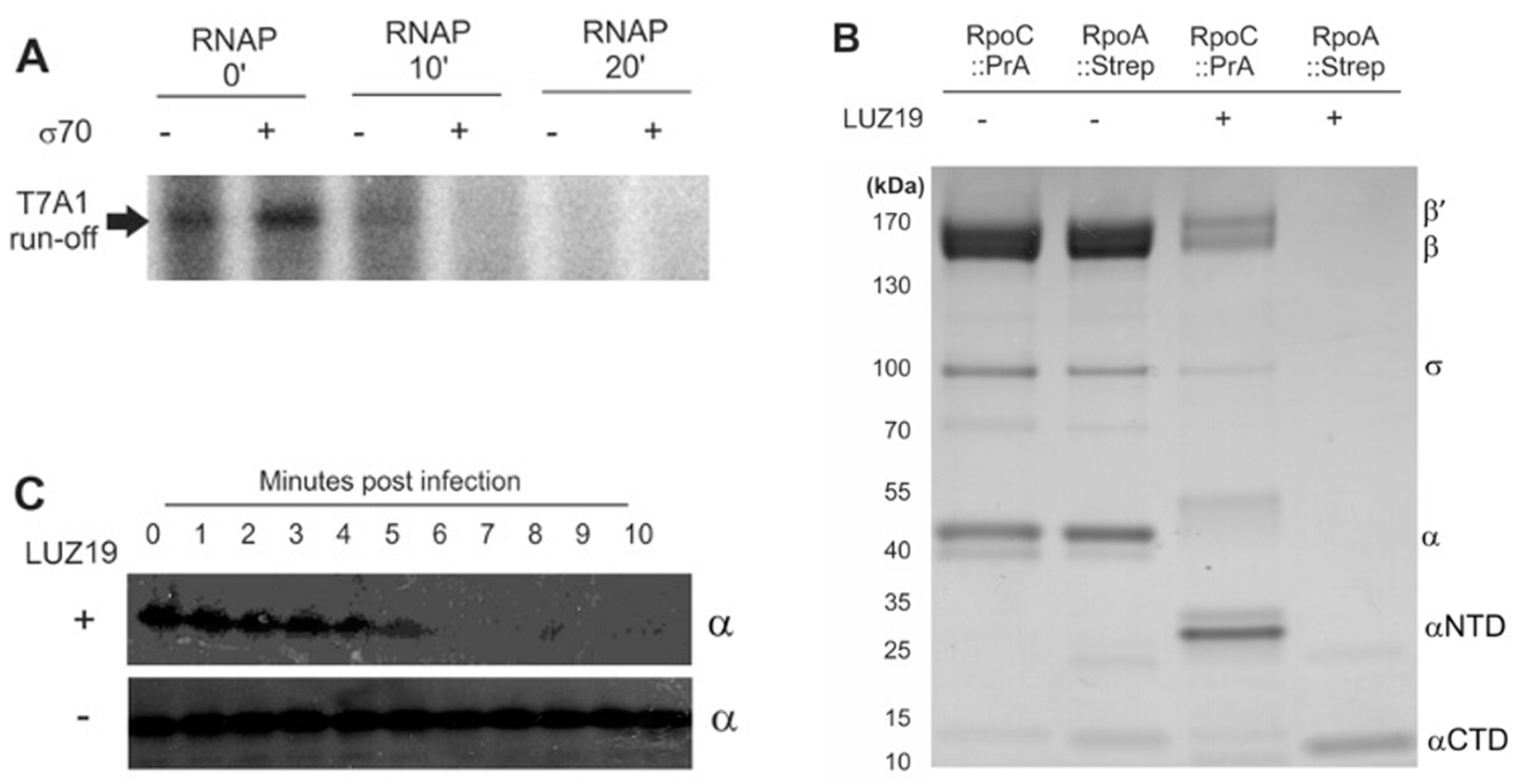
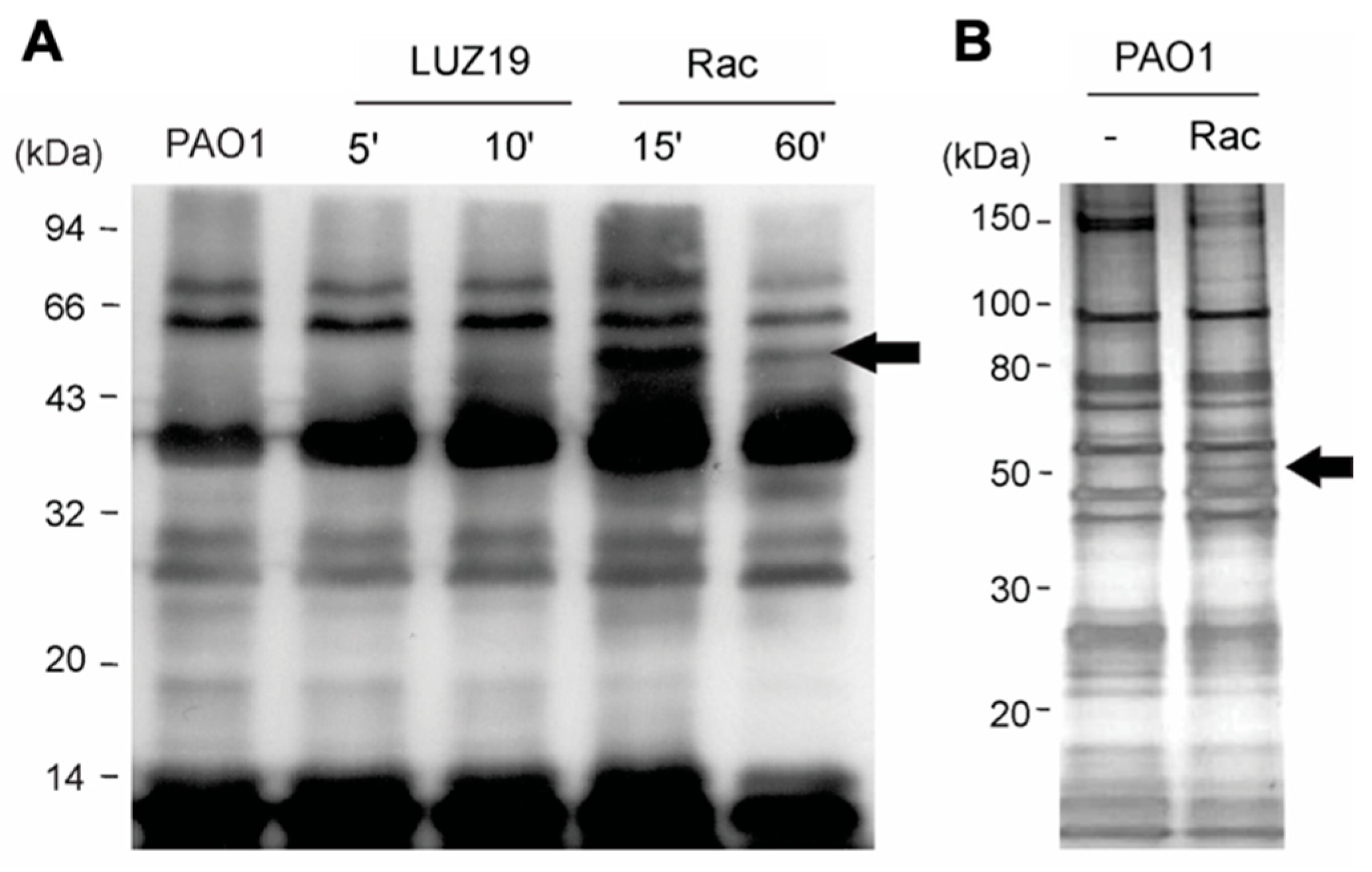
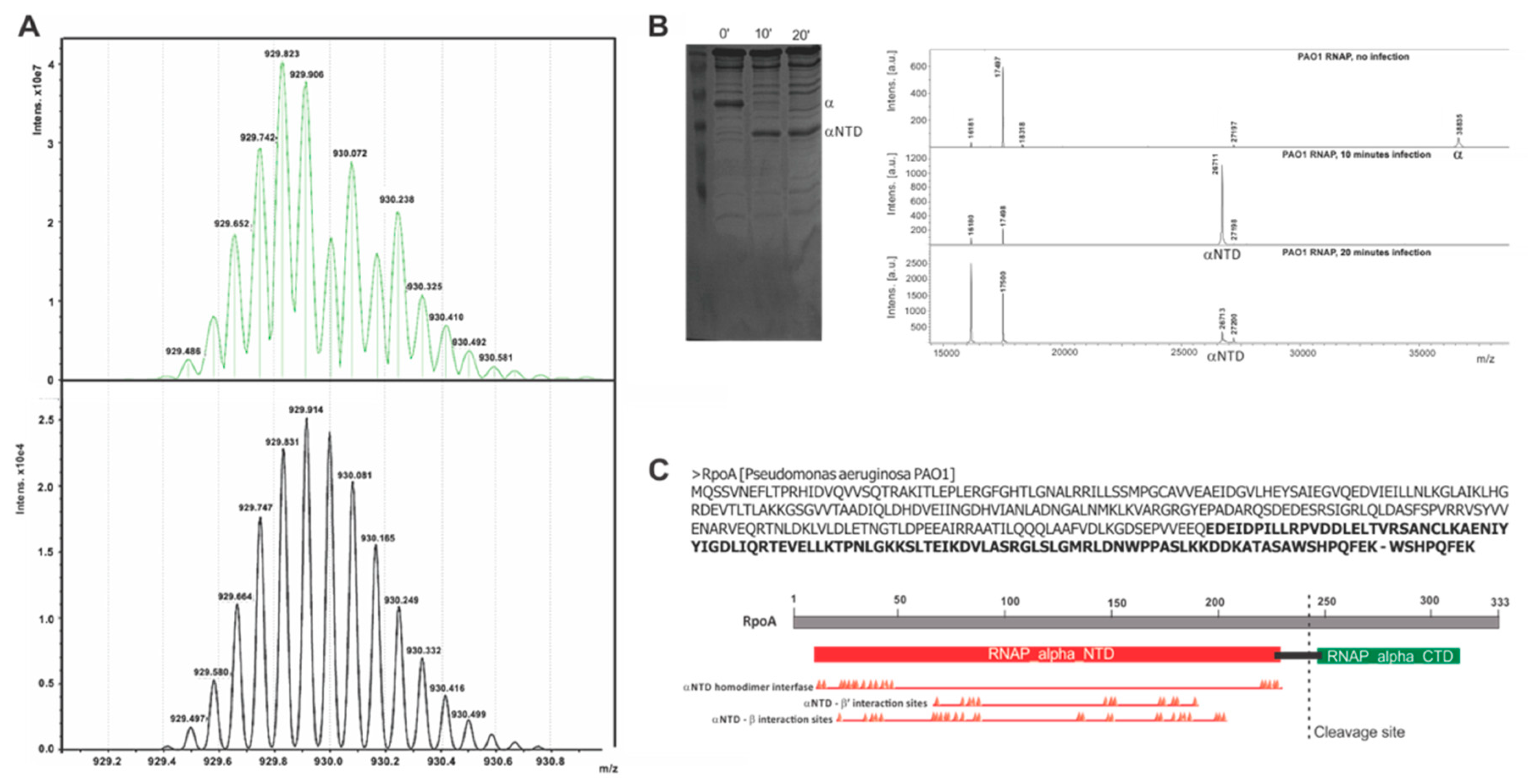
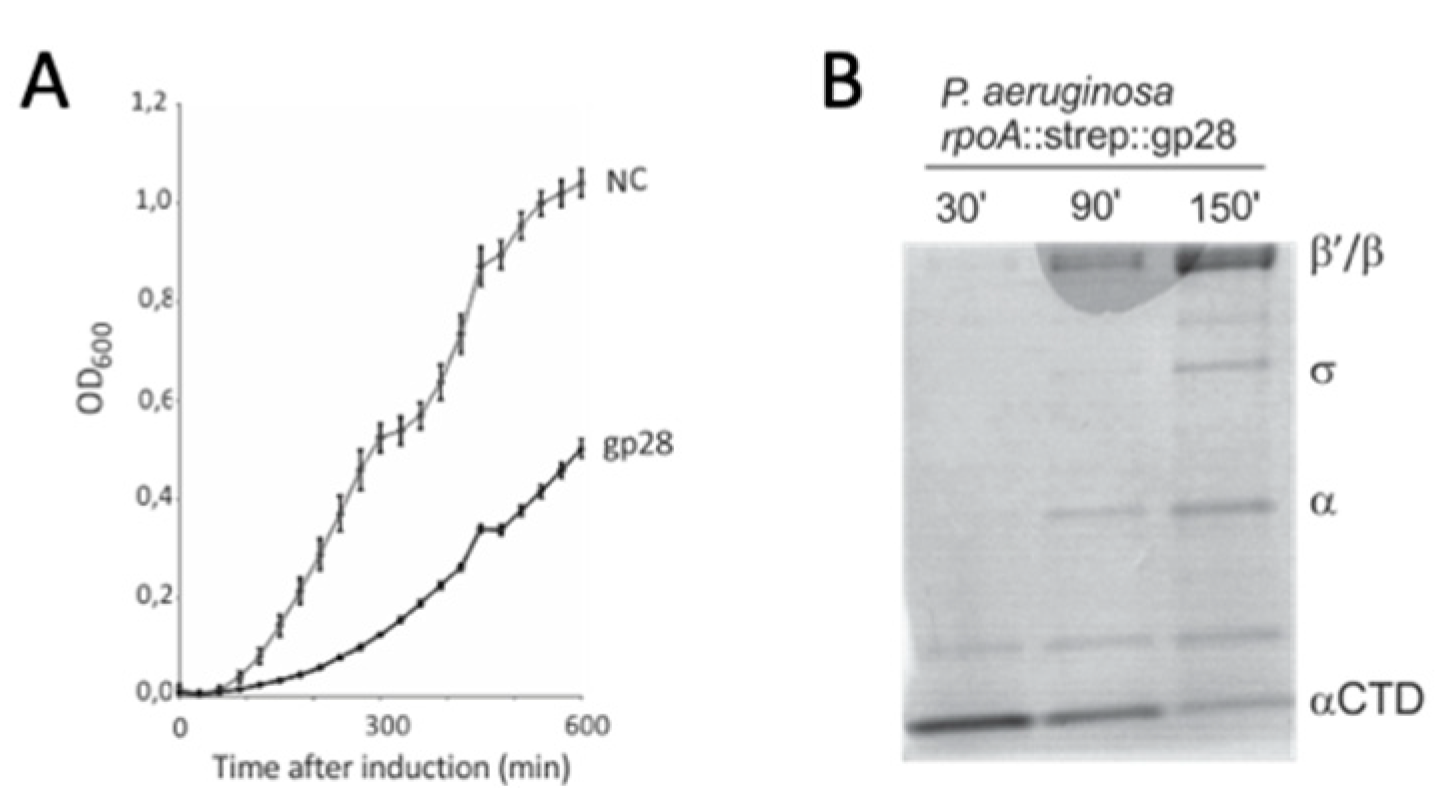
3.1. Bacteriophage LUZ19 Infection Leads to A Cleavage of the Host RNAP α Subunit
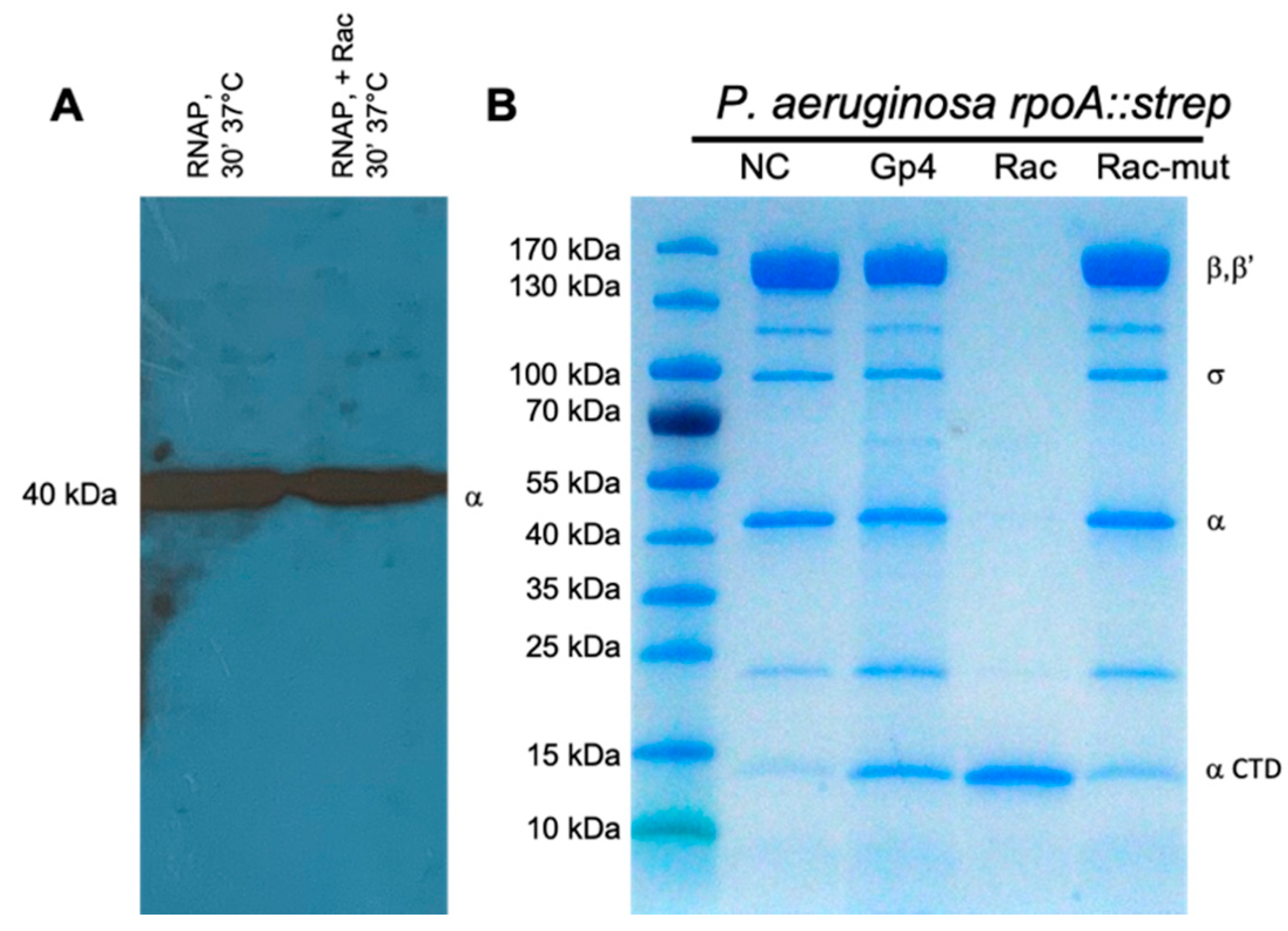
3.2. Gene Product 28 Provokes the RNAP Alpha Subunit Cleavage (Rac)
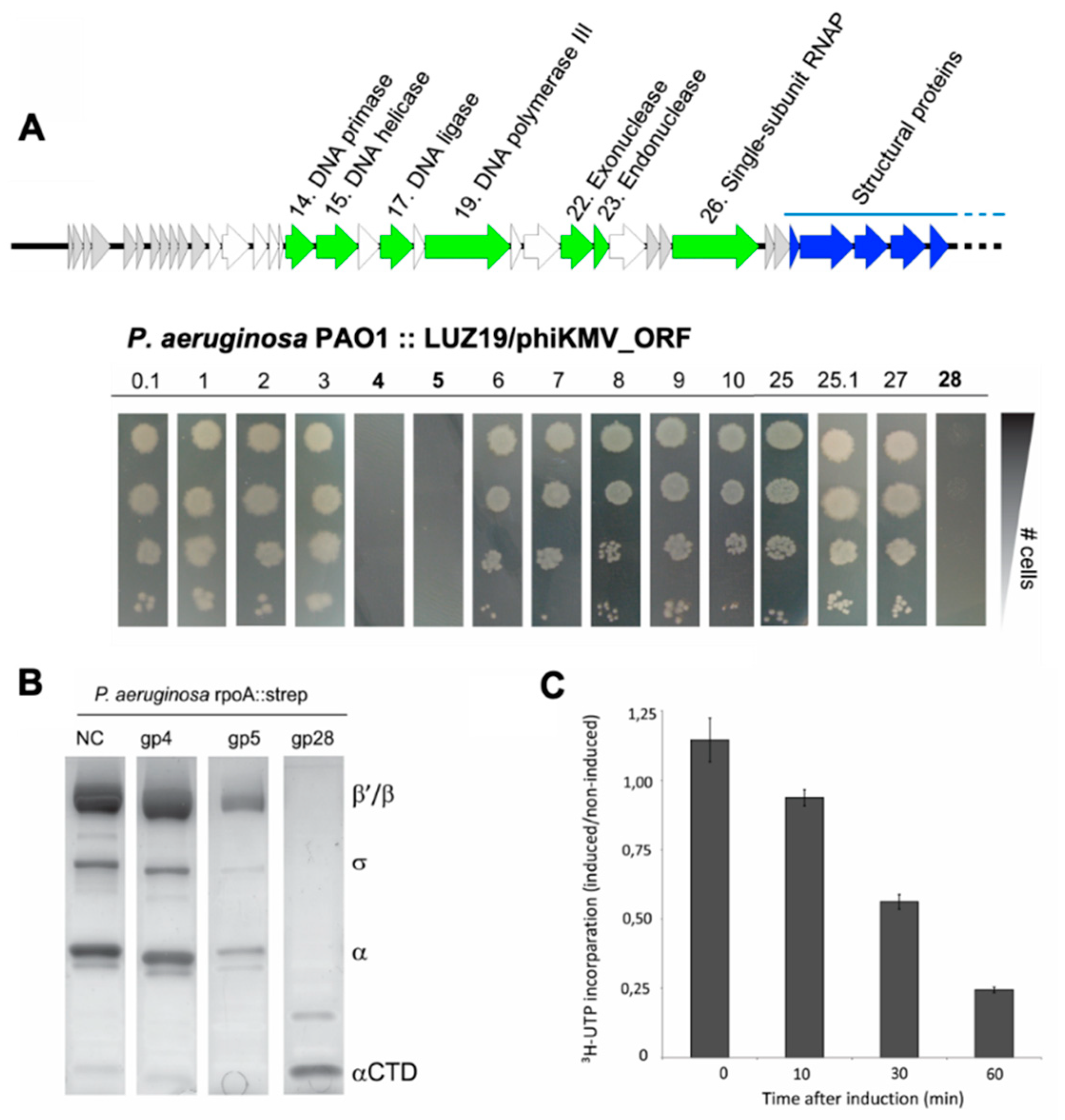
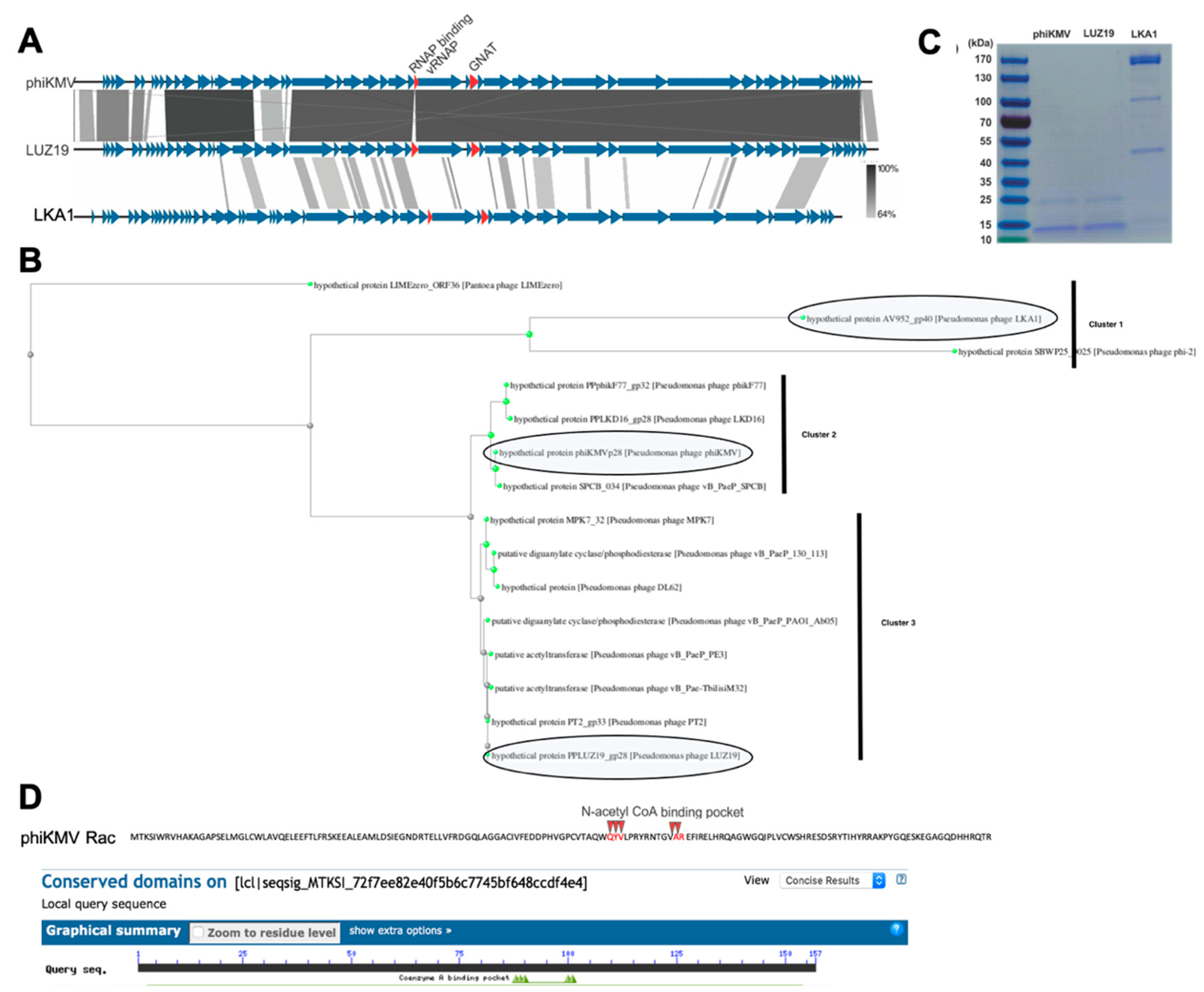
3.3. The RNAP α Subunit Cleavage Is Triggered by a Predicted Acetyltransferase Encoded by Many phiKMV-Related Viruses
4. Discussion
4.1. A Novel Mode of Phage-Induced Transcriptional Shutdown
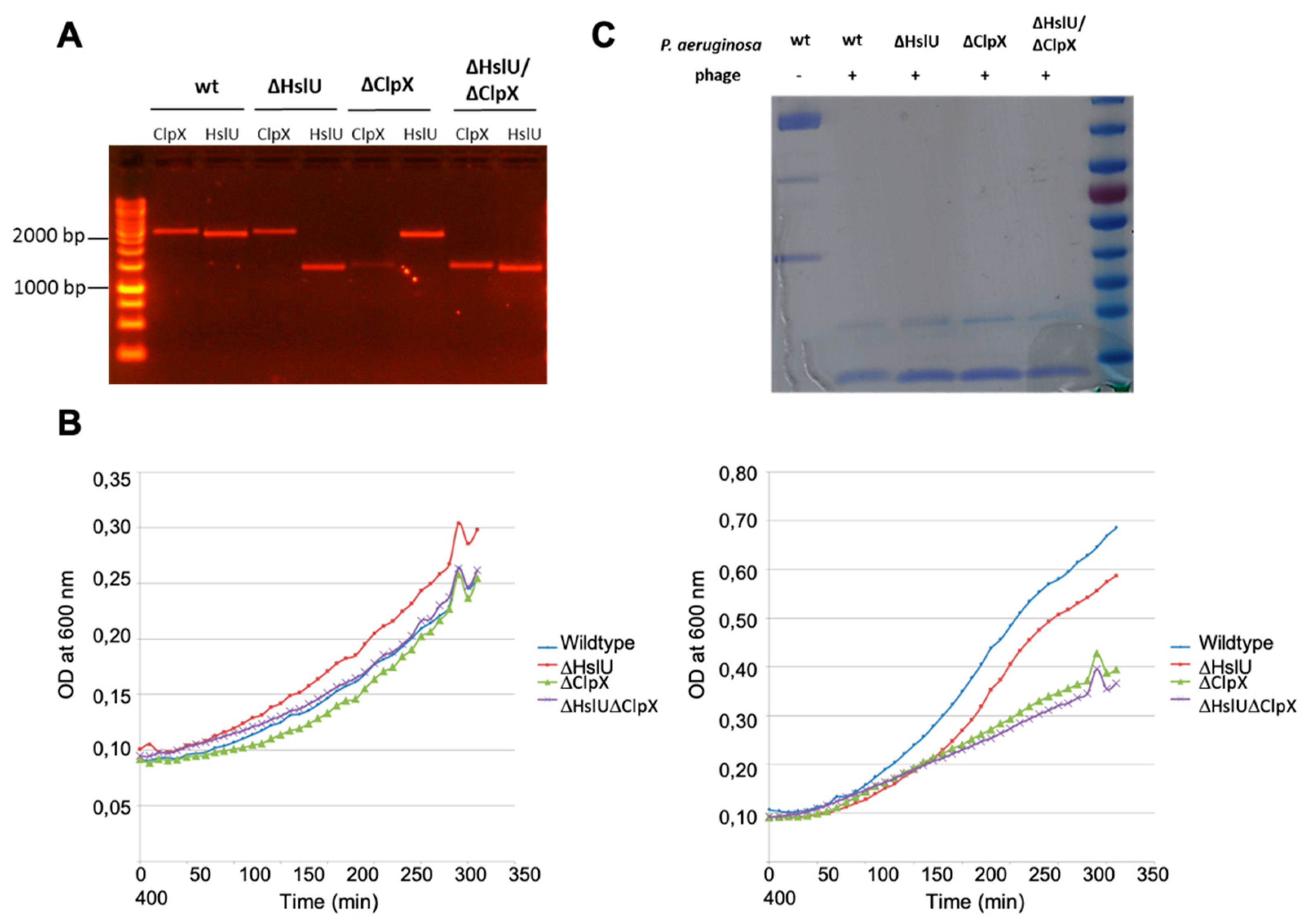
4.2. Consequences of α CTD Cleavage During Phage Infection
4.3. Posttranslational Control Mechanisms by Bacterial Viruses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hit | Query Cover | e-Value | Percent Identity | Accession |
|---|---|---|---|---|
| hypothetical protein PPLUZ19_gp28 (Pseudomonas phage LUZ19) | 100% | 4.00 × 10−120 | 100.00% | YP_001671973.1 |
| hypothetical protein PT2_gp33 (Pseudomonas phage PT2) | 100% | 2.00 × 10−119 | 100.00% | YP_002117812.1 |
| putative acetyltransferase (Pseudomonas phage vB_PaeP_PE3) | 100% | 4.00 × 10−119 | 99.36% | QHZ59694.1 |
| putative diguanylate cyclase/phosphodiesterase (Pseudomonas phage vB_PaeP_PAO1_Ab05) | 100% | 1.00 × 10−118 | 98.73% | YP_009125731.1 |
| putative acetyltransferase (Pseudomonas phage vB_Pae-TbilisiM32) | 100% | 2.00 × 10−118 | 99.36% | YP_006299952.1 |
| hypothetical protein MPK7_32 (Pseudomonas phage MPK7) | 100% | 2.00 × 10−118 | 98.09% | YP_008431340.1 |
| putative diguanylate cyclase/phosphodiesterase (Pseudomonas phage vB_PaeP_130_113) | 100% | 9.00 × 10−117 | 96.82% | AVX47641.1 |
| hypothetical protein (Pseudomonas phage DL62) | 100% | 1.00 × 10−115 | 96.18% | YP_009201889.1 |
| hypothetical protein phiKMVp28 (Pseudomonas phage phiKMV) | 100% | 2.00 × 10−113 | 92.99% | NP_877467.1 |
| hypothetical protein SPCB_034 (Pseudomonas phage vB_PaeP_SPCB) | 100% | 2.00 × 10−112 | 92.36% | QGJ86814.1 |
| hypothetical protein PPphikF77_gp32 (Pseudomonas phage phikF77) | 100% | 2.00 × 10−111 | 91.08% | YP_002727851.1 |
| hypothetical protein PPLKD16_gp28 (Pseudomonas phage LKD16) | 100% | 8.00 × 10−111 | 90.45% | YP_001522820.1 |
| hypothetical protein AV952_gp40 (Pseudomonas phage LKA1) | 73% | 1.00 × 10−21 | 37.39% | YP_001522880.1 |
| hypothetical protein SBWP25_0025 (Pseudomonas phage phi-2) | 68% | 6.00 × 10−15 | 31.78% | YP_003345490.1 |
| hypothetical protein LIMEzero_ORF36 (Pantoea phage LIMEzero) | 65% | 3.00 × 10−10 | 35.92% | YP_004539109.1 |
| Algorithm | Identification | Probability (%) | e-Value |
|---|---|---|---|
| PHYRE | Acyl-CoA N-acyltransferase (Nat) | 99.94 | 0.22 |
| HHPRED | Acetyltransferase (GNAT) | 90 | 1.8 × 10−24 |
| PFAM | Acetyltransferase 7 | NA | 0.0029 |
References
- Murakami, K.S. Structural biology of bacterial RNA polymerase. Biomolecules 2015, 5, 848–864. [Google Scholar] [CrossRef]
- Gourse, R.L.; Ross, W.; Gaal, T. UPs and downs in bacterial transcription initiation: The role of the alpha subunit of RNA polymerase in promoter recognition. Mol. Microbiol. 2000, 37, 687–695. [Google Scholar] [CrossRef]
- Ruff, E.F.; Thomas Record, M.; Artsimovitch, I. Initial events in bacterial transcription initiation. Biomolecules 2015, 5, 1035–1062. [Google Scholar] [CrossRef] [Green Version]
- Husnain, S.I.; Meng, W.; Busby, S.J.W.; Thomas, M.S. Escherichia coli can tolerate insertions of up to 16 amino acids in the RNA polymerase alpha subunit inter-domain linker. Biochim. Biophys. Acta 2004, 1678, 47–56. [Google Scholar] [CrossRef]
- Tiemann, B.; Depping, R.; Gineikiene, E.; Kaliniene, L.; Nivinskas, R.; Ruger, W. ModA and ModB, two ADP-ribosyltransferases encoded by bacteriophage T4: Catalytic properties and mutation analysis. J. Bacteriol. 2004, 186, 7262–7272. [Google Scholar] [CrossRef] [Green Version]
- Sommer, N.; Salniene, V.; Gineikiene, E.; Nivinskas, R.; Ruger, W. T4 early promoter strength probed in vivo with unribosylated and ADP-ribosylated Escherichia coli RNA polymerase: A mutation analysis. Microbiology 2000, 146 Pt 1, 2643–2653. [Google Scholar] [CrossRef] [Green Version]
- Tabib-Salazar, A.; Mulvenna, N.; Severinov, K.; Matthews, S.J.; Wigneshweraraj, S. Xenogeneic Regulation of the Bacterial Transcription Machinery. J. Mol. Biol. 2019, 431, 4078–4092. [Google Scholar] [CrossRef]
- De Smet, J.; Hendrix, H.; Blasdel, B.G.; Danis-Wlodarczyk, K.; Lavigne, R. Pseudomonas predators: Understanding and exploiting phage-host interactions. Nat. Rev. Microbiol. 2017, 15, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zheng, S.; Yang, J.S.; Chen, Y.; Cheng, Z. Comprehensive profiling of protein lysine acetylation in Escherichia coli. J. Proteome Res. 2013, 12, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Vellaichamy, A.; Wang, D.; Zamdborg, L.; Kelleher, N.L.; Huber, S.C.; Zhao, Y. Differential lysine acetylation profiles of Erwinia amylovora strains revealed by proteomics. J. Proteom. 2013, 79, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Yu, B.J.; Kim, J.A.; Lee, Y.-J.; Choi, S.-G.; Kang, S.; Pan, J.-G. The acetylproteome of Gram-positive model bacterium Bacillus subtilis. Proteomics 2013, 13, 1726–1736. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Yang, C.; Xiong, H.; Lin, Y.; Yao, J.; Li, H.; Xie, L.; Zhao, W.; Yao, Y.; et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010, 327, 1004–1007. [Google Scholar] [CrossRef] [Green Version]
- Gaviard, C.; Broutin, I.; Cosette, P.; Dé, E.; Jouenne, T.; Hardouin, J. Lysine Succinylation and Acetylation in Pseudomonas aeruginosa. J. Proteome Res. 2018, 17, 2449–2459. [Google Scholar] [CrossRef]
- Ouidir, T.; Cosette, P.; Jouenne, T.; Hardouin, J. Proteomic profiling of lysine acetylation in Pseudomonas aeruginosa reveals the diversity of acetylated proteins. Proteomics 2015, 15, 2152–2157. [Google Scholar] [CrossRef]
- Ouidir, T.; Kentache, T.; Hardouin, J. Protein lysine acetylation in bacteria: Current state of the art. Proteomics 2016, 16, 301–309. [Google Scholar] [CrossRef]
- Christensen, D.G.; Xie, X.; Basisty, N.; Byrnes, J.; McSweeney, S.; Schilling, B.; Wolfe, A.J. Post-translational Protein Acetylation: An elegant mechanism for bacteria to dynamically regulate metabolic functions. Front. Microbiol. 2019, 10, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Macek, B.; Forchhammer, K.; Hardouin, J.; Weber-Ban, E.; Grangeasse, C.; Mijakovic, I. Protein post-translational modifications in bacteria. Nat. Rev. Microbiol. 2019, 17, 651–664. [Google Scholar] [CrossRef]
- Reverdy, A.; Chen, Y.; Hunter, E.; Gozzi, K.; Chai, Y. Protein lysine acetylation plays a regulatory role in bacillus subtilis multicellularity. PLoS ONE 2018, 13, 1–25. [Google Scholar] [CrossRef]
- Thao, S.; Chen, C.S.; Zhu, H.; Escalante-Semerena, J.C. Nepsilon-lysine acetylation of a bacterial transcription factor inhibits Its DNA-binding activity. PLoS ONE 2010, 5, e15123. [Google Scholar] [CrossRef]
- Lima, B.P.; Thanh Huyen, T.T.; Basell, K.; Becher, D.; Antelmann, H.; Wolfe, A.J. Inhibition of acetyl phosphate-dependent transcription by an acetylatable lysine on RNA polymerase. J. Biol. Chem. 2012, 287, 32147–32160. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Sha, W.; Liu, Z.; Tang, T.; Liu, H.; Qin, L.; Cui, Z.; Chen, J.; Liu, F.; Zheng, R.; et al. Lysine acetylation of DosR regulates the hypoxia response of Mycobacterium tuberculosis. Emerg. Microbes Infect. 2018, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Choi, J.S.; Kim, J.S.; Cho, Y.H.; Roe, J.H. Lysine acetylation of the housekeeping sigma factor enhances the activity of the RNA polymerase holoenzyme. Nucleic Acids Res. 2020, 48, 2401–2411. [Google Scholar] [CrossRef]
- Lavigne, R.; Burkal’tseva, M.V.; Robben, J.; Sykilinda, N.N.; Kurochkina, L.P.; Grymonprez, B.; Jonckx, B.; Krylov, V.N.; Mesyanzhinov, V.V.; Volckaert, G. The genome of bacteriophage phiKMV, a T7-like virus infecting Pseudomonas aeruginosa. Virology 2003, 312, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Ceyssens, P.J.; Glonti, T.; Kropinski, N.M.; Lavigne, R.; Chanishvili, N.; Kulakov, L.; Lashkhi, N.; Tediashvili, M.; Merabishvili, M. Phenotypic and genotypic variations within a single bacteriophage species. Virol. J. 2011, 8, 134. [Google Scholar] [CrossRef] [Green Version]
- Klimuk, E.; Akulenko, N.; Makarova, K.S.; Ceyssens, P.J.; Volchenkov, I.; Lavigne, R.; Severinov, K. Host RNA polymerase inhibitors encoded by phiKMV-like phages of Pseudomonas. Virology 2013, 436, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Stover, C.K.; Pham, X.Q.; Erwin, A.L.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Van den Bossche, A.; Ceyssens, P.J.; De Smet, J.; Hendrix, H.; Bellon, H.; Leimer, N.; Wagemans, J.; Delattre, A.S.; Cenens, W.; Aertsen, A.; et al. Systematic Identification of Hypothetical Bacteriophage Proteins Targeting Key Protein Complexes of Pseudomonas aeruginosa. J. Proteome Res. 2014, 13, 4446–4456. [Google Scholar] [CrossRef]
- Jacobs, M.A.; Alwood, A.; Thaipisuttikul, I.; Spencer, D.; Haugen, E.; Ernst, S.; Will, O.; Kaul, R.; Raymond, C.; Levy, R.; et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 14339–14344. [Google Scholar] [CrossRef] [Green Version]
- Voisard, C.; Bull, C.T.; Keel, C.; Laville, J.; Maurhofer, M.; Schnider, U.; Défago, G.; Haas, D. Biocontrol of Root Diseases by Pseudomonas fluorescens CHA0: Current Concepts and Experimental Approaches. Mol. Ecol. Rhizosph. Microorg. 1994, 67–89. [Google Scholar] [CrossRef]
- Choi, K.H.; Gaynor, J.B.; White, K.G.; Lopez, C.; Bosio, C.M.; Karkhoff-Schweizer, R.R.; Schweizer, H.P. A Tn7-based broad-range bacterial cloning and expression system. Nat. Methods 2005, 2, 443–448. [Google Scholar] [CrossRef]
- Wagemans, J.; Blasdel, B.G.; Van den Bossche, A.; Uytterhoeven, B.; De Smet, J.; Paeshuyse, J.; Cenens, W.; Aertsen, A.; Uetz, P.; Delattre, A.S.; et al. Functional elucidation of antibacterial phage ORFans targeting Pseudomonas aeruginosa. Cell. Microbiol. 2014, 16, 1822–1835. [Google Scholar] [CrossRef]
- Wagemans, J.; Delattre, A.S.; Uytterhoeven, B.; De Smet, J.; Cenens, W.; Aertsen, A.; Ceyssens, P.J.; Lavigne, R.; Smet, J.D.; Cenens, W.; et al. Antibacterial phage ORFans of Pseudomonas aeruginosa phage LUZ24 reveal a novel MvaT inhibiting protein. Front. Microbiol. 2015, 6, 1242. [Google Scholar] [CrossRef] [Green Version]
- Ceyssens, P.J.; Lavigne, R.; Mattheus, W.; Chibeu, A.; Hertveldt, K.; Mast, J.; Robben, J.; Volckaert, G. Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: Establishment of the phiKMV subgroup within the T7 supergroup. J. Bacteriol. 2006, 188, 6924–6931. [Google Scholar] [CrossRef] [Green Version]
- Kashlev, M.; Nudler, E.; Severinov, K.; Borukhov, S.; Komissarova, N.; Goldfarb, A. Histidine-tagged RNA polymerase of Escherichia coli and transcription in solid phase. In RNA Polymerase and Associated Factors, Part B; Academic Press: Cambridge, MA, USA, 1996; Volume 274, pp. 326–334. ISBN 0076-6879. [Google Scholar]
- Tang, H.; Kim, Y.; Severinov, K.; Goldfarb, A.; Ebright, R. Escherichia coli RNA polymerase holoenzyme: Rapid reconstitution from recombinant α, β, β′, and σ subunits. In RNA Polymerase and Associated Factors Part A; Academic Press: Cambridge, MA, USA, 1996; Volume 273, pp. 130–134. ISBN 0076-6879. [Google Scholar]
- Ceyssens, P.J.; Minakhin, L.; Van den Bossche, A.; Yakunina, M.; Klimuk, E.; Blasdel, B.; De Smet, J.; Noben, J.P.; Blasi, U.; Severinov, K.; et al. Development of Giant Bacteriophage KZ Is Independent of the Host Transcription Apparatus. J. Virol. 2014, 88, 10501–10510. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, R.; Lecoutere, E.; Wagemans, J.; Cenens, W.; Aertsen, A.; Schoofs, L.; Landuyt, B.; Paeshuyse, J.; Scheer, M.; Schobert, M.; et al. A multifaceted study of Pseudomonas aeruginosa shutdown by virulent podovirus LUZ19. mBio 2013, 4, e00061-13. [Google Scholar] [CrossRef] [Green Version]
- Gourse, R.L.; Gaal, T.; Bartlett, M.S.; Appleman, J.A.; Ross, W. rRNA transcription and growth rate-dependent regulation of ribosome synthesis in Escherichia coli. Annu. Rev. Microbiol. 1996, 50, 645–677. [Google Scholar] [CrossRef]
- Bleves, S.; Viarre, V.; Salacha, R.; Michel, G.P.F.; Filloux, A.; Voulhoux, R. Protein secretion systems in Pseudomonas aeruginosa: A wealth of pathogenic weapons. Int. J. Med. Microbiol. 2010, 300, 534–543. [Google Scholar] [CrossRef]
- Lewenza, S.; Falsafi, R.K.; Winsor, G.; Gooderham, W.J.; McPhee, J.B.; Brinkman, F.S.; Hancock, R.E. Construction of a mini-Tn5-luxCDABE mutant library in Pseudomonas aeruginosa PAO1: A tool for identifying differentially regulated genes. Genome Res. 2005, 15, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Sousa, M.C.; Trame, C.B.; Tsuruta, H.; Wilbanks, S.M.; Reddy, V.S.; McKay, D.B. Crystal and solution structures of an HslUV protease-chaperone complex. Cell 2000, 103, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulos, J.A.; Guarne, A.; Ortega, J. ClpP: A structurally dynamic protease regulated by AAA+ proteins. J. Struct. Biol. 2012, 179, 202–210. [Google Scholar] [CrossRef]
- Sauer, R.T.; Bolon, D.N.; Burton, B.M.; Burton, R.E.; Flynn, J.M.; Grant, R.A.; Hersch, G.L.; Joshi, S.A.; Kenniston, J.A.; Levchenko, I.; et al. Sculpting the proteome with AAA(+) proteases and disassembly machines. Cell 2004, 119, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Flynn, J.M.; Neher, S.B.; Kim, Y.I.; Sauer, R.T.; Baker, T.A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 2003, 11, 671–683. [Google Scholar] [CrossRef]
- Burton, R.E.; Baker, T.A.; Sauer, R.T. Nucleotide-dependent substrate recognition by the AAA+ HslUV protease. Nat. Struct. Mol. Biol. 2005, 12, 245–251. [Google Scholar] [CrossRef]
- Koodathingal, P.; Jaffe, N.E.; Kraut, D.A.; Prakash, S.; Fishbain, S.; Herman, C.; Matouschek, A. ATP-dependent proteases differ substantially in their ability to unfold globular proteins. J. Biol. Chem. 2009, 284, 18674–18684. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.M.; Sauer, R.T.; Baker, T.A. Role of the processing pore of the ClpX AAA+ ATPase in the recognition and engagement of specific protein substrates. Genes Dev. 2004, 18, 369–374. [Google Scholar] [CrossRef] [Green Version]
- Farrell, C.M.; Baker, T.A.; Sauer, R.T. Altered specificity of a AAA+ protease. Mol. Cell 2007, 25, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Camara, B.; Liu, M.; Reynolds, J.; Shadrin, A.; Liu, B.; Kwok, K.; Simpson, P.; Weinzierl, R.; Severinov, K.; Cota, E.; et al. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymerase by antagonizing stable DNA strand separation near the transcription start site. Proc. Natl. Acad. Sci. USA 2010, 107, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- James, E.; Liu, M.; Sheppard, C.; Mekler, V.; Camara, B.; Liu, B.; Simpson, P.; Cota, E.; Severinov, K.; Matthews, S.; et al. Structural and mechanistic basis for the inhibition of Escherichia coli RNA polymerase by T7 Gp2. Mol. Cell 2012, 47, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Osmundson, J.; Montero-Diez, C.; Westblade, L.F.; Hochschild, A.; Darst, S.A. Promoter-specific transcription inhibition in Staphylococcus aureus by a phage protein. Cell 2012, 151, 1005–1016. [Google Scholar] [CrossRef] [Green Version]
- Wood, L.F.; Tszine, N.Y.; Christie, G.E. Activation of P2 late transcription by P2 Ogr protein requires a discrete contact site on the C terminus of the alpha subunit of Escherichia coli RNA polymerase. J. Mol. Biol. 1997, 274, 1–7. [Google Scholar] [CrossRef]
- Hirvonen, C.A.; Ross, W.; Wozniak, C.E.; Marasco, E.; Anthony, J.R.; Aiyar, S.E.; Newburn, V.H.; Gourse, R.L. Contributions of UP elements and the transcription factor FIS to expression from the seven rrn P1 promoters in Escherichia coli. J. Bacteriol. 2001, 183, 6305–6314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelman, J.T.; Gourse, R.L. Open complex DNA scrunching: A key to transcription start site selection and promoter escape. Bioessays 2017, 39, 1600193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrem, S.T.; Ross, W.; Gaal, T.; Chen, Z.W.; Niu, W.; Ebright, R.H.; Gourse, R.L. Bacterial promoter architecture: Subsite structure of UP elements and interactions with the carboxy-terminal domain of the RNA polymerase alpha subunit. Genes Dev. 1999, 13, 2134–2147. [Google Scholar] [CrossRef] [PubMed]
- Ross, W.; Gourse, R.L. Sequence-independent upstream DNA-alphaCTD interactions strongly stimulate Escherichia coli RNA polymerase-lacUV5 promoter association. Proc. Natl. Acad. Sci. USA 2005, 102, 291–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montero-Diez, C.; Deighan, P.; Osmundson, J.; Darst, S.A.; Hochschild, A. Phage-encoded inhibitor of Staphylococcus aureus transcription exerts context-dependent effects on promoter function in a modified Escherichia coli-based transcription system. J. Bacteriol. 2013, 195, 3621–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deutscher, M.P. Degradation of stable RNA in bacteria. J. Biol. Chem. 2003, 278, 45041–45044. [Google Scholar] [CrossRef] [Green Version]
- Kochanowski, K.; Sauer, U.; Noor, E. Posttranslational regulation of microbial metabolism. Curr. Opin. Microbiol. 2015, 27, 10–17. [Google Scholar] [CrossRef]
- Trentini, D.B.; Suskiewicz, M.J.; Heuck, A.; Kurzbauer, R.; Deszcz, L.; Mechtler, K.; Clausen, T. Arginine phosphorylation marks proteins for degradation by a Clp protease. Nature 2016, 539, 48–53. [Google Scholar] [CrossRef]
- Lima, B.P.; Antelmann, H.; Gronau, K.; Chi, B.K.; Becher, D.; Brinsmade, S.R.; Wolfe, A.J. Involvement of protein acetylation in glucose-induced transcription of a stress-responsive promoter. Mol. Microbiol. 2011, 81, 1190–1204. [Google Scholar] [CrossRef] [Green Version]
- Liang, W.; Malhotra, A.; Deutscher, M.P. Acetylation regulates the stability of a bacterial protein: Growth stage-dependent modification of RNase R. Mol. Cell 2011, 44, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Robertson, E.S.; Nicholson, A.W. Phosphorylation of Escherichia coli translation initiation factors by the bacteriophage T7 protein kinase. Biochemistry 1992, 31, 4822–4827. [Google Scholar] [CrossRef] [PubMed]
- Depping, R.; Lohaus, C.; Meyer, H.E.; Ruger, W. The mono-ADP-ribosyltransferases Alt and ModB of bacteriophage T4: Target proteins identified. Biochem. Biophys. Res. Commun. 2005, 335, 1217–1223. [Google Scholar] [CrossRef]
- Alawneh, A.M.; Qi, D.; Yonesaki, T.; Otsuka, Y. An ADP-ribosyltransferase Alt of bacteriophage T4 negatively regulates the Escherichia coliMazF toxin of a toxin-antitoxin module. Mol. Microbiol. 2016, 99, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Guan, X.; Li, N.; Zhang, F.; Zhu, Y.; Ren, K.; Yu, L.; Zhou, F.; Han, Z.; Gao, N.; et al. An anti-CRISPR protein disables type V Cas12a by acetylation. Nat. Struct. Mol. Biol. 2019, 26, 308–314. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceyssens, P.-J.; De Smet, J.; Wagemans, J.; Akulenko, N.; Klimuk, E.; Hedge, S.; Voet, M.; Hendrix, H.; Paeshuyse, J.; Landuyt, B.; et al. The Phage-Encoded N-Acetyltransferase Rac Mediates Inactivation of Pseudomonas aeruginosa Transcription by Cleavage of the RNA Polymerase Alpha Subunit. Viruses 2020, 12, 976. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090976
Ceyssens P-J, De Smet J, Wagemans J, Akulenko N, Klimuk E, Hedge S, Voet M, Hendrix H, Paeshuyse J, Landuyt B, et al. The Phage-Encoded N-Acetyltransferase Rac Mediates Inactivation of Pseudomonas aeruginosa Transcription by Cleavage of the RNA Polymerase Alpha Subunit. Viruses. 2020; 12(9):976. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090976
Chicago/Turabian StyleCeyssens, Pieter-Jan, Jeroen De Smet, Jeroen Wagemans, Natalia Akulenko, Evgeny Klimuk, Subray Hedge, Marleen Voet, Hanne Hendrix, Jan Paeshuyse, Bart Landuyt, and et al. 2020. "The Phage-Encoded N-Acetyltransferase Rac Mediates Inactivation of Pseudomonas aeruginosa Transcription by Cleavage of the RNA Polymerase Alpha Subunit" Viruses 12, no. 9: 976. https://0-doi-org.brum.beds.ac.uk/10.3390/v12090976